
Cell-Free DNA Variant Sequencing Using Plasma and AR-V7 Testing of Circulating Tumor Cells in Prostate Cancer Patients

 ,
,  ,
, 

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Tumor Material
2.2. Sampling of Blood, Isolation of CTCs, and Processing of Plasma
2.3. Library Construction and Sequencing
2.4. Data Analysis and Bioinformatics Analysis
2.5. Isolation of mRNA and qRT–PCR
2.6. Statistical Analysis
3. Results
3.1. Analysis of Plasma-Derived cfDNA
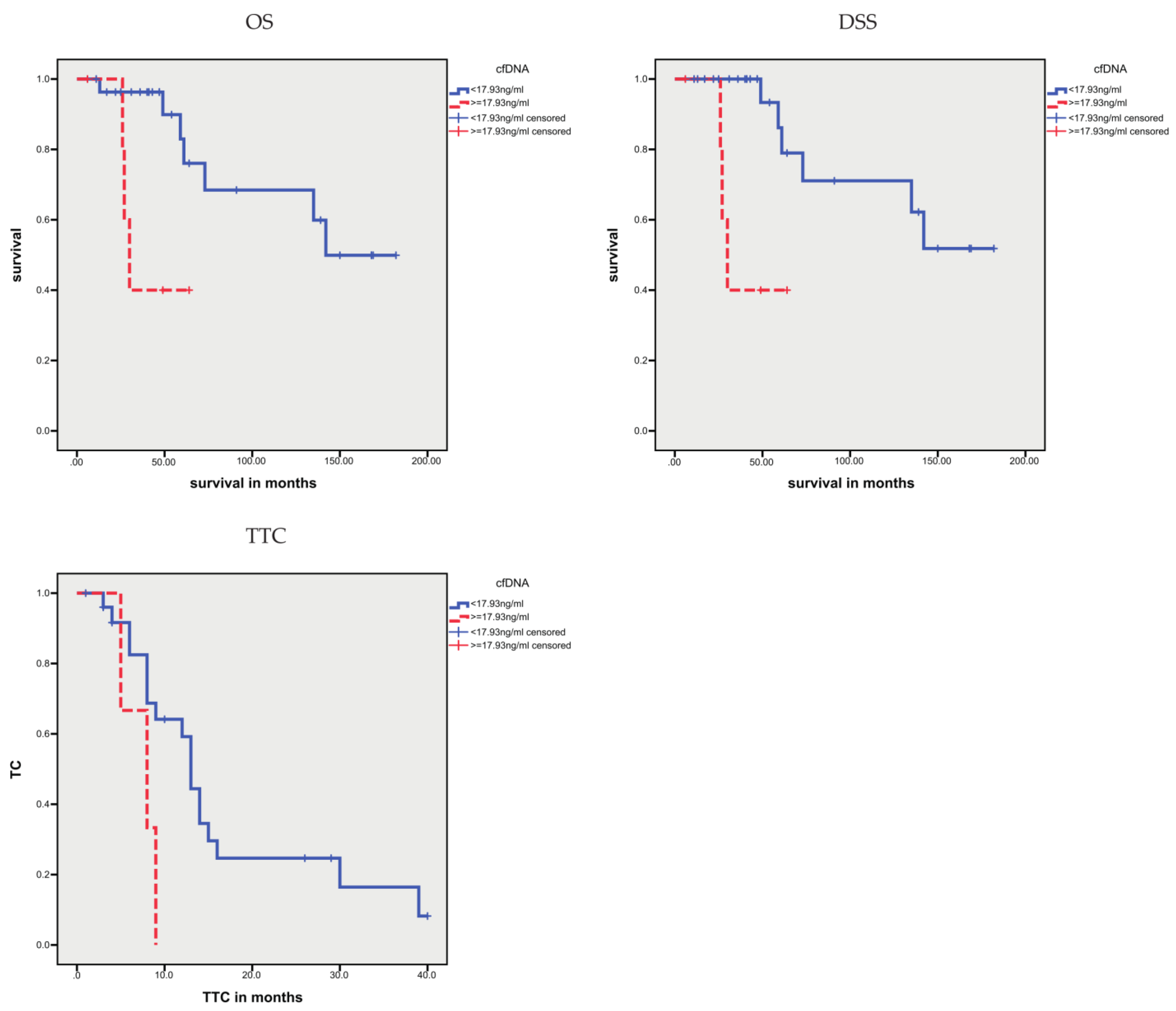
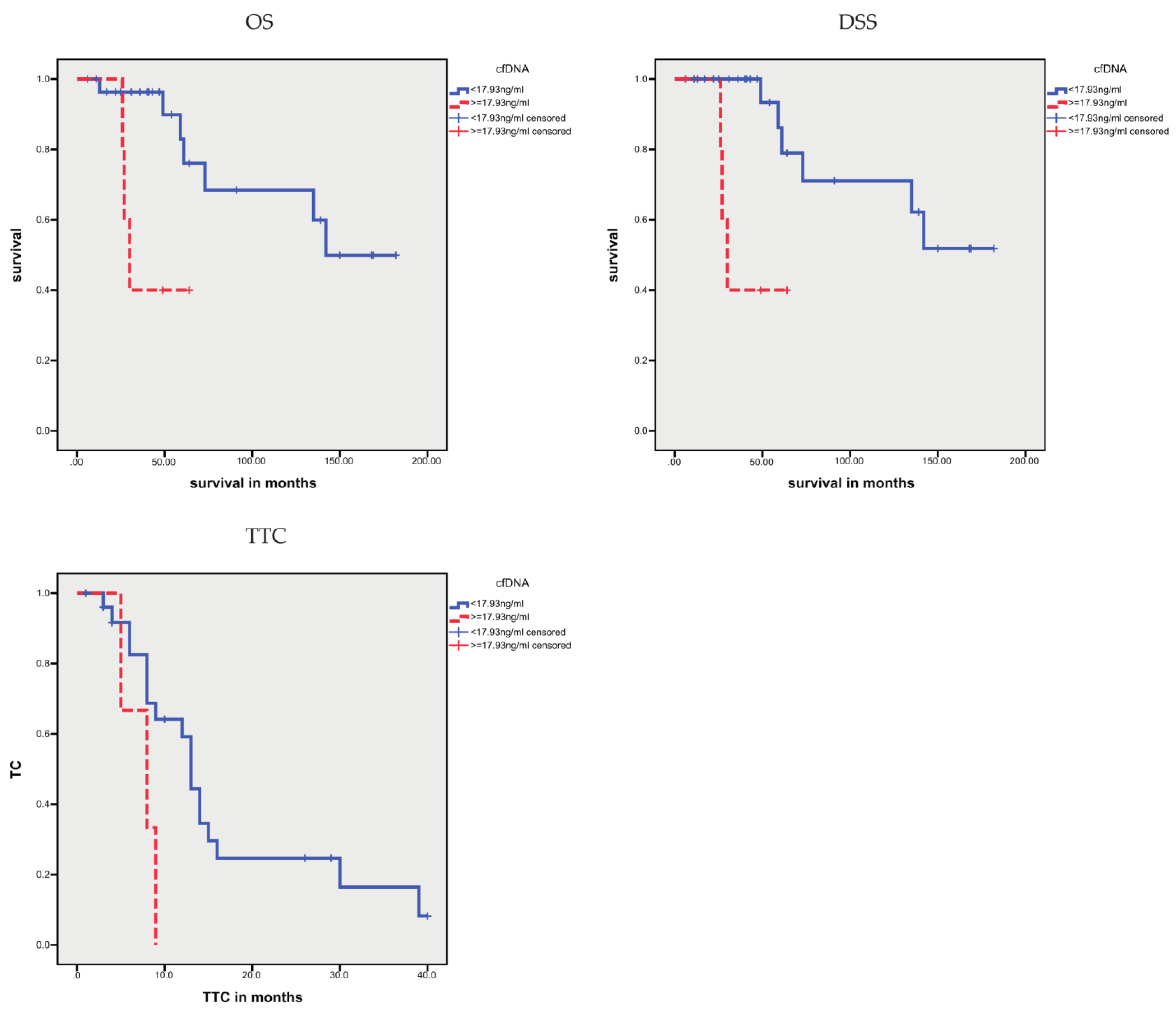
3.1.1. Correlation of cfDNA Concentration with Clinicopathological and Molecular Data
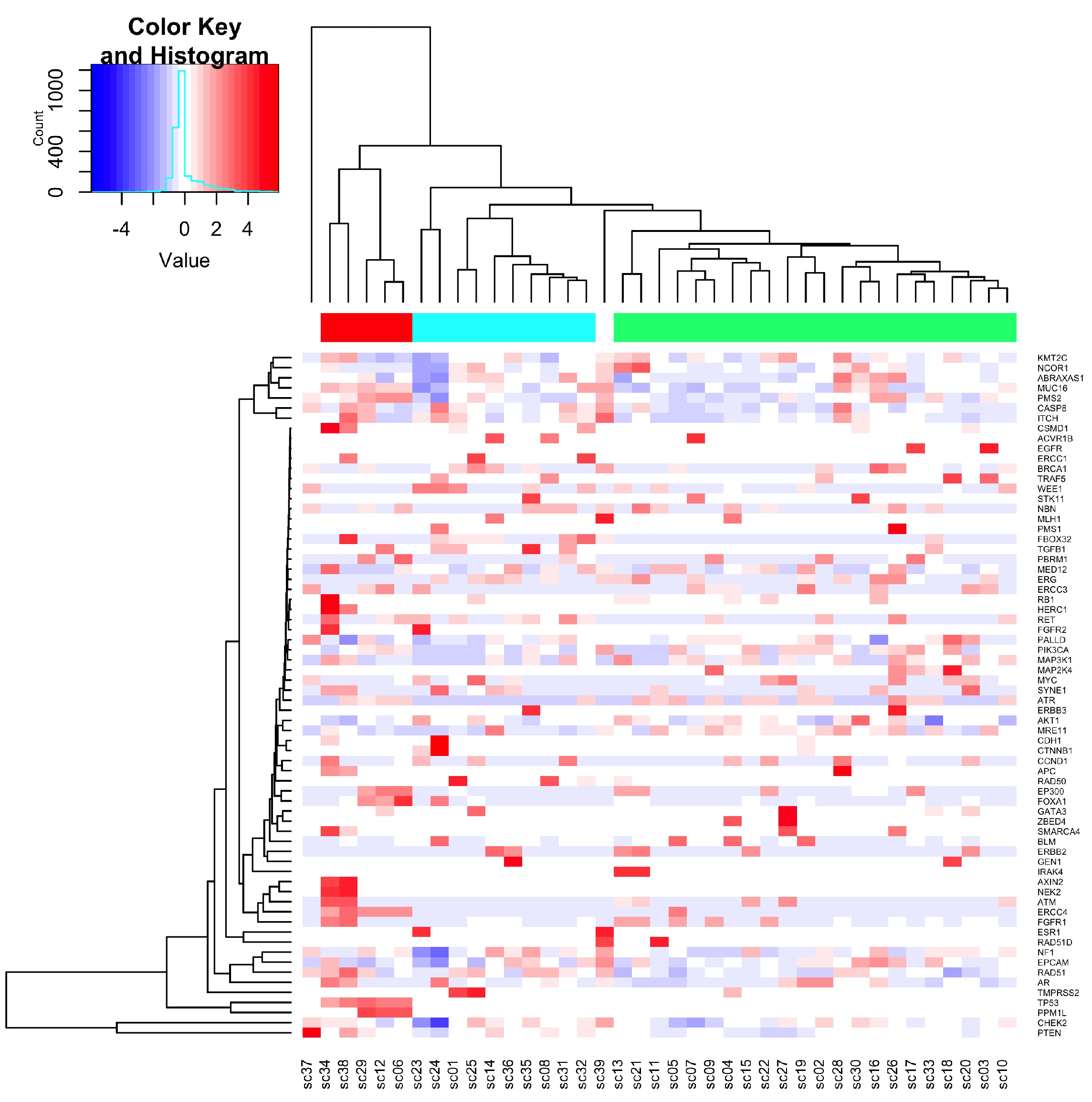
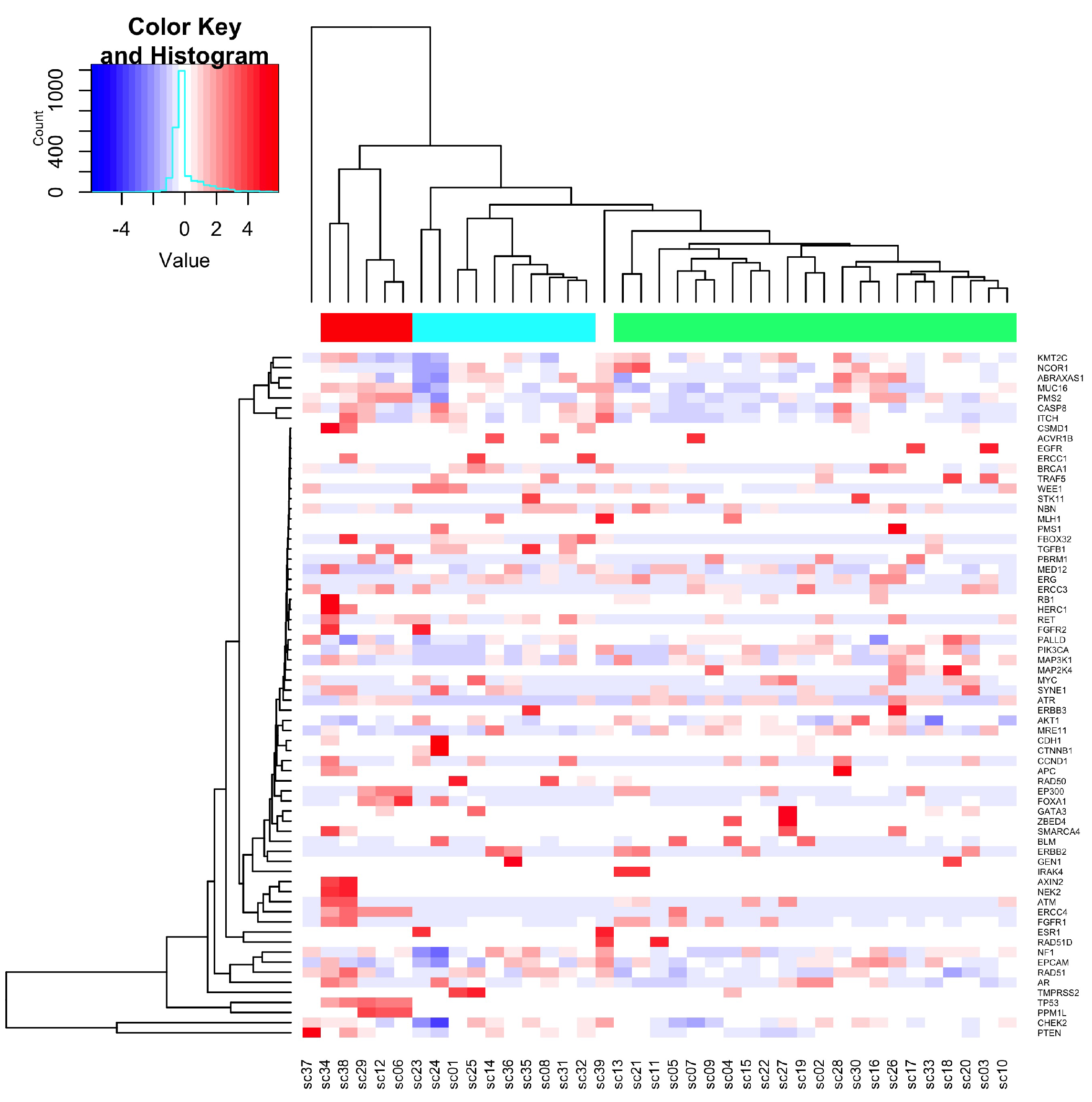
3.1.2. Sequence Variants Detected in cfDNA
3.1.3. Frequency of Variants
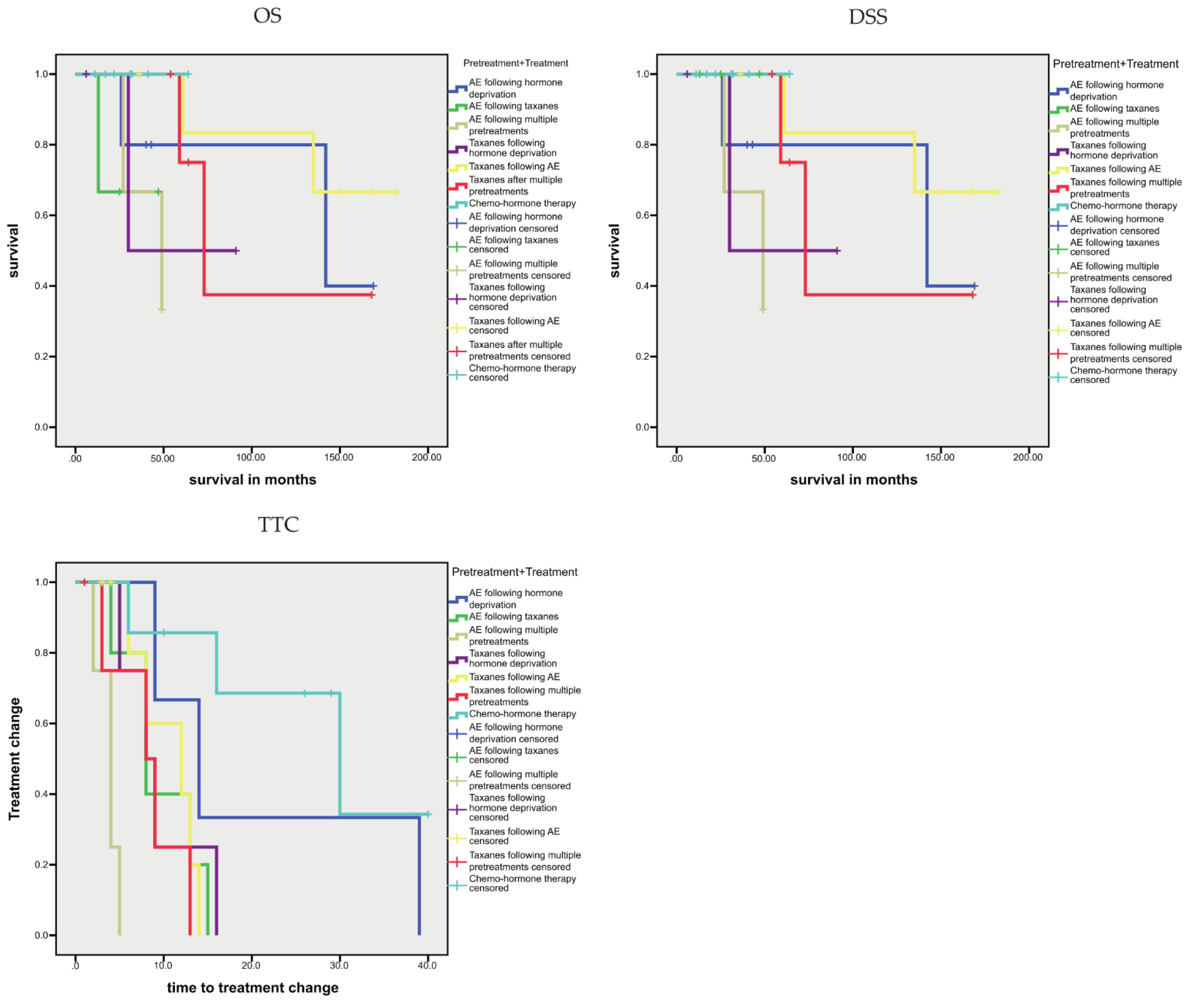
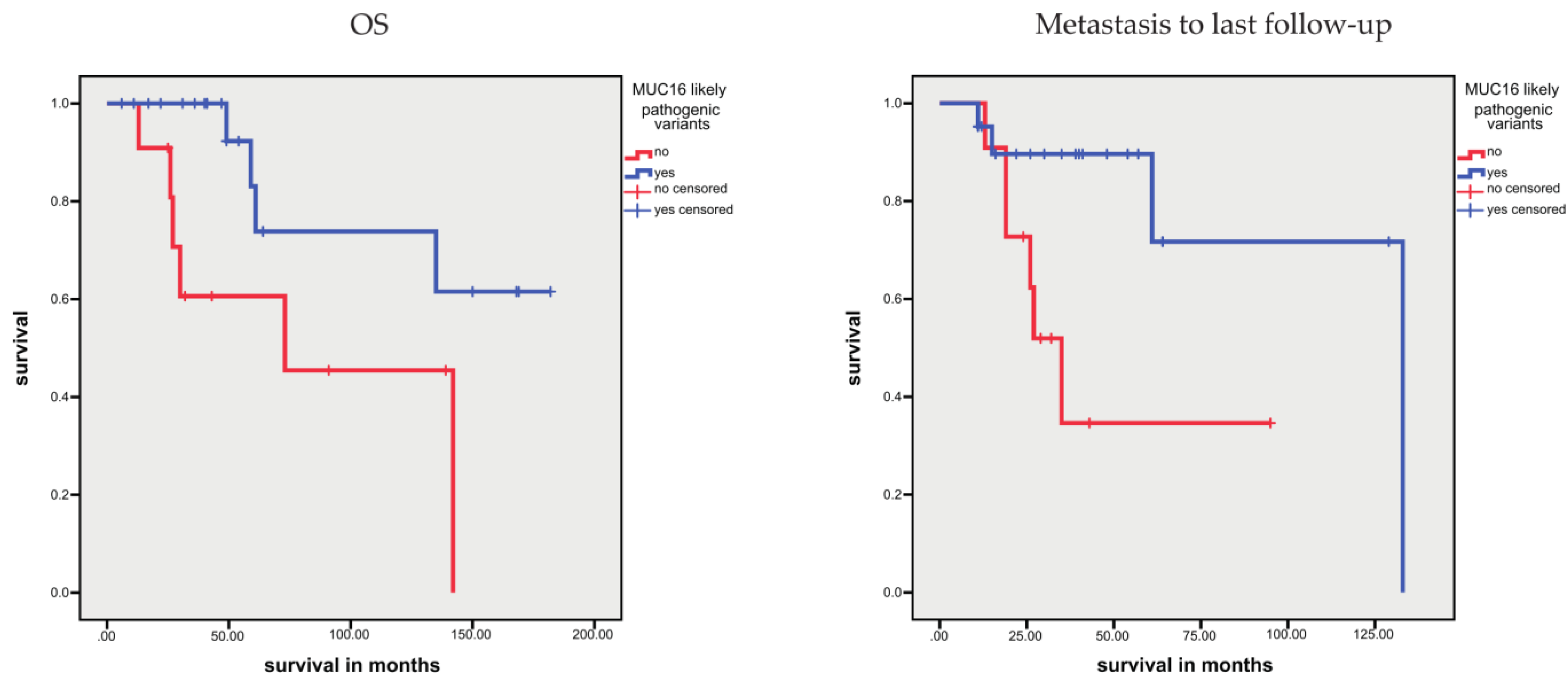
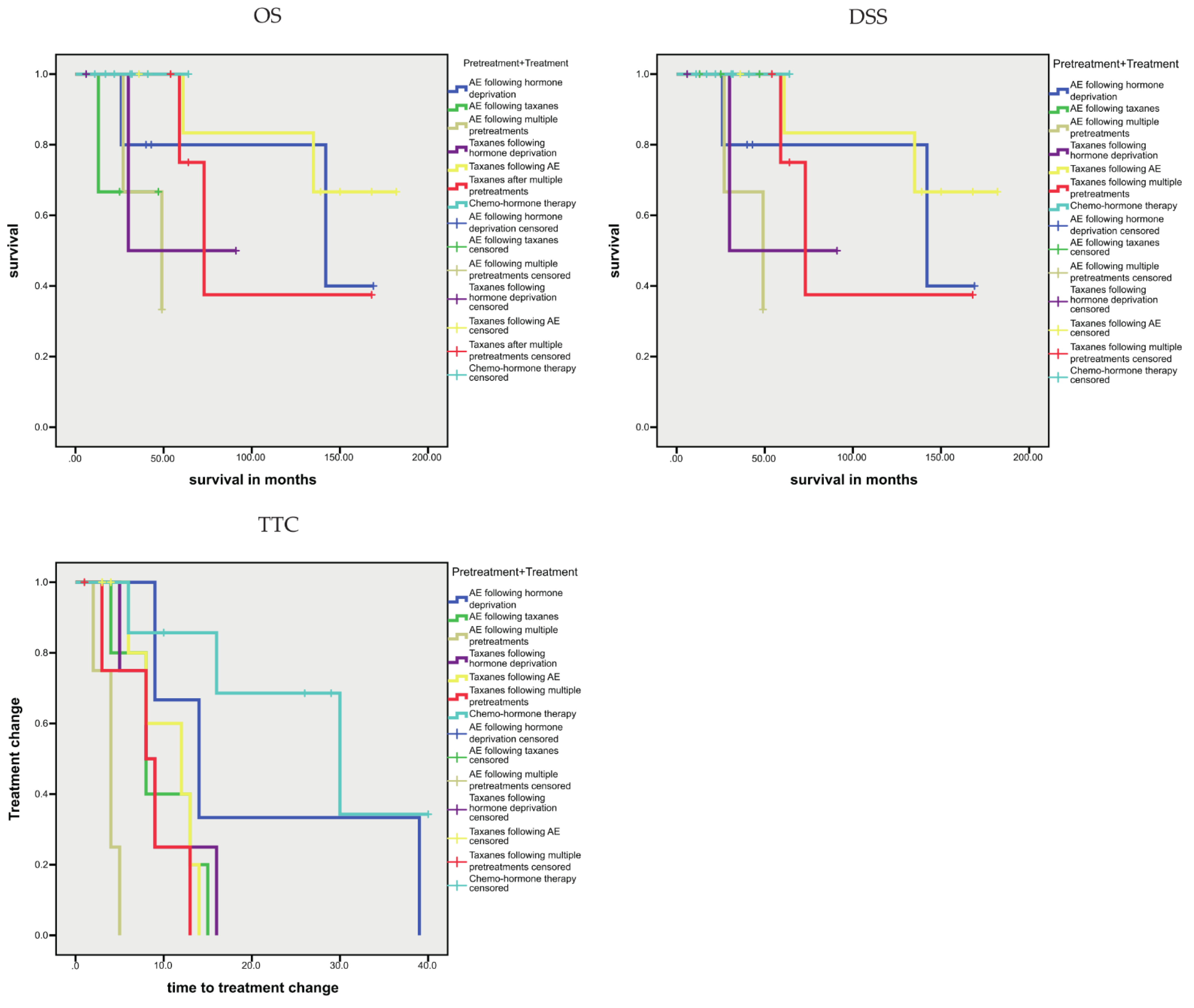
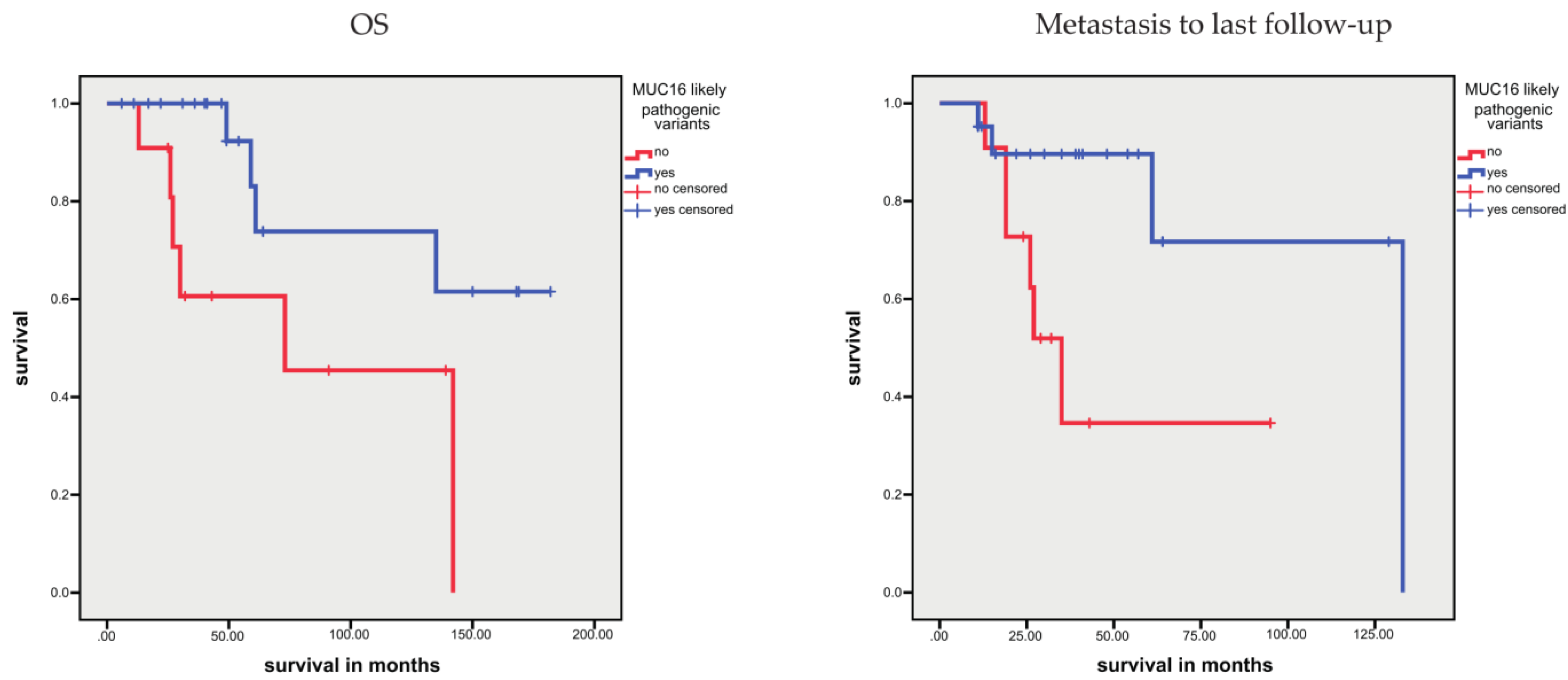
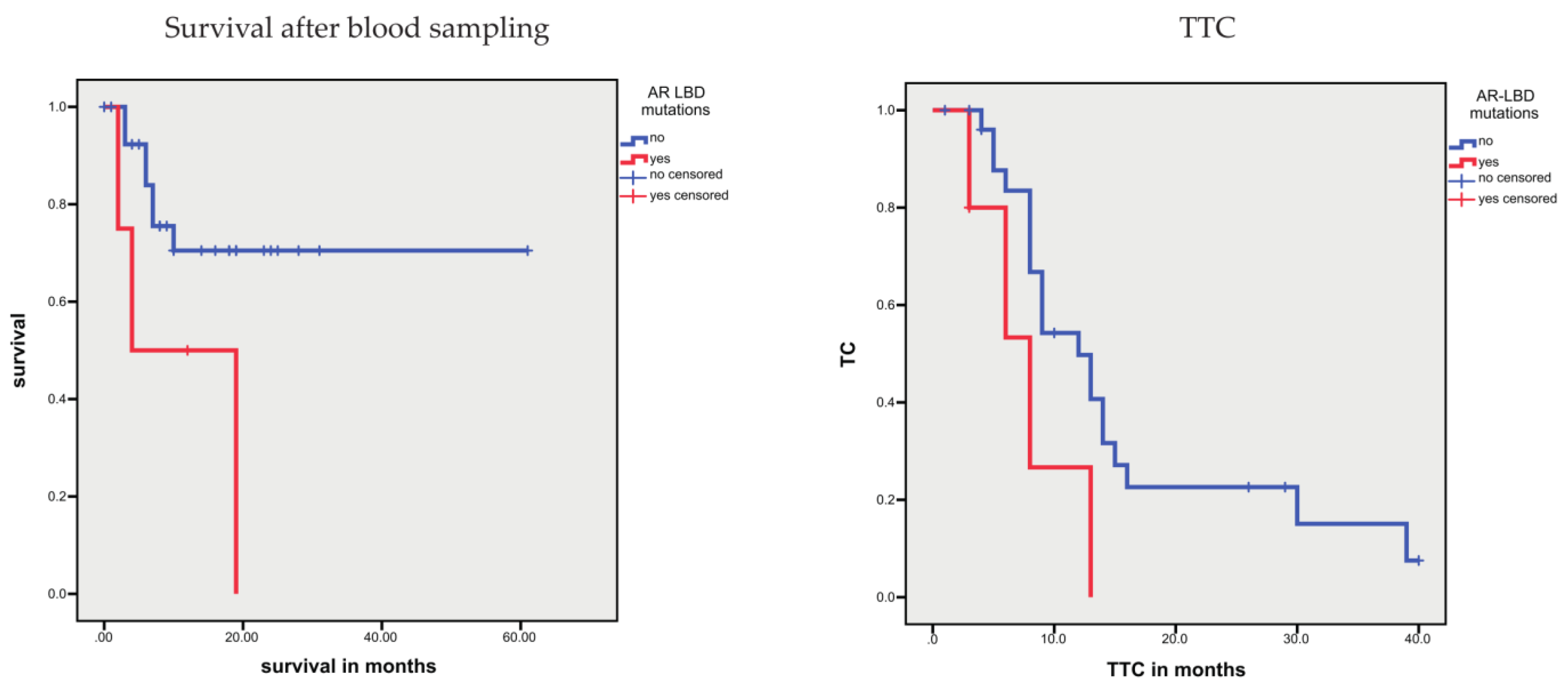
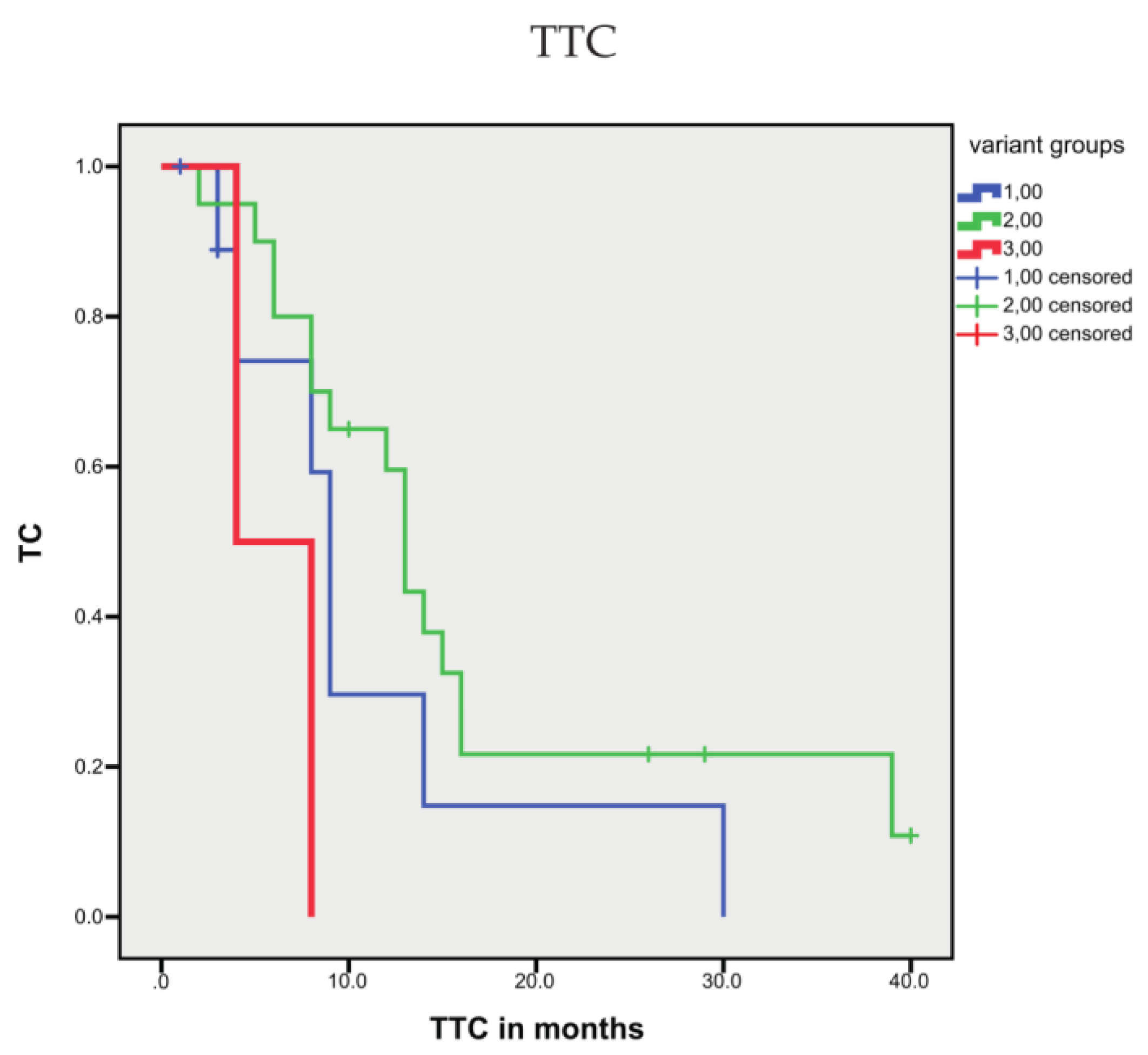
3.1.4. Association between Sequence Variants and Prognosis
3.2. Analysis of CTCs
3.2.1. Isolation of CTCs and Correlation with Clinicopathological and Molecular Data
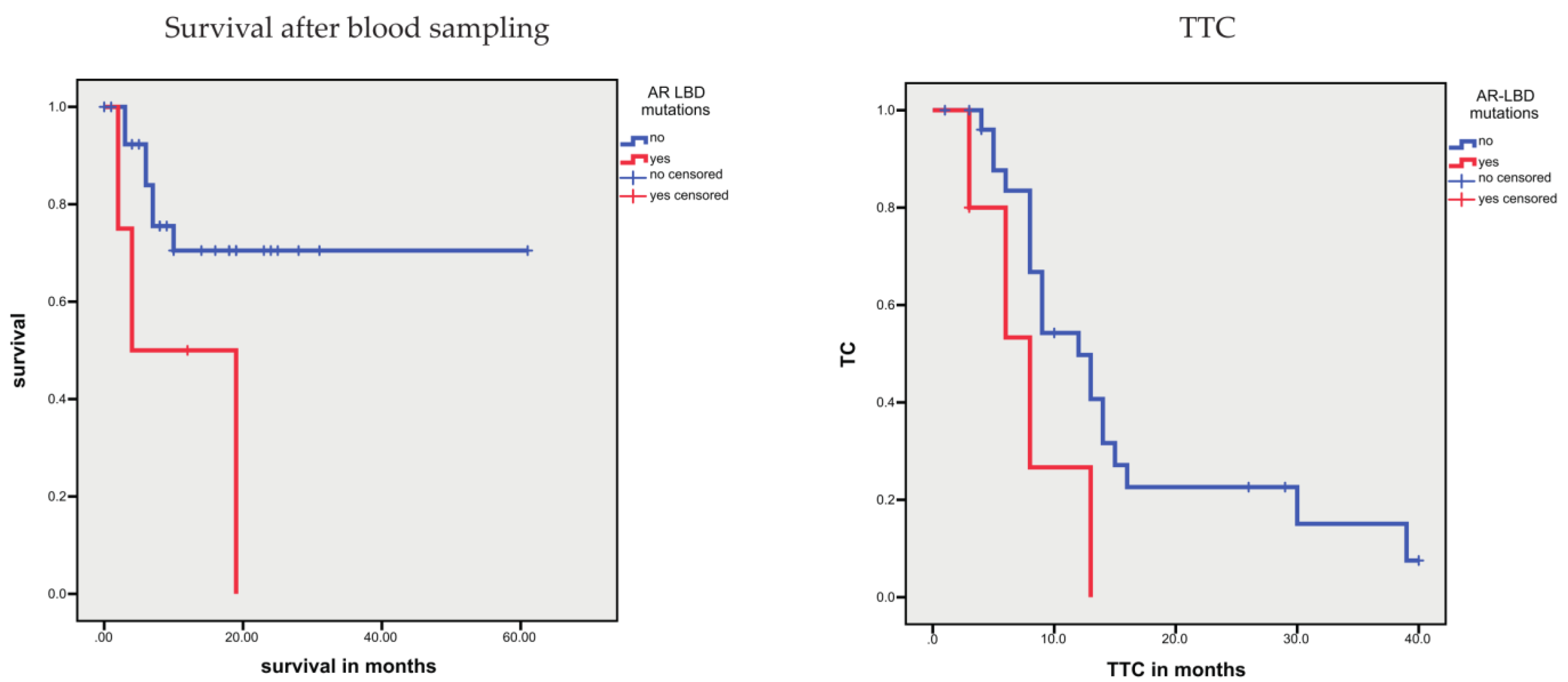
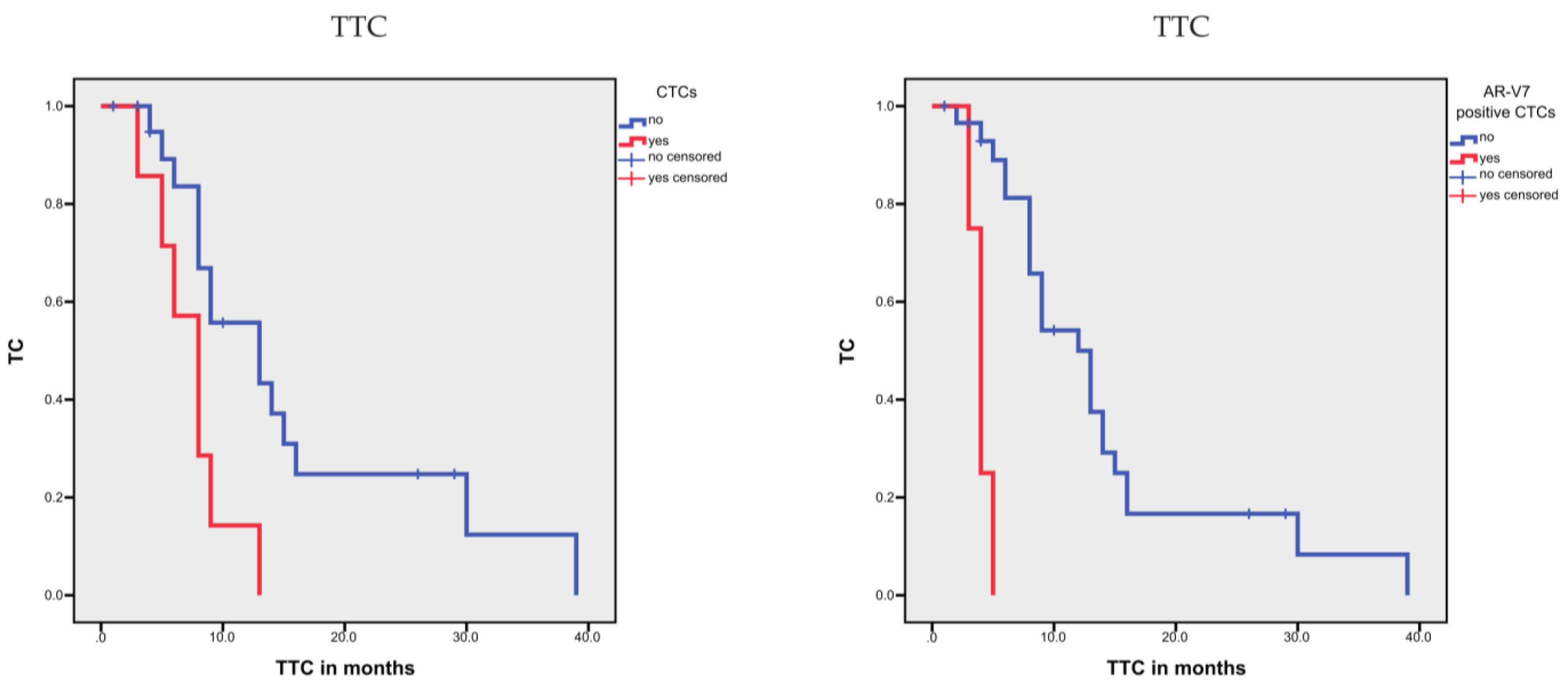
3.2.2. Associations between CTCs and Prognosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inamura, K. Prostatic cancers: Understanding their molecular pathology and the 2016 WHO classification. Oncotarget 2018, 9, 14723–14737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.T.; Delijani, K.; Mecum, A.; Goldkorn, A. Current status of liquid biopsies for the detection and management of prostate cancer. Cancer Manag. Res. 2019, 11, 5271–5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyen, J.; Alix-Panabières, C.; Hofman, P.; Parks, S.K.; Chamorey, E.; Naman, H.; Hannoun-Lévi, J.M. Circulating tumor cells in prostate cancer: A potential surrogate marker of survival. Crit. Rev. Oncol. Hematol. 2012, 81, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Eisenberger, M.A.; Halabi, S.; Oudard, S.; Nanus, D.M.; Petrylak, D.P.; Sartor, A.O.; Scher, H.I. Biomarkers in the management and treatment of men with metastatic castration-resistant prostate cancer. Eur. Urol. 2012, 61, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Diamond, E.; Lee, G.Y.; Akhtar, N.H.; Kirby, B.J.; Giannakakou, P.; Tagawa, S.T.; Nanus, D.M. Isolation and characterization of circulating tumor cells in prostate cancer. Front. Oncol. 2012, 2, 131. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, A.S.; Ferraldeschi, R.; Krupa, R.; Landers, M.; Graf, R.; Louw, J.; Jendrisak, A.; Bales, N.; Marrinucci, D.; Zafeiriou, Z.; et al. Phenotypic diversity of circulating tumour cells in patients with metastatic castration-resistant prostate cancer. BJU Int. 2017, 120, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Hille, C.; Scher, H.I. Circulating Tumor Cells in Prostate Cancer: From Discovery to Clinical Utility. Clin. Chem. 2019, 65, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Mandel, P.C.; Huland, H.; Tiebel, A.; Haese, A.; Salomon, G.; Budäus, L.; Tilki, D.; Chun, F.; Heinzer, H.; Graefen, M.; et al. Enumeration and Changes in Circulating Tumor Cells and Their Prognostic Value in Patients Undergoing Cytoreductive Radical Prostatectomy for Oligometastatic Prostate Cancer-Translational Research Results from the Prospective ProMPT trial. Eur. Urol. Focus. 2019, 7, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Broncy, L.; Paterlini-Bréchot, P. Clinical Impact of Circulating Tumor Cells in Patients with Localized Prostate Cancer. Cells 2019, 8, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefsson, A.; Larsson, K.; Månsson, M.; Björkman, J.; Rohlova, E.; Åhs, D.; Brisby, H.; Damber, J.E.; Welén, K. Circulating tumor cells mirror bone metastatic phenotype in prostate cancer. Oncotarget 2018, 9, 29403–29413. [Google Scholar] [CrossRef] [PubMed]
- Gorges, T.M.; Tinhofer, I.; Drosch, M.; Röse, L.; Zollner, T.M.; Krahn, T.; von Ahsen, O. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer 2012, 12, 178. [Google Scholar] [CrossRef] [Green Version]
- Keup, C.; Storbeck, M.; Hauch, S.; Hahn, P.; Sprenger-Haussels, M.; Tewes, M.; Mach, P.; Hoffmann, O.; Kimmig, R.; Kasimir-Bauer, S. Cell-Free DNA Variant Sequencing Using CTC-Depleted Blood for Comprehensive Liquid Biopsy Testing in Metastatic Breast Cancer. Cancers 2019, 11, 238. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.; Welti, J.C.; Lambros, M.B.K.; Dolling, D.; Rodrigues, D.N.; Pope, L.; Aversa, C.; Figueiredo, I.; Fraser, J.; Ahmad, Z.; et al. Clinical Utility of Circulating Tumour Cell Androgen Receptor Splice Variant-7 Status in Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2019, 76, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, A.J.; Halabi, S.; Luo, J.; Nanus, D.M.; Giannakakou, P.; Szmulewitz, R.Z.; Danila, D.C.; Healy, P.; Anand, M.; Rothwell, C.J.; et al. Prospective Multicenter Validation of Androgen Receptor Splice Variant 7 and Hormone Therapy Resistance in High-Risk Castration-Resistant Prostate Cancer: The PROPHECY Study. J. Clin. Oncol. 2019, 37, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, A.; Linder, A.; Flondell Site, D.; Canesin, G.; Stiehm, A.; Anand, A.; Bjartell, A.; Damber, J.E.; Welén, K. Circulating Tumor Cells as a Marker for Progression-free Survival in Metastatic Castration-naïve Prostate Cancer. Prostate 2017, 77, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, A.; Damber, J.E.; Welén, K. AR-V7 expression in circulating tumor cells as a potential prognostic marker in metastatic hormone-sensitive prostate cancer. Acta Oncol. 2019, 58, 1660–1664. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; Jayaram, A.; Winquist, E.; McLaughlin, B.; Lu, D.; Fleisher, M.; Orr, S.; Lowes, L.; et al. Assessment of the Validity of Nuclear-Localized Androgen Receptor Splice Variant 7 in Circulating Tumor Cells as a Predictive Biomarker for Castration-Resistant Prostate Cancer. JAMA Oncol. 2018, 4, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Bernemann, C.; Schnoeller, T.J.; Luedeke, M.; Steinestel, K.; Boegemann, M.; Schrader, A.J.; Steinestel, J. Expression of AR-V7 in Circulating Tumour Cells Does Not Preclude Response to Next Generation Androgen Deprivation Therapy in Patients with Castration Resistant Prostate Cancer. Eur. Urol. 2017, 71, 1–3. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.; Butt, A.; Cahill, D.; Wheeler, M.; Popert, R.; Swaminathan, R. Role of cell-free plasma DNA as a diagnostic marker for prostate cancer. Ann. N. Y. Acad. Sci. 2004, 1022, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Altimari, A.; Grigioni, A.D.; Benedettini, E.; Gabusi, E.; Schiavina, R.; Martinelli, A.; Morselli-Labate, A.M.; Martorana, G.; Grigioni, W.F.; Fiorentino, M. Diagnostic role of circulating free plasma DNA detection in patients with localized prostate cancer. Am. J. Clin. Pathol. 2008, 129, 756–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinger, J.; Bastian, P.J.; Haan, K.I.; Heukamp, L.C.; Buettner, R.; Fimmers, R.; Mueller, S.C.; von Ruecker, A. Noncancerous PTGS2 DNA fragments of apoptotic origin in sera of prostate cancer patients qualify as diagnostic and prognostic indicators. Int. J. Cancer 2008, 122, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Gang, F.; Li, X.; Jin, T.; Houbao, H.; Yu, C.; Guorong, L. Plasma cell-free DNA and its DNA integrity as biomarker to distinguish prostate cancer from benign prostatic hyperplasia in patients with increased serum prostate-specific antigen. Int. Urol. Nephrol. 2013, 45, 1023–1028. [Google Scholar] [CrossRef]
- Wroclawski, M.L.; Serpa-Neto, A.; Fonseca, F.L.; Castro-Neves-Neto, O.; Pompeo, A.S.; Machado, M.T.; Pompeo, A.C.; del Giglio, A. Cell-free plasma DNA as biochemical biomarker for the diagnosis and follow-up of prostate cancer patients. Tumour. Biol. 2013, 34, 2921–2927. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Annala, M.; Aggarwal, R.; Beja, K.; Feng, F.; Youngren, J.; Foye, A.; Lloyd, P.; Nykter, M.; Beer, T.M.; et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J. Natl. Cancer Inst. 2017, 109, djx118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Luo, J.; Wu, S.; Si, H.; Gao, C.; Xu, W.; Abdullah, S.E.; Higgs, B.W.; Dennis, P.A.; van der Heijden, M.S.; et al. Prognostic and Predictive Impact of Circulating Tumor DNA in Patients with Advanced Cancers Treated with Immune Checkpoint Blockade. Cancer Discov. 2020, 10, 1842–1853. [Google Scholar] [CrossRef]
- González-Billalabeitia, E.; Conteduca, V.; Wetterskog, D.; Jayaram, A.; Attard, G. Circulating tumor DNA in advanced prostate cancer: Transitioning from discovery to a clinically implemented test. Prostate Cancer Prostatic Dis. 2019, 22, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Wetterskog, D.; Sharabiani, M.T.A.; Grande, E.; Fernandez-Perez, M.P.; Jayaram, A.; Salvi, S.; Castellano, D.; Romanel, A.; Lolli, C.; et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Ann. Oncol. 2017, 28, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [Green Version]
- Vandekerkhove, G.; Struss, W.J.; Annala, M.; Kallio, H.M.L.; Khalaf, D.; Warner, E.W.; Herberts, C.; Ritch, E.; Beja, K.; Loktionova, Y.; et al. Circulating Tumor DNA Abundance and Potential Utility in De Novo Metastatic Prostate Cancer. Eur. Urol. 2019, 75, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Butler, T.M.; Johnson-Camacho, K.; Peto, M.; Wang, N.J.; Macey, T.A.; Korkola, J.E.; Koppie, T.M.; Corless, C.L.; Gray, J.W.; Spellman, P.T. Exome Sequencing of Cell-Free DNA from Metastatic Cancer Patients Identifies Clinically Actionable Mutations Distinct from Primary Disease. PLoS ONE. 2015, 10, e0136407. [Google Scholar] [CrossRef] [Green Version]
- Keup, C.; Benyaa, K.; Hauch, S.; Sprenger-Haussels, M.; Tewes, M.; Mach, P.; Bittner, A.K.; Kimmig, R.; Hahn, P.; Kasimir-Bauer, S. Targeted deep sequencing revealed variants in cell-free DNA of hormone receptor-positive metastatic breast cancer patients. Cell Mol. Life Sci. 2020, 77, 497–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Taubert, H.; Meye, A.; Würl, P. Soft tissue sarcomas and p53 mutations. Mol. Med. 1998, 4, 365–372. [Google Scholar] [CrossRef] [Green Version]
- Boerrigter, E.; Groen, L.N.; Van Erp, N.P.; Verhaegh, G.W.; Schalken, J.A. Clinical utility of emerging biomarkers in prostate cancer liquid biopsies. Expert Rev. Mol. Diagn. 2020, 20, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Perkins, G.; Yap, T.A.; Pope, L.; Cassidy, A.M.; Dukes, J.P.; Riisnaes, R.; Massard, C.; Cassier, P.A.; Miranda, S.; Clark, J.; et al. Multi-purpose utility of circulating plasma DNA testing in patients with advanced cancers. PLoS ONE 2012, 7, e47020. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, Y.; Xu, Y.; Li, L.; Gong, Y.; Zhang, K.; Zhang, M.; Guan, Y.; Chang, L.; Xia, X.; et al. Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients. Nat. Commun. 2021, 12, 11. [Google Scholar] [CrossRef]
- Fettke, H.; Kwan, E.M.; Bukczynska, P.; Ng, N.; Nguyen-Dumont, T.; Southey, M.C.; Davis, I.D.; Mant, A.; Parente, P.; Pezaro, C.; et al. Prognostic Impact of Total Plasma Cell-free DNA Concentration in Androgen Receptor Pathway Inhibitor-treated Metastatic Castration-resistant Prostate Cancer. Eur. Urol. Focus 2020, in press. [Google Scholar] [CrossRef]
- Mehra, N.; Dolling, D.; Sumanasuriya, S.; Christova, R.; Pope, L.; Carreira, S.; Seed, G.; Yuan, W.; Goodall, J.; Hall, E.; et al. Plasma Cell-free DNA Concentration and Outcomes from Taxane Therapy in Metastatic Castration-resistant Prostate Cancer from Two Phase III Trials (FIRSTANA and PROSELICA). Eur. Urol. 2018, 74, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, M.; Tan, W.; Zheng, T.; Wang, A.; Montesinos, C.; Wong, C.; Du, P.; Jia, S.; Yadav, S.; Horvath, L.G.; et al. Clinical and genomic insights into circulating tumor DNA-based alterations across the spectrum of metastatic hormone-sensitive and castrate-resistant prostate cancer. EBioMedicine 2020, 54, 102728. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.N.; Boysen, G.; Sumanasuriya, S.; Seed, G.; Marzo, A.M.; de Bono, J. The molecular underpinnings of prostate cancer: Impacts on management and pathology practice. J. Pathol. 2017, 241, 173–182. [Google Scholar] [CrossRef]
- Annala, M.; Vandekerkhove, G.; Khalaf, D.; Taavitsainen, S.; Beja, K.; Warner, E.W.; Sunderland, K.; Kollmannsberger, C.; Eigl, B.J.; Finch, D.; et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torquato, S.; Pallavajjala, A.; Goldstein, A.; Toro, P.V.; Silberstein, J.L.; Lee, J.; Nakazawa, M.; Waters, I.; Chu, D.; Shinn, D.; et al. Genetic Alterations Detected in Cell-Free DNA Are Associated With Enzalutamide and Abiraterone Resistance in Castration-Resistant Prostate Cancer. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef]
- De Laere, B.; Oeyen, S.; Mayrhofer, M.; Whitington, T.; van Dam, P.J.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; et al. TP53 Outperforms Other Androgen Receptor Biomarkers to Predict Abiraterone or Enzalutamide Outcome in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 1766–1773. [Google Scholar] [CrossRef] [Green Version]
- Teroerde, M.; Nientiedt, C.; Duensing, A.; Hohenfellner, M.; Stenzinger, A.; Duensing, S. Revisiting the Role of p53 in Prostate Cancer. In Prostate Cancer; Bott, S.R.J., Ng, K.L., Eds.; Exon Publications: Brisbane, Australia, 2021. Available online:https://www.ncbi.nlm.nih.gov/books/NBK571319/ (accessed on 20 October 2021). [CrossRef]
- Li, X.; Pasche, B.; Zhang, W.; Chen, K. Association of MUC16 Mutation With Tumor Mutation Load and Outcomes in Patients With Gastric Cancer. JAMA Oncol. 2018, 4, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lin, D.; Li, A.; Chen, Y.; Ou, Q.; Hu, H.; Yao, H. Association of Immune Checkpoint Inhibitor Therapy With Survival in Patients With Cancers With MUC16 Variants. JAMA Netw. Open. 2020, 3, e205837. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Herberts, C.; Murtha, A.J.; Fu, S.; Wang, G.; Schönlau, E.; Xue, H.; Lin, D.; Gleave, A.; Yip, S.; Angeles, A. Activating AKT1 and PIK3CA Mutations in Metastatic Castration-Resistant Prostate Cancer. Eur. Urol. 2020, 78, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Madison, R.W.; Chung, J.H.; Gjoerup, O.; Severson, E.A.; Dennis, L.; Fendler, B.J.; Morley, S.; Zhong, L.; Graf, R.P.; et al. Genomic analysis of circulating tumor DNA in 3,334 patients with advanced prostate cancer identifies targetable BRCA alterations and AR resistance mechanisms. Clin. Cancer Res. 2021, 27, 3094–3105. [Google Scholar] [CrossRef]
- Barata, P.; Agarwal, N.; Nussenzveig, R.; Gerendash, B.; Jaeger, E.; Hatton, W.; Ledet, E.; Lewis, B.; Layton, J.; Babiker, H.; et al. Clinical activity of pembrolizumab in metastatic prostate cancer with microsatellite instability high (MSI-H) detected by circulating tumor DNA. J. Immunother. Cancer 2020, 8, e001065. [Google Scholar] [CrossRef]
- Sumanasuriya, S.; Omlin, A.; Armstrong, A.; Attard, G.; Chi, K.N.; Bevan, C.L.; Shibakawa, A.; IJzerman, M.J.; De Laere, B.; Lolkema, M.; et al. Consensus Statement on Circulating Biomarkers for Advanced Prostate Cancer. Eur. Urol. Oncol. 2018, 1, 151–159. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar] [CrossRef]
- Lozano, R.; Lorente, D.; Aragon, I.M.; Romero-Laorden, N.; Nombela, P.; Mateo, J.; Reid, A.H.M.; Cendón, Y.; Bianchini, D.; Llacer, C.; et al. Value of Early Circulating Tumor Cells Dynamics to Estimate Docetaxel Benefit in Metastatic Castration-Resistant Prostate Cancer (mCRPC) Patients. Cancers 2021, 13, 2334. [Google Scholar] [CrossRef]
- Heidrich, I.; Ačkar, L.; Mossahebi Mohammadi, P.; Pantel, K. Liquid biopsies: Potential and challenges. Int J. Cancer 2021, 148, 528–545. [Google Scholar] [CrossRef]
- Fan, L.; Zhang, F.; Xu, S.; Cui, X.; Hussain, A.; Fazli, L.; Gleave, M.; Dong, X.; Qi, J. Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, 4584–4593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesmann, N.; Strozynski, J.; Beck, C.; Zimmermann, N.; Mendler, S.; Gieringer, R.; Schmidtmann, I.; Brieger, J. Knockdown of hnRNPK leads to increased DNA damage after irradiation and reduces survival of tumor cells. Carcinogenesis 2017, 38, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, N.K.; Kim, J.; Cinar, B.; Ramachandran, A.; Hager, M.H.; Di Vizio, D.; Adam, R.M.; Rubin, M.A.; Raychaudhuri, P.; De Benedetti, A.; et al. Heterogeneous nuclear ribonucleoprotein K is a novel regulator of androgen receptor translation. Cancer Res. 2009, 69, 2210–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar-Hoyos, L.F.; Penson, A.; Kannan, R.; Cho, H.; Pan, C.H.; Singh, R.K.; Apken, L.H.; Hobbs, G.A.; Luo, R.; Lecomte, N.; et al. Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 2020, 38, 198–211. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal. Transduct Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | n |
|---|---|
| Patients | 34 |
| Samples | 39 |
| Age, median (range) in years | 67 (48–89) |
| Metastasis | |
| cM0 | 3 |
| cM1 | 31 |
| cfDNA, mean (range) in ng/mL | 17.93 (2.37–206.25) |
| CTC-positive patients (samples) | 9 (11) |
| AR-V7-CTC-positive patients (samples) | 3 (4) |
| Survival time, median (range) in months | 48 (6–182) |
| Overall survival | |
| Alive | 24 |
| Dead | 10 |
| Disease-specific survival | |
| Yes | 25 |
| No | 9 |
| Time from blood sampling to last contact, median (range) | 10 (0–61) |
| Time from metastasis to last follow up, median (range) | 31 (11–133) |
| Time to treatment change | 9 (1–40) |
| Treatment change | |
| Yes | 24 |
| No | 8 |
| Not known | 2 |
| Pretreatment | |
| None | 6 |
| Depriv | 10 |
| AE | 8 |
| Tax | 3 |
| Multiple | 7 |
| Treatment | |
| AE | 10 |
| Tax | 18 |
| Chemotherapy/hormone | 6 |
| Pretreatment + treatment | |
| None + chemotherapy/hormone | 6 |
| Depriv + AE | 5 |
| Tax + AE | 4 |
| Mult + AE | 2 |
| Depriv + Tax | 4 |
| AE + Tax | 8 |
| Mult + Tax | 5 |
| Parameter | n |
|---|---|
| cfDNA, mean (median; range) in ng/mL | 17.93 (10.56; 2.37–206.25) |
| CTC-positive patients (samples) | 9 (11) |
| AR-V7-CTC-positive patients (samples) | 3 (4) |
| Spearman Rho Test | CTC | ARV7-CTC | Time from Blood Sampling to Last Information Collection | Time to Treatment Change | OS | DSS | Variant Groups | TP53 Sequencing Groups | |
|---|---|---|---|---|---|---|---|---|---|
| cfDNA | correl. coeff. | 0.227 | 0.346 | −0.333 | −0.304 | 0.174 | 0.109 | 0.113 | 0.168 |
| Sig. (2-side) | 0.176 | 0.036 | 0.047 | 0.071 | 0.324 | 0.540 | 0.505 | 0.307 | |
| CTCs | correl. coeff. | 0.535 | −0.287 | −0.187 | 0.188 | 0.236 | 0.472 | 0.309 | |
| Sig. (2-side) | 0.001 | 0.100 | 0.289 | 0.294 | 0.186 | 0.004 | 0.063 | ||
| ARV7-positive CTCs | correl. coeff. | −0.377 | −0.378 | 0.250 | 0.280 | 0.219 | 0.523 | ||
| Sig. (2-side) | 0.028 | 0.028 | 0.160 | 0.115 | 0.206 | 0.001 | |||
| Time from blood sampling to last information collection | correl. coeff. | 0.857 | −0.464 | −0.441 | 0.102 | −0.246 | |||
| Sig. (2-side) | <0.001 | 0.007 | 0.012 | 0.565 | 0.147 | ||||
| Time to treatment change | correl. coeff. | −0.521 | −0.517 | 0.131 | −0.177 | ||||
| Sig. (2-side) | 0.002 | 0.002 | 0.459 | 0.301 | |||||
| OS | correl. coeff. | 0.930 | −0.300 | 0.339 | |||||
| Sig. (2-side) | <0.001 | 0.095 | 0.050 | ||||||
| DSS | correl. coeff. | −0.345 | 0.404 | ||||||
| Sig. (2-side) | 0.053 | 0.018 | |||||||
| Variant groups | correl. coeff. | 0.217 | |||||||
| Sig. (2-side) | 0.197 | ||||||||
| Parameter | Kaplan–Meier analysis | |||||
|---|---|---|---|---|---|---|
| OS | DSS | TTC | ||||
| Months | p | Months | p | Months | p | |
| cfDNA, higher vs. lower levels | 42.2 vs. 134.0 | 0.014 | 42.2 vs. 138.7 | 0.004 | 6.3 vs. 16.2 | 0.001 |
| CTCs, yes vs. no | n.s. | n.s. | 7.6 vs. 15.1 | 0.031 | ||
| AR-V7-positive CTCs, yes vs. no | n.s. | 57.7 vs. 134.8 | 0.097 | 4.0 vs. 14.2 | <0.001 | |
| Parameter | Univariate Cox’s Regression Analysis | |||||
|---|---|---|---|---|---|---|
| OS | DSS | TTC | ||||
| RR (95%CI) | p | RR (95%CI) | p | RR (95%CI | p | |
| cfDNA, higher vs. lower levels | 5.53 (1.20–25.31) | 0.027 | 7.87 (1.55–40.0453.86) | 0.013 | 4.11 (1.57–10.77) | 0.004 |
| CTCs, yes vs. no | n.s. | n.s. | 2.32 (1.02–5.30) | 0.046 | ||
| AR-V7-positive CTCs, yes vs. no | n.s. | n.s. | 14.75 (3.15–69.1) | 0.001 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lieb, V.; Abdulrahman, A.; Weigelt, K.; Hauch, S.; Gombert, M.; Guzman, J.; Bellut, L.; Goebell, P.J.; Stöhr, R.; Hartmann, A.; et al. Cell-Free DNA Variant Sequencing Using Plasma and AR-V7 Testing of Circulating Tumor Cells in Prostate Cancer Patients. Cells 2021, 10, 3223. https://doi.org/10.3390/cells10113223
Lieb V, Abdulrahman A, Weigelt K, Hauch S, Gombert M, Guzman J, Bellut L, Goebell PJ, Stöhr R, Hartmann A, et al. Cell-Free DNA Variant Sequencing Using Plasma and AR-V7 Testing of Circulating Tumor Cells in Prostate Cancer Patients. Cells. 2021; 10(11):3223. https://doi.org/10.3390/cells10113223
Chicago/Turabian StyleLieb, Verena, Amer Abdulrahman, Katrin Weigelt, Siegfried Hauch, Michael Gombert, Juan Guzman, Laura Bellut, Peter J. Goebell, Robert Stöhr, Arndt Hartmann, and et al. 2021. "Cell-Free DNA Variant Sequencing Using Plasma and AR-V7 Testing of Circulating Tumor Cells in Prostate Cancer Patients" Cells 10, no. 11: 3223. https://doi.org/10.3390/cells10113223
APA StyleLieb, V., Abdulrahman, A., Weigelt, K., Hauch, S., Gombert, M., Guzman, J., Bellut, L., Goebell, P. J., Stöhr, R., Hartmann, A., Wullich, B., Taubert, H., & Wach, S. (2021). Cell-Free DNA Variant Sequencing Using Plasma and AR-V7 Testing of Circulating Tumor Cells in Prostate Cancer Patients. Cells, 10(11), 3223. https://doi.org/10.3390/cells10113223