
Next- and Third-Generation Sequencing Outperforms Culture-Based Methods in the Diagnosis of Ascitic Fluid Bacterial Infections of ICU Patients


 ,
, 
Abstract
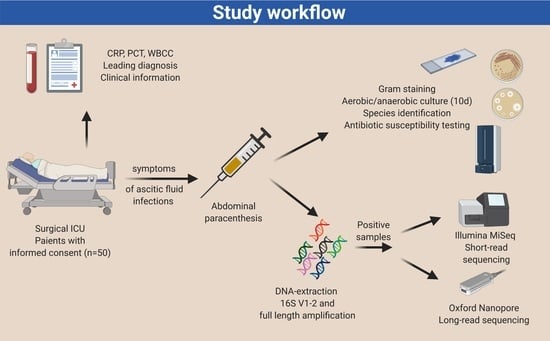
1. Introduction
2. Methods
2.1. Study Design and Ethics Statement
2.2. Clinical Information Acquisition
2.3. Microbiological Culture-Based Methods and Microscopy
2.4. Microbial Genomic DNA Preparation
2.5. Bacterial Sequencing Using Short-Read 16S rDNA Sequencing
2.6. Bacterial Sequencing Using Long-Read 16S rDNA Sequencing
2.7. Sequencing Analysis Pipeline
2.8. Data Visualization and Statistics
3. Results
3.1. Characteristics of the Study Cohort
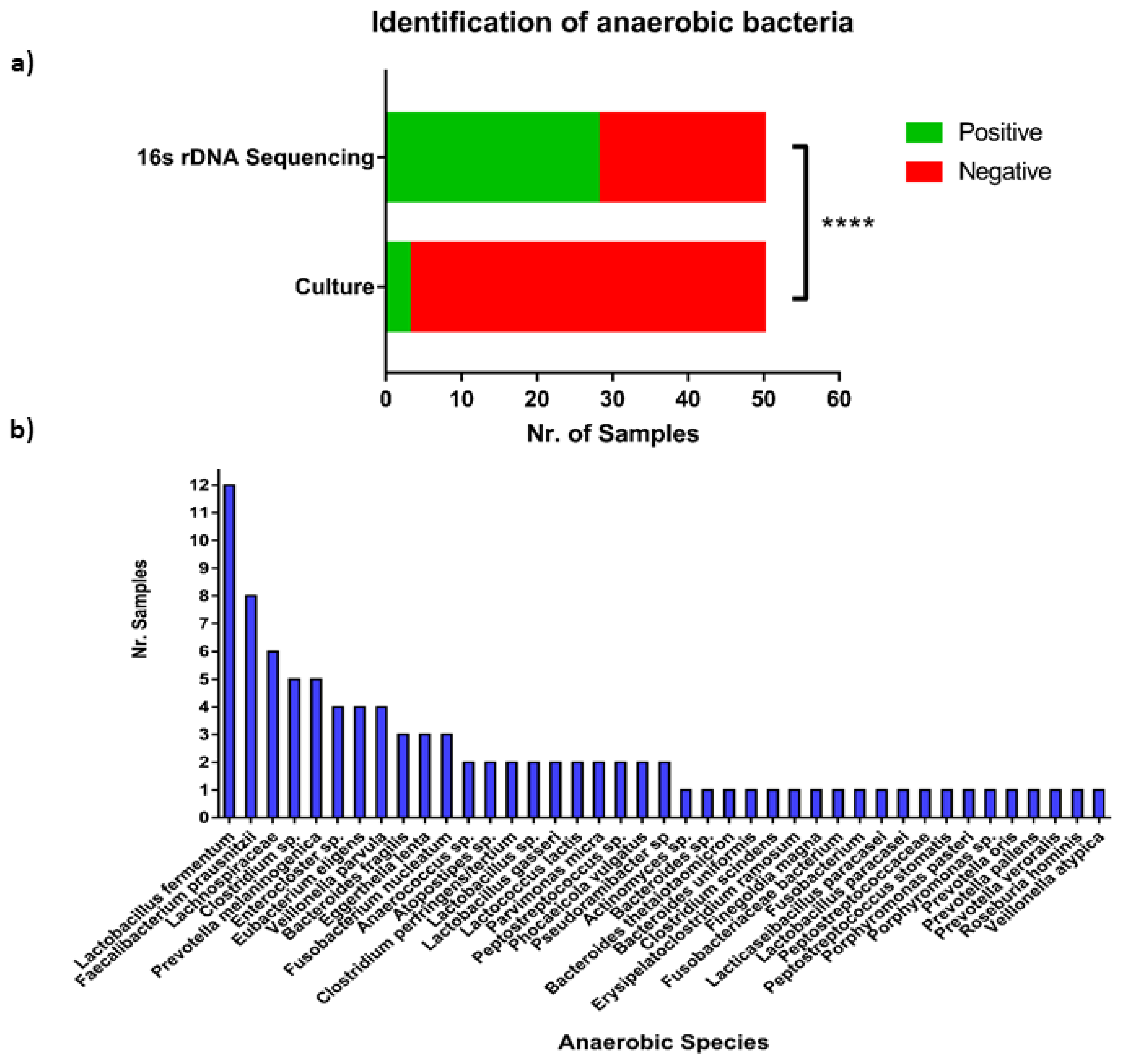
3.2. Culture of Ascites Samples
3.3. Generation of 16S rRNA Short and Long Read Sequencing Data
3.4. Clinical Evalution of Short- and Long-Read Sequencing Output Compared with Standard Microbiology Culture Results
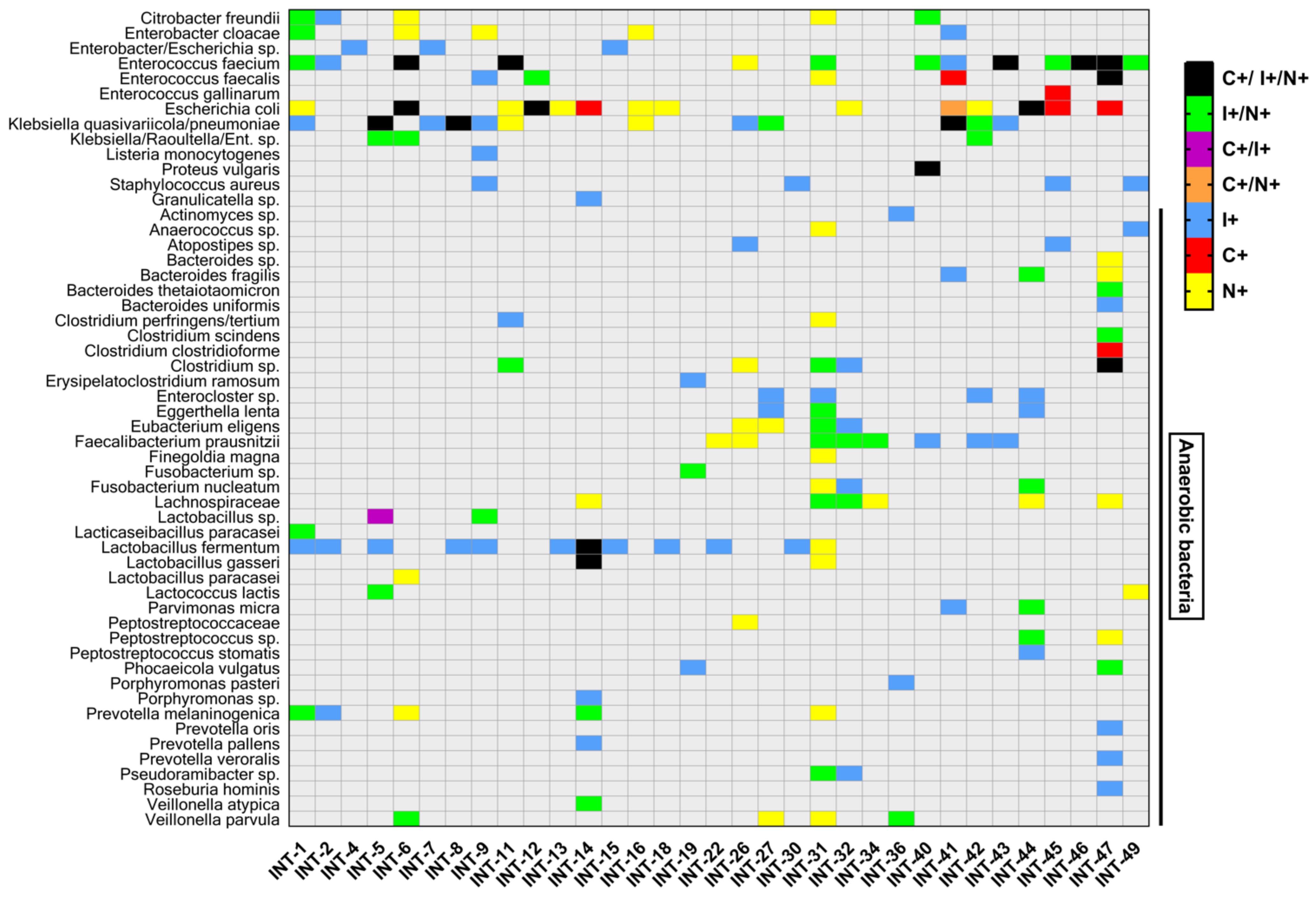
3.5. Anaerobic Bacteria Identification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pedersen, J.S.; Bendtsen, F.; Møller, S. Management of cirrhotic ascites. Ther. Adv. Chronic Dis. 2015, 6, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Ginés, P.; Quintero, E.; Arroyo, V.; Terés, J.; Bruguera, M.; Rimola, A.; Caballería, J.; Rodés, J.; Rozman, C. Compensated cirrhosis: Natural history and prognostic factors. Hepatology 1987, 7, 122–128. [Google Scholar] [CrossRef]
- Sarker, J.A.; Alam, M.S.; Khan, M.; Mahtab, M.-A.; Shahed Ashraf, M.; Ahmad Khondaker, F. Variant of Ascitic Fluid Bacterial Infections in Patients of Liver Cirrhosis. Euroasian J. Hepatogastroenterol. 2015, 5, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.-L.; Rello, J.; Marshall, J.; Silva, E.; Anzueto, A.; Martin, C.D.; Moreno, R.; Lipman, J.; Gomersall, C.; Sakr, Y.; et al. International study of the prevalence and outcomes of infection in intensive care units. JAMA 2009, 302, 2323–2329. [Google Scholar] [CrossRef] [PubMed]
- Montravers, P.; Gauzit, R.; Muller, C.; Marmuse, J.P.; Fichelle, A.; Desmonts, J.M. Emergence of Antibiotic-Resistant Bacteria in Cases of Peritonitis After Intraabdominal Surgery Affects the Efficacy of Empirical Antimicrobial Therapy. Clin. Infect. Dis. 1996, 23, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.C.; Innes, M. Intensive care unit management of intra-abdominal infection. Crit. Care Med. 2003, 31, 2228–2237. [Google Scholar] [CrossRef] [PubMed]
- MacIntosh, T. Emergency Management of Spontaneous Bacterial Peritonitis—A Clinical Review. Cureus 2018, 10, e2253. [Google Scholar] [CrossRef] [PubMed]
- Sartelli, M.; Viale, P.; Catena, F.; Ansaloni, L.; Moore, E.; Malangoni, M.; Moore, F.A.; Velmahos, G.; Coimbra, R.; Ivatury, R.; et al. 2013 WSES guidelines for management of intra-abdominal infections. World J. Emerg. Surg. 2013, 8, 3. [Google Scholar] [CrossRef]
- Orman, E.S.; Hayashi, P.H.; Bataller, R.; Barritt, A.S. Paracentesis is Associated with Reduced Mortality in Patients Hospitalized with Cirrhosis and Ascites. Clin. Gastroenterol. Hepatol. 2014, 12, 496–503.e1. [Google Scholar] [CrossRef]
- Gaieski, D.F.; Mikkelsen, M.E.; Band, R.A.; Pines, J.M.; Massone, R.; Furia, F.F.; Frances, S.S.; Munish, G. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit. Care Med. 2010, 38, 1045–1053. [Google Scholar] [CrossRef]
- Carey-Ann, B.D.; Carroll, K.C. Diagnosis of Clostridium difficile Infection: An Ongoing Conundrum for Clinicians and for Clinical Laboratories. Clin. Microbiol. Rev. 2013, 26, 604–630. [Google Scholar] [CrossRef]
- European Association for The Study of the Liver. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J. Hepatol. 2010, 53, 397–417. [Google Scholar] [CrossRef]
- Barreales, M.; Fernández, I. Spontaneous bacterial peritonitis. Rev. Esp. Enferm. Dig. 2011, 103, 255–263. [Google Scholar] [CrossRef][Green Version]
- Rogers, G.B.; Russell, L.E.; Preston, P.G.; Marsh, P.; Collins, J.E.; Saunders, J.; Sutton, J.; Fine, D.; Bruce, K.D.; Wright, M. Characterisation of bacteria in ascites—Reporting the potential of culture-independent, molecular analysis. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 533–541. [Google Scholar] [CrossRef][Green Version]
- Wiest, R.; Garcia-Tsao, G. Bacterial translocation (BT) in cirrhosis. Hepatology 2005, 41, 422–433. [Google Scholar] [CrossRef]
- Walker, A.W.; Duncan, S.H.; Louis, P.; Flint, H.J. Phylogeny, culturing, and metagenomics of the human gut microbiota. Trends Microbiol. 2014, 22, 267–274. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, C.-L.; Chen, T.-H.; Liang, Y.-H.; Chen, H.-L.; Lin, C.-Y.; Chiu, C.-H. Application of next-generation sequencing to study ascitic microbiome in cirrhotic patients with or without spontaneous bacterial peritonitis. J. Microbiol. Immunol. Infect. 2015, 48, 504–509. [Google Scholar] [CrossRef]
- Santiago, A.; Pozuelo, M.; Poca, M.; Gely, C.; Nieto, J.C.; Torras, X.; Román, E.; Campos, D.; Sarrabayrouse, G.; Vidal, S. Alteration of the serum microbiome composition in cirrhotic patients with ascites. Sci. Rep. 2016, 6, 25001. [Google Scholar] [CrossRef]
- Ascitic Microbiota Composition Is Correlated with Clinical Severity in Cirrhosis with Portal Hypertension. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3783492/ (accessed on 5 May 2019).
- Jeong, J.; Yun, K.; Mun, S.; Chung, W.-H.; Choi, S.-Y.; Nam, Y.; Lim, M.Y.; Hong, C.P.; Park, C.H.; Ahn, Y.J.; et al. The effect of taxonomic classification by full-length 16S rRNA sequencing with a synthetic long-read technology. Sci. Rep. 2021, 11, 1727. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]
- A Portable System for Rapid Bacterial Composition Analysis Using a Nanopore-Based Sequencer and Laptop Computer. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5516037/ (accessed on 23 September 2018).
- Gu, W.; Deng, X.; Lee, M.; Sucu, Y.D.; Arevalo, S.; Stryke, D.; Federman, S.; Gopez, A.; Reyes, K.; Zorn, K.; et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat. Med. 2020, 27, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Charalampous, T.; Kay, G.L.; Richardson, H.; Aydin, A.; Baldan, R.; Jeanes, C.; Rae, D.; Grundy, S.; Turner, D.J.; Wain, J.; et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 2019, 37, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Taxt, A.M.; Avershina, E.; Frye, S.A.; Naseer, U.; Ahmad, R. Rapid identification of pathogens, antibiotic resistance genes and plasmids in blood cultures by nanopore sequencing. Sci. Rep. 2020, 10, 7622. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.S.; Au, C.H.; Leung, S.M.; Ho, D.N.; Wong, E.Y.L.; To, M.Y.; Ng, M.K.; Chan, T.L.; Ma, E.S.K.; Tang, B.S.F. Potential utility of targeted Nanopore sequencing for improving etiologic diagnosis of bacterial and fungal respiratory infection. Diagn. Pathol. 2020, 15, 41. [Google Scholar] [CrossRef]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.-E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC A Quality Control tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 May 2018).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Callahan, B.J.; Sankaran, K.; Fukuyama, J.A.; McMurdie, P.J.; Holmes, S.P. Bioconductor workflow for microbiome data analysis: From raw reads to community analyses. F1000Research 2016, 5, 1492. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.-A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Fan, J.; Huang, S.; Chorlton, S.D. BugSeq: A highly accurate cloud platform for long-read metagenomic analyses. BMC Bioinform. 2021, 22, 160. [Google Scholar] [CrossRef]
- Jung, A.; Chorlton, S.D. BugSeq 16S: NanoCLUST with Improved Consensus Sequence Classification. March 2021, p. 2021.03.16.434153. Available online: https://www.biorxiv.org/content/10.1101/2021.03.16.434153v1 (accessed on 2 September 2021).
- Kolde, R. Pheatmap: Pretty Heatmaps. 2019. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 15 September 2021).
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Use R! Springer: New York, NY, USA, 2009; Available online: https://www.springer.com/de/book/9780387981413 (accessed on 15 September 2020).
- Arvaniti, V.; D’Amico, G.; Fede, G.; Manousou, P.; Tsochatzis, E.; Pleguezuelo, M.; Burroughs, A.K. Infections in Patients with Cirrhosis Increase Mortality Four-Fold and Should Be Used in Determining Prognosis. Gastroenterology 2010, 139, 1246–1256.e5. [Google Scholar] [CrossRef]
- Runyon, B.A. Management of adult patients with ascites due to cirrhosis: An update. Hepatology 2009, 49, 2087–2107. [Google Scholar] [CrossRef]
- Weyrich, L.S.; Farrer, A.G.; Eisenhofer, R.; Arriola, L.A.; Young, J.; Selway, C.A.; Handsley-Davis, M.; Adler, C.J.; Breen, J.; Cooper, A. Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 2019, 19, 982–996. [Google Scholar] [CrossRef]
- Selway, C.A.; Eisenhofer, R.; Weyrich, L.S. Microbiome applications for pathology: Challenges of low microbial biomass samples during diagnostic testing. J. Pathol. Clin. Res. 2020, 6, 97–106. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef]
- Laurence, M.; Hatzis, C.; Brash, D.E. Common Contaminants in Next-Generation Sequencing That Hinder Discovery of Low-Abundance Microbes. PLoS ONE 2014, 9, e97876. [Google Scholar]
- Tay, P.W.L.; Xiao, J.; Tan, D.J.H.; Ng, C.; Lye, Y.N.; Lim, W.H.; Teo, V.X.Y.; Heng, R.R.Y.; Yeow, M.W.X.; Lum, L.H.W.; et al. An Epidemiological Meta-Analysis on the Worldwide Prevalence, Resistance, and Outcomes of Spontaneous Bacterial Peritonitis in Cirrhosis. Front. Med. 2021, 8, 693652. [Google Scholar] [CrossRef]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. Msphere 2021, 6, e01202-20. [Google Scholar] [CrossRef]
- Dever, J.B.; Sheikh, M.Y. Review article: Spontaneous bacterial peritonitis—Bacteriology, diagnosis, treatment, risk factors and prevention. Aliment. Pharmacol. Ther. 2015, 41, 1116–1131. [Google Scholar] [CrossRef]
- Codoñer, F.M.; Ramírez-Bosca, A.; Climent, E.; Carrión-Gutierrez, M.; Guerrero, M.; Pérez-Orquín, J.M.; de la Parte, J.H.; Genovés, S.; Ramón, D.; Navarro-López, V.; et al. Gut microbial composition in patients with psoriasis. Sci. Rep. 2018, 8, 3812. [Google Scholar] [CrossRef]
- Iljazovic, A.; Roy, U.; Gálvez, E.J.C.; Lesker, T.R.; Zhao, B.; Gronow, A.; Amend, L.; Will, S.E.; Hofmann, J.D.; Pils, M.; et al. Perturbation of the gut microbiome by Prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal Immunol. 2021, 14, 113–124. [Google Scholar] [CrossRef]
- Meta-Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer—Nature Medicine. Available online: https://www.nature.com/articles/s41591-019-0406-6 (accessed on 12 August 2021).
- Badr, M.T.; Blümel, B.; Baumgartner, S.; Komp, J.M.A.; Häcker, G. Antimicrobial Susceptibility Patterns and Wild-Type MIC Distributions of Anaerobic Bacteria at a German University Hospital: A Five-Year Retrospective Study (2015–2019). Antibiotics 2020, 9, 823. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Culture/16s-pos | Culture-neg/16s-pos | Culture/16s-neg | p-Value | |
|---|---|---|---|---|
| N | 13 | 22 | 14 | |
| Age (years) | 63 (52.5–73) | 72 (53.75–79) | 62 (55.5–71) | 0.45 |
| Sex (male) | 10 (77%) | 12 (55%) | 11 (79%) | 0.23 |
| Leucocytes (Tsd) | 17.84 (9.85–25.98) | 12.03 (8.733–22.1) | 15.95 (10.63–17.79) | 0.74 |
| CRP | 126 (61.65–293.9) | 141.1 (78.4–197.2) | 103.3 (61.15–144.8) | 0.47 |
| PCT | 1.02 (0.715–1.715) | 2.575 (0.415–7.983) | 1.35 (0.3875–4.323) | 0.56 |
| Alcoholism | 1 (8%) | 6 (32%) | 2 (17%) | 0.27 |
| Smoking | 3 (23%) | 9 (45%) | 2 (18%) | 0.22 |
| Granulocytes (microscopic) | 3 (1.5–3) | 2.5 (1–3) | 1.5 (1–2.25) | 0.22 |
| Hospital stay after paracenthesis (d) | 27.5 (10.5–35) | 14.5 (10.75–29.5) | 12.5 (8.75–28) | 0.48 |
| ICU stay after paracenthesis (d) | 4 (1.5–8.5) | 4 (1.75–12) | 2 (0.75–5.75) | 0.33 |
| 6-day evaluation | 3.5 (3–4) | 4 (3–4) | 3 (3–3.25) | 0.31 |
| ICU discharge (alive) | 10 (77%) | 17 (77%) | 14 (100%) | 0.15 |
| Intestinal ischemia | 2 (15%) | 6 (27%) | 0 (0%) | 0.1 |
| Tumor | 6 (46%) | 13 (59%) | 9 (64%) | 0.62 |
| Peritonitis | 8 (62%) | 7 (32%) | 1 (7%) | 0.01 |
| Cirrhosis | 1 (8%) | 1 (5%) | 2 (14%) | 0.58 |
| Antibiotictherapy (+5 d) | 11 (92%) | 12 (63%) | 9 (64%) | 0.19 |
| Blood culture positivity (±5 d) | 4 (40%) | 5 (29%) | 1 (13%) | 0.44 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goelz, H.; Wetzel, S.; Mehrbarzin, N.; Utzolino, S.; Häcker, G.; Badr, M.T. Next- and Third-Generation Sequencing Outperforms Culture-Based Methods in the Diagnosis of Ascitic Fluid Bacterial Infections of ICU Patients. Cells 2021, 10, 3226. https://doi.org/10.3390/cells10113226
Goelz H, Wetzel S, Mehrbarzin N, Utzolino S, Häcker G, Badr MT. Next- and Third-Generation Sequencing Outperforms Culture-Based Methods in the Diagnosis of Ascitic Fluid Bacterial Infections of ICU Patients. Cells. 2021; 10(11):3226. https://doi.org/10.3390/cells10113226
Chicago/Turabian StyleGoelz, Hanna, Simon Wetzel, Negin Mehrbarzin, Stefan Utzolino, Georg Häcker, and Mohamed Tarek Badr. 2021. "Next- and Third-Generation Sequencing Outperforms Culture-Based Methods in the Diagnosis of Ascitic Fluid Bacterial Infections of ICU Patients" Cells 10, no. 11: 3226. https://doi.org/10.3390/cells10113226
APA StyleGoelz, H., Wetzel, S., Mehrbarzin, N., Utzolino, S., Häcker, G., & Badr, M. T. (2021). Next- and Third-Generation Sequencing Outperforms Culture-Based Methods in the Diagnosis of Ascitic Fluid Bacterial Infections of ICU Patients. Cells, 10(11), 3226. https://doi.org/10.3390/cells10113226