
Current Therapeutic Strategies and Prospects for EGFR Mutation-Positive Lung Cancer Based on the Mechanisms Underlying Drug Resistance


Abstract
:1. Introduction
2. Advances in the Treatment of EGFR Mutation-Positive Lung Cancer
2.1. First and Second-Generation EGFR-TKI Monotherapy
2.2. Treatment of EGFR T790M Acquired Resistance
2.3. Third-Generation EGFR-TKI Monotherapy
2.4. EGFR-TKI Combination Therapy
2.5. Immunotherapy for EGFR-Mutated Lung Cancer
3. Mechanisms Underlying Acquired Resistance to EGFR-TKIs
3.1. Mechanisms Underlying Development of Resistance to First- and Second-Generation EGFR-TKIs
3.2. Mechanisms Underlying Development of Resistance to Third-Generation EGFR-TKIs
4. Possibility of Combination Therapy with EGFR-TKI and Other Drugs
4.1. Combination Therapy with Cytotoxic Anticancer Agents
4.2. Combination Therapy with Angiogenesis Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non—Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib Versus Crizotinib in Untreated ALK-positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Shaw, A.T.; Riely, G.J.; Bang, Y.J.; Kim, D.W.; Camidge, D.R.; Solomon, B.J.; Varella-Garcia, M.; Iafrate, A.J.; Shapiro, G.I.; Usari, T.; et al. Crizotinib in ROS1-Rearranged Advanced Non-Small-Cell Lung Cancer (NSCLC): Updated Results, Including Overall Survival, From PROFILE 1001. Ann. Oncol. 2019, 30, 1121–1126. [Google Scholar] [CrossRef]
- Planchard, D.; Smit, E.F.; Groen, H.J.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib Plus Trametinib in Patients with Previously Untreated BRAF (V600E)-Mutant Metastatic Non-Small-Cell Lung Cancer: An Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non–Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Kohno, T.; Nakaoku, T.; Tsuta, K.; Tsuchihara, K.; Matsumoto, S.; Yoh, K.; Goto, K. Beyond ALK-RET, ROS1 and Other On-cogene Fusions in Lung Cancer. Transl. Lung Cancer Res. 2015, 4, 156–164. [Google Scholar]
- Kobayashi, Y.; Mitsudomi, T. Not All Epidermal Growth Factor Receptor Mutations in Lung Cancer Are Created Equal: Per-spectives for Individualized Treatment Strategy. Cancer Sci. 2016, 107, 1179–1186. [Google Scholar] [CrossRef]
- Yasuda, H.; Kobayashi, S.; Costa, D. EGFR exon 20 insertion mutations in non-small-cell lung cancer: Preclinical data and clinical implications. Lancet Oncol. 2012, 13, e23–e31. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.-H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.-H.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Feng, P.-H.; Karaseva, N.; Kim, S.-W.; Kim, T.; Lee, C.; Poltoratskiy, A.; Yanagitani, N.; Marshall, R.; Huang, X.; et al. Osimertinib plus platinum–pemetrexed in newly diagnosed epidermal growth factor receptor mutation-positive advanced/metastatic non-small-cell lung cancer: Safety run-in results from the FLAURA2 study. ESMO Open 2021, 6, 100271. [Google Scholar] [CrossRef]
- Kenmotsu, H.; Wakuda, K.; Mori, K.; Kato, T.; Sugawara, S.; Kirita, K.; Okamoto, I.; Azuma, K.; Nishino, K.; Teraoka, S.; et al. Primary Results of a Randomized Phase II Study of Osimertinib Plus Bevacizumab Versus Osimertinib Monotherapy for Un-treated Patients with Non-Squamous Non-Small Cell Lung Cancer Harboring EGFR Mutations: WJOG9717L Study. Ann. Oncol. 2021, 32, LBA44. [Google Scholar] [CrossRef]
- Hosomi, Y.; Morita, S.; Sugawara, S.; Kato, T.; Fukuhara, T.; Gemma, A.; Takahashi, K.; Fujita, Y.; Harada, T.; Minato, K.; et al. Gefitinib Alone Versus Gefitinib Plus Chemotherapy for Non–Small-Cell Lung Cancer with Mutated Epidermal Growth Factor Receptor: NEJ009 Study. J. Clin. Oncol. 2020, 38, 115–123. [Google Scholar] [CrossRef]
- Noronha, V.; Patil, V.M.; Joshi, A.; Menon, N.; Chougule, A.; Mahajan, A.; Janu, A.; Purandare, N.; Kumar, R.; More, S.; et al. Gefitinib Versus Gefitinib Plus Pemetrexed and Carboplatin Chemotherapy in EGFR-Mutated Lung Cancer. J. Clin. Oncol. 2020, 38, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Fukuhara, T.; Furuya, N.; Watanabe, K.; Sugawara, S.; Iwasawa, S.; Tsunezuka, Y.; Yamaguchi, O.; Okada, M.; Yoshimori, K.; et al. Erlotinib Plus Bevacizumab Versus Erlotinib Alone in Patients With EGFR-Positive Advanced Non-Squamous Non-Small-Cell Lung Cancer (NEJ026): Interim Analysis of an Open-Label, Randomised, Multicentre, phase 3 Trial. Lancet Oncol. 2019, 20, 625–635. [Google Scholar] [CrossRef]
- Kawashima, Y.; Fukuhara, T.; Saito, H.; Furuya, N.; Watanabe, K.; Sugawara, S.; Iwasawa, S.; Tsunezuka, Y.; Yamaguchi, O.; Okada, M.; et al. Bevacizumab plus erlotinib versus erlotinib alone in Japanese patients with advanced, metastatic, EGFR-mutant non-small-cell lung cancer (NEJ026): Overall survival analysis of an open-label, randomised, multicentre, phase 3 trial. Lancet Respir. Med. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Aix, S.P.; Paz-Ares, L.; Chiu, C.-H.; Park, K.; Novello, S.; Nadal, E.; et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Wang, F.; Wang, L.; Wong, S.C.C.; Cho, W.C.S.; Chan, L.W.C. MiR-30a-5p Overexpression May Overcome EGFR-Inhibitor Resistance through Regulating PI3K/AKT Signaling Pathway in Non-small Cell Lung Cancer Cell Lines. Front. Genet. 2016, 7, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleu-kin-6/STAT3 Signaling as a Promising Target to Improve the Efficacy of Cancer Immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Ota, K.; Azuma, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Tanizaki, J.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; et al. Induction of PD-L1 Expression by the EML4–ALK Oncoprotein and Downstream Signaling Pathways in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 4014–4021. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-Y.; Liao, W.-Y.; Ho, C.-C.; Chen, K.-Y.; Tsai, T.-H.; Hsu, C.-L.; Su, K.-Y.; Chang, Y.-L.; Wu, C.-T.; Liao, B.-C.; et al. Association between programmed death-ligand 1 expression, immune microenvironments, and clinical outcomes in epidermal growth factor receptor mutant lung adenocarcinoma patients treated with tyrosine kinase inhibitors. Eur. J. Cancer 2020, 124, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Haratani, K.; Hayashi, H.; Tanaka, T.; Kaneda, H.; Togashi, Y.; Sakai, K.; Hayashi, K.; Tomida, S.; Chiba, Y.; Yonesaka, K.; et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation-positive non-small-cell lung cancer based on T790M status after disease progression during EGFR-TKI treatment. Ann. Oncol. 2017, 28, 1532–1539. [Google Scholar] [CrossRef]
- Isomoto, K.; Haratani, K.; Hayashi, H.; Shimizu, S.; Tomida, S.; Niwa, T.; Yokoyama, T.; Fukuda, Y.; Chiba, Y.; Kato, R.; et al. Impact of EGFR-TKI Treatment on the Tumor Immune Microenvironment in EGFR Mutation–Positive Non–Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2037–2046. [Google Scholar] [CrossRef]
- Inomata, M.; Azechi, K.; Takata, N.; Hayashi, K.; Tokui, K.; Taka, C.; Okazawa, S.; Kambara, K.; Imanishi, S.; Miwa, T.; et al. Association of Tumor PD-L1 Expression with the T790M Mutation and Progression-Free Survival in Patients with EGFR-Mutant Non-Small Cell Lung Cancer Receiving EGFR-TKI Therapy. Diagnostics 2020, 10, 1006. [Google Scholar] [CrossRef]
- Kobayashi, K.; Seike, M.; Zou, F.; Noro, R.; Chiba, M.; Ishikawa, A.; Kunugi, S.; Kubota, K.; Gemma, A. Prognostic Significance of NSCLC and Response to EGFR-TKIs of EGFR-Mutated NSCLC Based on PD-L1 Expression. Anticancer. Res. 2018, 38, 753–762. [Google Scholar] [PubMed]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Mok, T.S.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab Plus Bevacizumab and Chemotherapy in Non-Small-Cell Lung Cancer (IMpower150): Key Subgroup Analyses of Patients with EGFR Mutations or Baseline Liver Me-tastases in a Randomised, Open-Label Phase 3 Trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Hayashi, H.; Sugawara, S.; Fukuda, Y.; Sato, Y.; Miura, S.; Ota, K.; Ozawa, Y.; Hara, S.; Tanizaki, J.; Azuma, K.; et al. A randomized phase II study comparing nivolumab (NIVO) with carboplatin-pemetrexed (CbPEM) for patients (pts) with EGFR mutation-positive non-small cell lung cancer (NSCLC) who acquire resistance to tyrosine kinase inhibitors (TKIs) not due to a secondary T790M mutation (WJOG8515L). J. Clin. Oncol. 2021, 39, 9037. [Google Scholar]
- Nguyen, K.-S.H.; Kobayashi, S.; Costa, D.B. Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non–Small-Cell Lung Cancers Dependent on the Epidermal Growth Factor Receptor Pathway. Clin. Lung Cancer 2009, 10, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors—Impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Aparicio, S.; Caldas, C. The Implications of Clonal Genome Evolution for Cancer Medicine. N. Engl. J. Med. 2013, 368, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Kawaguchi, T.; Isa, S.-I.; Ando, M.; Tamiya, A.; Kubo, A.; Saka, H.; Takeo, S.; Adachi, H.; Tagawa, T.; et al. Ultra-Sensitive Detection of the Pretreatment EGFR T790M Mutation in Non—Small Cell Lung Cancer Patients with an EGFR-Activating Mutation Using Droplet Digital PCR. Clin. Cancer Res. 2015, 21, 3552–3560. [Google Scholar] [CrossRef] [Green Version]
- Arcila, M.E.; Oxnard, G.R.; Nafa, K.; Riely, G.J.; Solomon, S.B.; Zakowski, M.F.; Kris, M.G.; Pao, W.; Miller, V.A.; Ladanyi, M. Rebiopsy of Lung Cancer Patientswith Acquired Resistance to EGFR Inhibitors and Enhanced Detection of the T790M Mutation Using a Locked Nucleic Acid-Based Assay. Clin. Cancer Res. 2011, 17, 1169–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, K.; Mizuuchi, H.; Maehara, Y.; Mitsudomi, T. Acquired resistance mechanisms to tyrosine kinase inhibitors in lung cancer with activating epidermal growth factor receptor mutation—diversity, ductility, and destiny. Cancer Metastasis Rev. 2012, 31, 807–814. [Google Scholar] [CrossRef]
- Jacobsen, K.; Bertran-Alamillo, J.; Molina, M.A.; Teixidó, C.; Karachaliou, N.; Pedersen, M.H.; Castellvi, J.; Garzón, M.; Servat, C.C.; Codony-Servat, J.; et al. Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients with EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Yun, C.-H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Bauml, J.; Cho, B.C.; Park, K.; Lee, K.H.; Cho, E.K.; Kim, D.-W.; Kim, S.-W.; Haura, E.B.; Sabari, J.K.; Sanborn, R.E.; et al. Amivantamab in combination with lazertinib for the treatment of osimertinib-relapsed, chemotherapy-naïve EGFR mutant (EGFRm) non-small cell lung cancer (NSCLC) and potential biomarkers for response. J. Clin. Oncol. 2021, 39, 9006. [Google Scholar] [CrossRef]
- Janne, P.A.; Baik, C.S.; Su, W.C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated (EGFRm) Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2021, 39, 9007. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Tsubata, Y.; Watanabe, K.; Saito, R.; Nakamura, A.; Yoshioka, H.; Morita, M.; Honda, R.; Kanaji, N.; Ohizumi, S.; Jingu, D.; et al. Osimertinib in poor performance status patients with T790M-positive advanced non-small-cell lung cancer after progression of first- and second-generation EGFR-TKI treatments (NEJ032B). Int. J. Clin. Oncol. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Fukuhara, T.; Saito, H.; Furuya, N.; Watanabe, K.; Sugawara, S.; Iwasawa, S.; Tsunezuka, Y.; Yamaguchi, O.; Okada, M.; Yoshimori, K.; et al. Evaluation of plasma EGFR mutation as an early predictor of response of erlotinib plus bevacizumab treatment in the NEJ026 study. EBioMedicine 2020, 57, 102861. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive Detection of Response and Resistance in EGFR-Mutant Lung Cancer Using Quanti-tative Next-Generation Genotyping of Cell-Free Plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [Green Version]
- Ebert, E.B.F.; McCulloch, T.; Hansen, K.H.; Linnet, H.; Sorensen, B.; Meldgaard, P. Clearing of circulating tumour DNA predicts clinical response to osimertinib in EGFR mutated lung cancer patients. Lung Cancer 2020, 143, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.-E.C.; Radhakrishna, V.K.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Tanino, R.; Tsubata, Y.; Harashima, N.; Harada, M.; Isobe, T. Novel drug-resistance mechanisms of pemetrexed-treated non-small cell lung cancer. Oncotarget 2018, 9, 16807–16821. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-C.; Yang, T.-Y.; Wu, C.-C.; Cheng, C.-C.; Hsu, S.-L.; Hung, H.-W.; Chen, J.-W.; Chang, G.-C. Pemetrexed Induces S-Phase Arrest and Apoptosis via a Deregulated Activation of Akt Signaling Pathway. PLoS ONE 2014, 9, e97888. [Google Scholar] [CrossRef]
- Tong, X.; Tanino, R.; Sun, R.; Tsubata, Y.; Okimoto, T.; Takechi, M.; Isobe, T. Protein tyrosine kinase 2: A novel therapeutic target to overcome acquired EGFR-TKI resistance in non-small cell lung cancer. Respir. Res. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Suda, K.; Bunn, P.A., Jr.; Rivard, C.J.; Mitsudomi, T.; Hirsch, F.R. Primary Double-Strike Therapy for Cancers to Overcome EGFR Kinase Inhibitor Resistance: Proposal from the Bench. J. Thorac. Oncol. 2017, 12, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to Anti-Angiogenic Therapy in Cancer—Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haibe, Y.; Kreidieh, M.; Haji, H.E.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to An-ti-angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akamatsu, H.; Toi, Y.; Hayashi, H.; Fujimoto, D.; Tachihara, M.; Furuya, N.; Otani, S.; Shimizu, J.; Katakami, N.; Azuma, K.; et al. Efficacy of Osimertinib Plus Bevaci-zumab vs Osimertinib in Patients with EGFR T790M-Mutated Non-Small Cell Lung Cancer Previously Treated with Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor: West Japan Oncology Group 8715L Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 386–394. [Google Scholar] [PubMed]
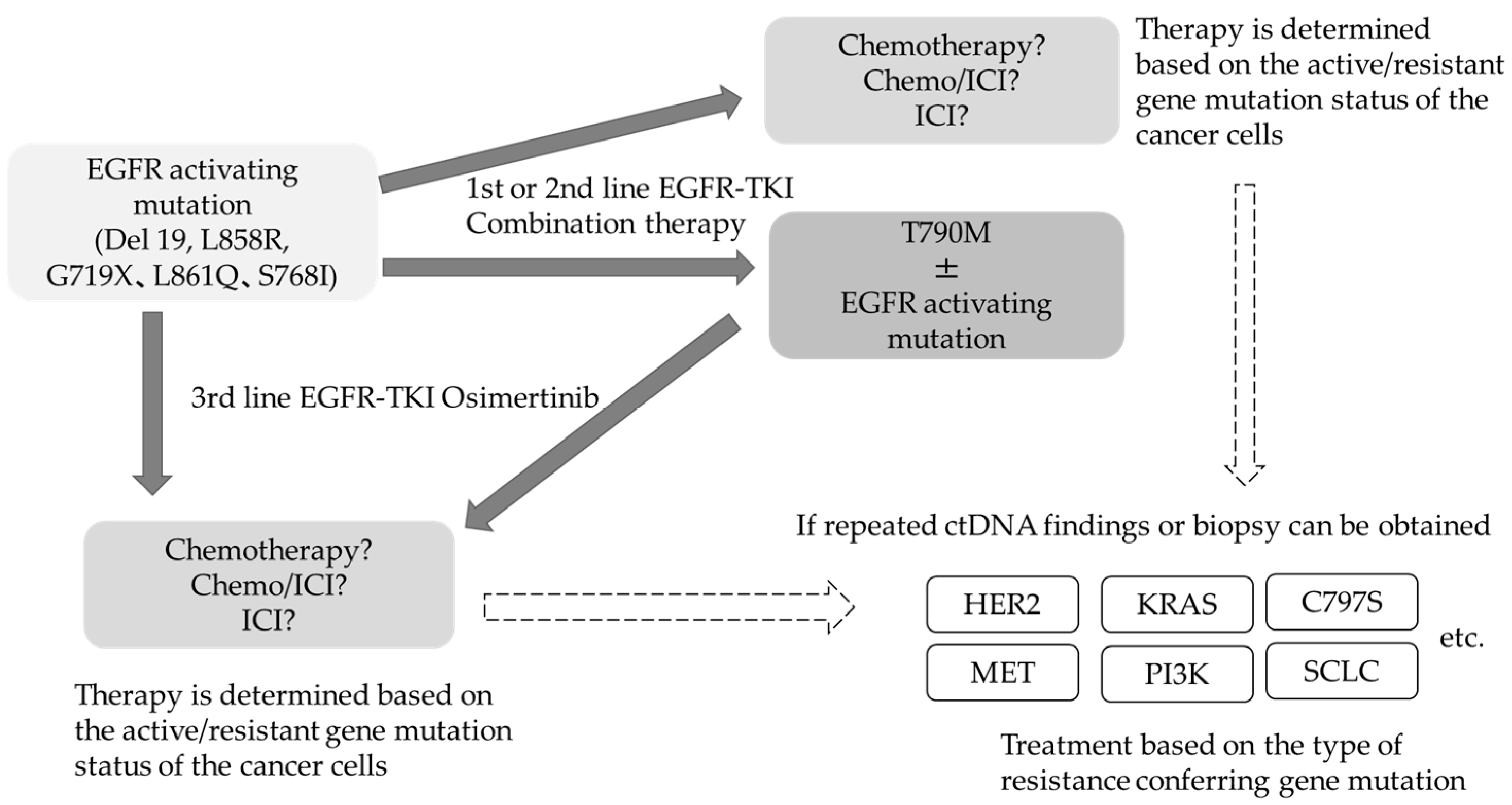

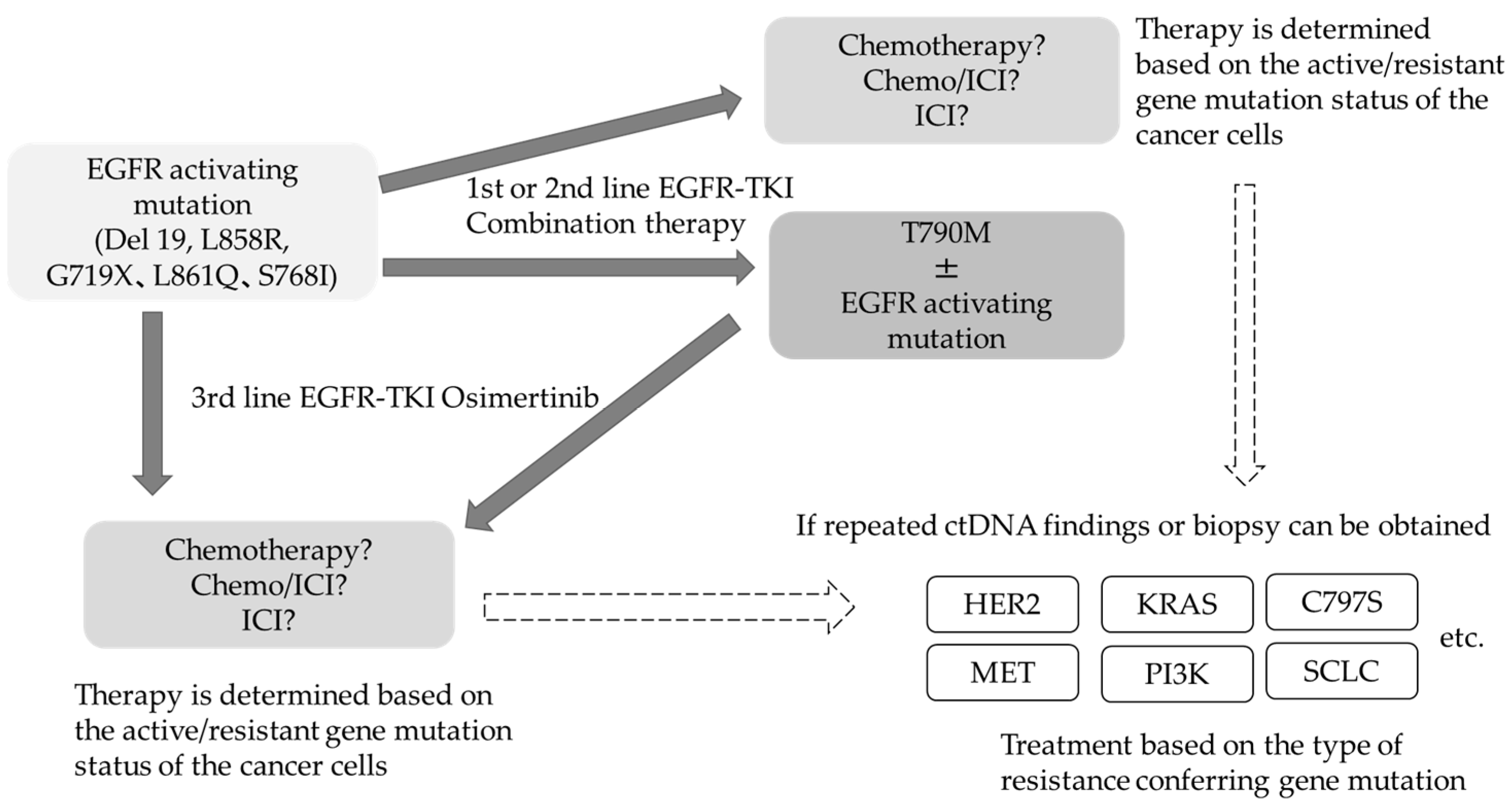{kind=link}
| Trial (Reference) | Target EGFR Mutation | Experimental Therapy | Control Therapy | PFS 1 (Primary Outcome) | Overall Survival | ||||
|---|---|---|---|---|---|---|---|---|---|
| Months | HR 4 (95% C.I. 5) | Months | HR (95% C.I.) | ||||||
| Exp 2 | Ctrl 3 | Exp | Ctrl | ||||||
| NEJ002(2) | Sensitizing mutation | Gefitinib | Cb 6/PTX | 10.8 | 5.4 | 0.33 | 30.5 | 23.6 | - |
| (0.22–0.41) | |||||||||
| WJTOG 3405(3) | Ex19_del Ex21_L858R | Gefitinib | CDDP 7/DTX 8 | 9.2 | 6.3 | 0.489 | 30.9 | NR 9 | 1.638 |
| (0.336–0.710) | (0.749–3.582) | ||||||||
| EURTAC(13) | Ex19_del Ex21_L858R | Erlotinib | CDDP | 9.7 | 5.2 | - | - | - | |
| EURTAC(13) | Ex19_del Ex21_L858R | Erlotinib | Cb/DTX | 9.7 | 5.2 | 0.37 | - | - | - |
| GEM 10 | (0.25–0.54) | ||||||||
| OPTIMAL (14) | Ex19_del Ex21_L858R | Erlotinib | Cb/GEM | 13.1 | 4.6 | 0.16 | - | - | - |
| OPTIMAL (14) | Ex19_del Ex21_L858R | Erlotinib | Cb/GEM | 13.1 | 4.6 | (0.10–0.26) | - | - | - |
| LUX-Lung 7 (15) | Ex19_del Ex21_L858R | Afatinib | Gefitinib | 11 | 10.9 | 0.74 | 27.9 | 24.5 | 0.86 |
| LUX-Lung 7 (15) | Ex19_del Ex21_L858R | Afatinib | Gefitinib | 11 | 10.9 | (0.57–0.95) | 27.9 | 24.5 | (0.66–1.12) |
| ARCHER 1050 (16) | Ex19_del Ex21_L858R | Dacomitinib | Gefitinib | 14.7 | 9.2 | 0.59 | 34.1 | 26.8 | 0.76 |
| ARCHER 1050 (16) | Ex19_del Ex21_L858R | Dacomitinib | Gefitinib | 14.7 | 9.2 | (0.47–0.74) | 34.1 | 26.8 | (0.582–0.993) |
| FLAURA (17) | Ex19_del Ex21_L858R | Osimertinib | Gefitinib | 18.9 | 10.2 | 0.46 | 38.6 | 31.8 | 0.799 |
| FLAURA (17) | Ex19_del Ex21_L858R | Osimertinib | Erlotinib | 18.9 | 10.2 | (0.37–0.57) | 38.6 | 31.8 | (0.641–0.997) |
| Trial (Reference) | Target EGFR Mutation | Experimental Therapy | Control Therapy | PFS 1 (Primary Outcome †) | Overall Survival | ||||
|---|---|---|---|---|---|---|---|---|---|
| Months | HR 4 (95% C.I. 5) | Months | HR (95% C.I.) | ||||||
| Exp 2 | Ctrl 3 | Exp | Ctrl | ||||||
| NEJ009 (20) | Sensitizing mutation | Cb 6/PEM 7/ Gefitinib | Gefitinib | 20.9 | 11.9 | 0.49 | 50.9 | 28.8 | 0.72 |
| (0.39–0.62) | (0.55–0.95) | ||||||||
| Noronha trial (21) | Sensitizing mutation | Cb/PEM/ Gefitinib | Gefitinib | 16.0 | 8.0 | 0.51 | NR 8 | 17 | 0.45 |
| (0.39–0.66) | (0.31–0.65) | ||||||||
| NEJ026 (22,23) | Ex19_del Ex21_L858R | Erlotinib/ Bevacizumab | Erlotinib | 16.9 | 13.3 | 0.605 | 50.7 | 46.2 | 1.007 |
| (0.417–0.877) | (0.681–1.49) | ||||||||
| RELAY (24) | Ex19_del Ex21_L858R | Erlotinib/ Bevacizumab | Erlotinib | 19.4 | 12.4 | 0.59 | NR | NR | 0.83 |
| (0.46–0.76) | (0.53–1.30) | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsubata, Y.; Tanino, R.; Isobe, T. Current Therapeutic Strategies and Prospects for EGFR Mutation-Positive Lung Cancer Based on the Mechanisms Underlying Drug Resistance. Cells 2021, 10, 3192. https://doi.org/10.3390/cells10113192
Tsubata Y, Tanino R, Isobe T. Current Therapeutic Strategies and Prospects for EGFR Mutation-Positive Lung Cancer Based on the Mechanisms Underlying Drug Resistance. Cells. 2021; 10(11):3192. https://doi.org/10.3390/cells10113192
Chicago/Turabian StyleTsubata, Yukari, Ryosuke Tanino, and Takeshi Isobe. 2021. "Current Therapeutic Strategies and Prospects for EGFR Mutation-Positive Lung Cancer Based on the Mechanisms Underlying Drug Resistance" Cells 10, no. 11: 3192. https://doi.org/10.3390/cells10113192
APA StyleTsubata, Y., Tanino, R., & Isobe, T. (2021). Current Therapeutic Strategies and Prospects for EGFR Mutation-Positive Lung Cancer Based on the Mechanisms Underlying Drug Resistance. Cells, 10(11), 3192. https://doi.org/10.3390/cells10113192