
Bench Research Informed by GWAS Results


{kind=link}
{kind=link}
Abstract
:1. Introduction
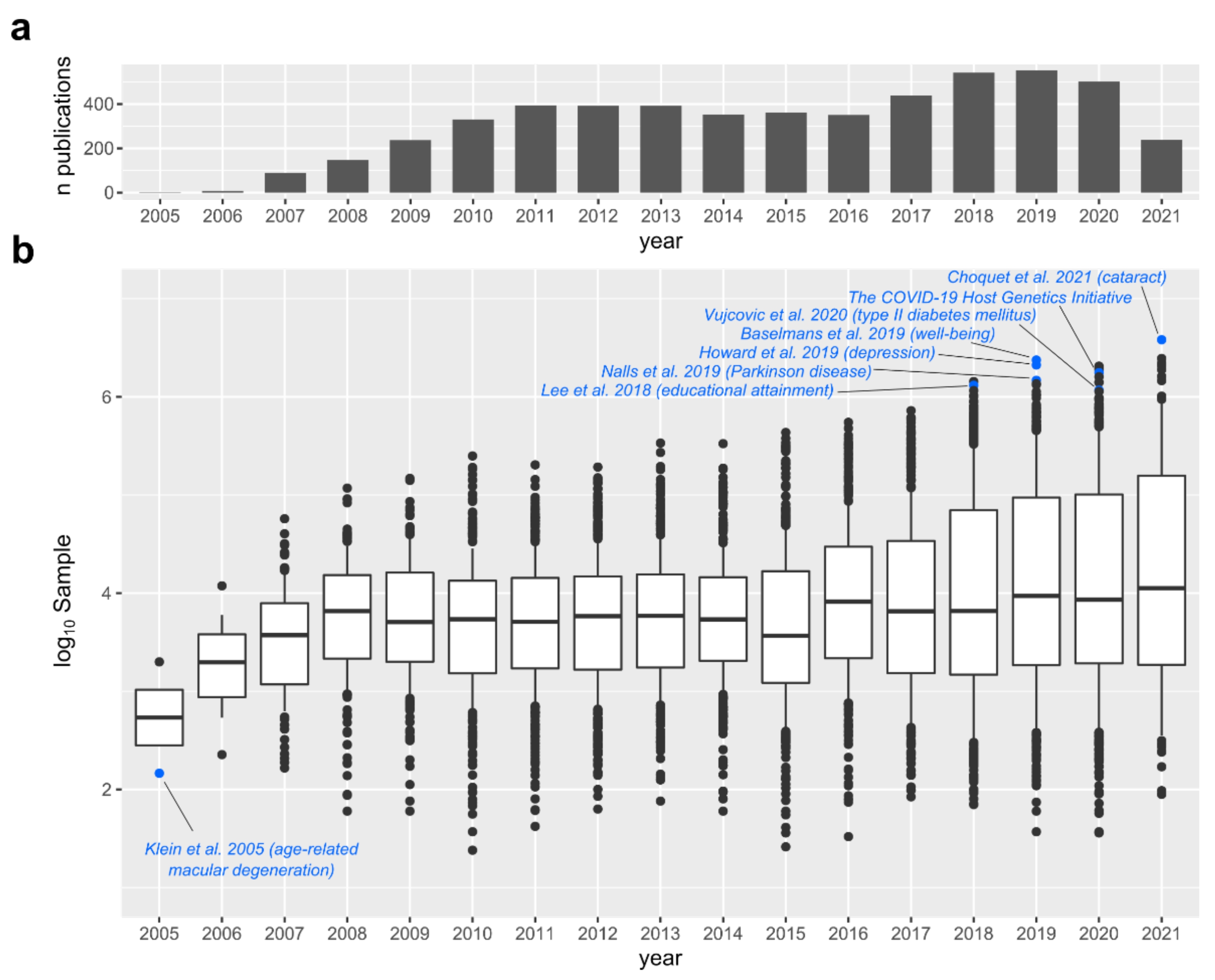
2. GWAS Is a Major Tool for the Genetics of Complex Traits
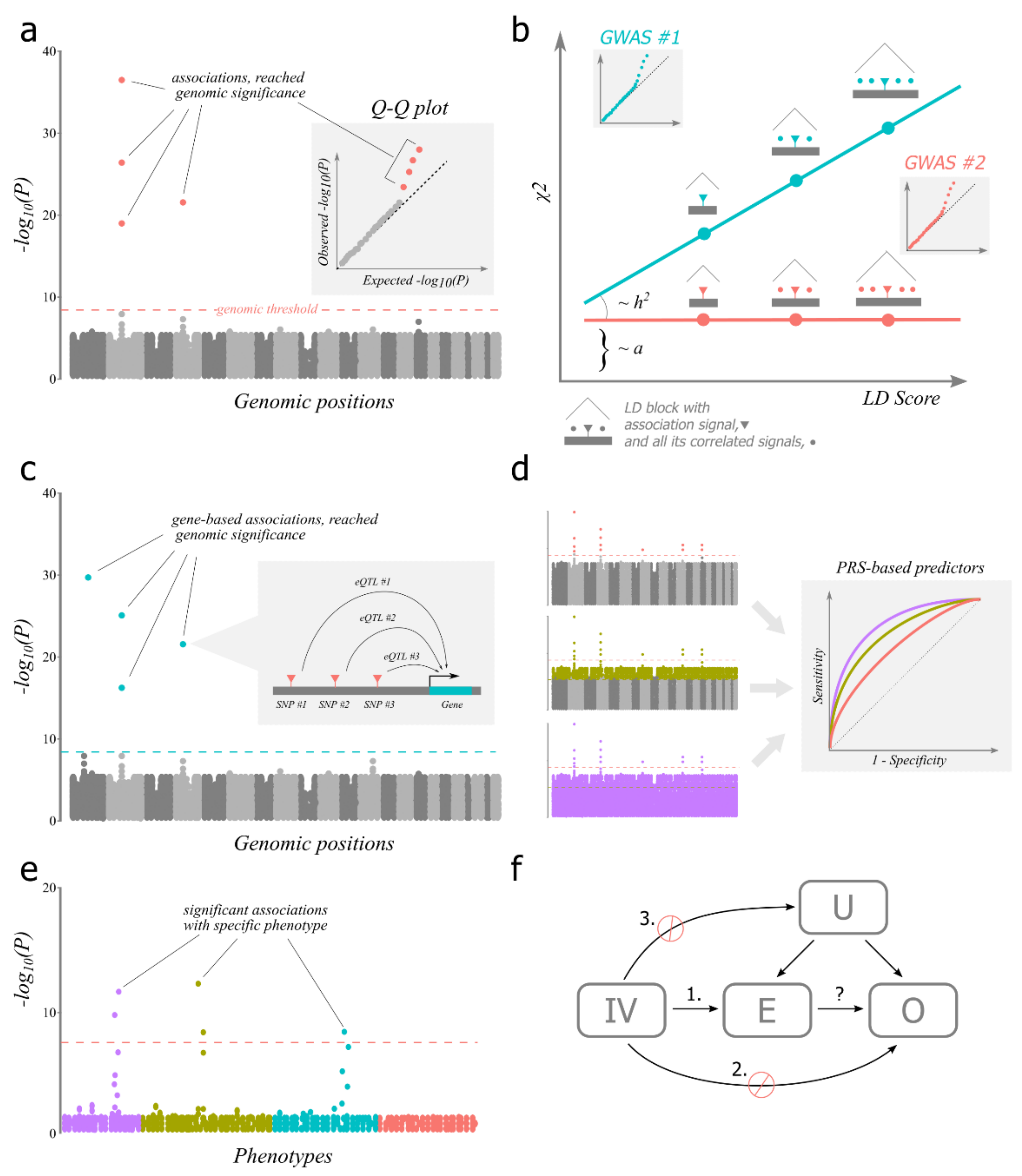
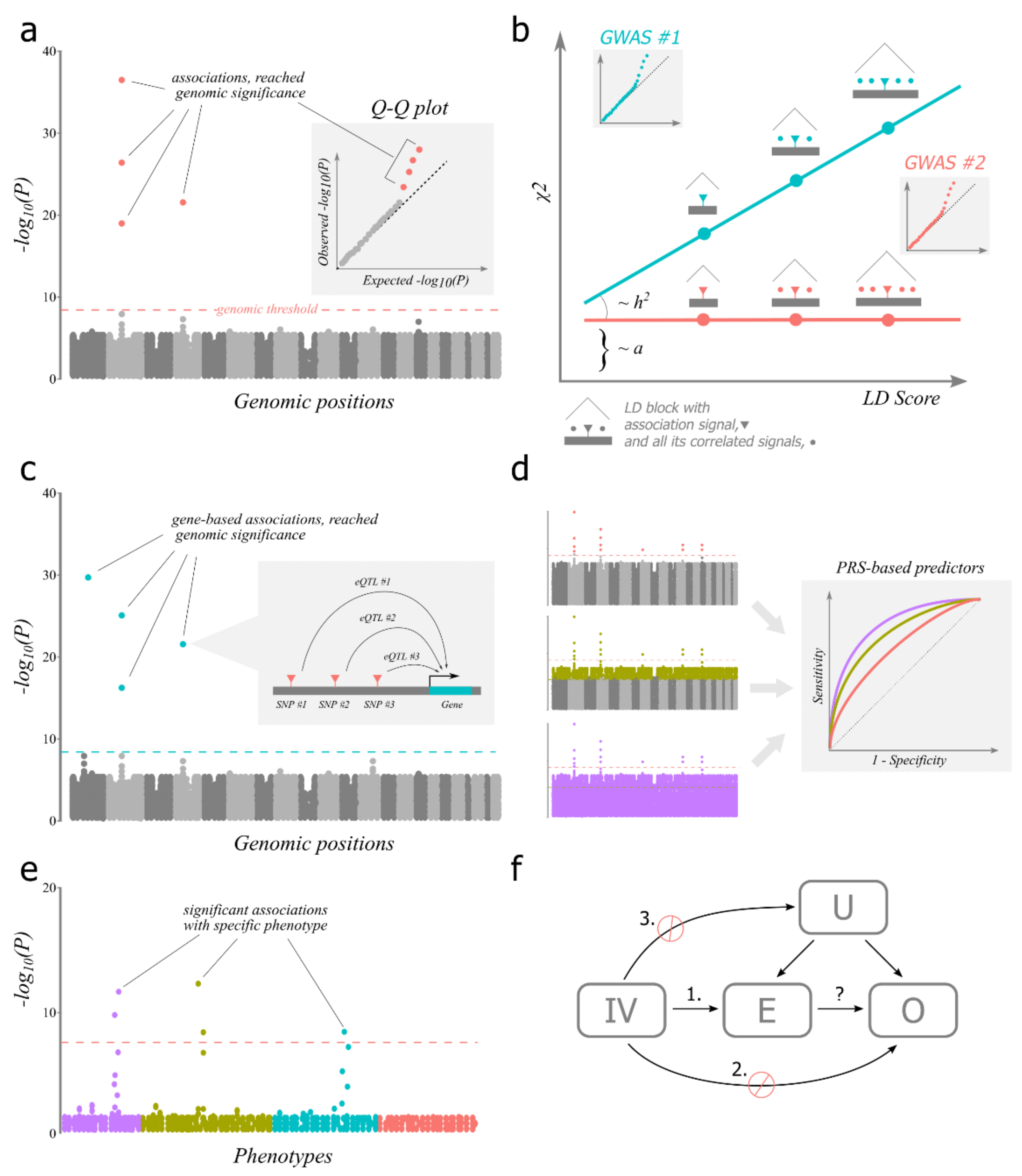
3. Assumptions of GWAS
3.1. Heritability of Complex Traits
3.2. Population Structure
3.3. Common Additive Variation
4. Arguments for GWAS
4.1. Reproducibility
4.2. Interpretability
4.3. Utility
4.4. Interoperability
5. Bench Use of GWAS
5.1. Narrow-Focus Follow-Up Studies
5.2. Interpretation of Functional Annotations Using GWAS Results
5.3. Use of PRS in Experimental Biology
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GWAS | genome-wide association study or GWA study |
| SNP | single-nucleotide polymorphism |
| PRS | polygenic risk score |
| LD | linkage disequilibrium |
| LDSC | LD score regression |
| S-LDSC | stratified LD score regression |
| IV | instrumental variable |
| ROC | receiver operating characteristic |
| AUC | area under the receiver operating characteristic curve |
| eQTL | expression quantitative trait loci |
| TWAS | transcriptome-wide association study |
| eGFR | estimated glomerular filtration rate |
| AD | Alzheimer’s disease |
References
- Sulkava, S.; Ollila, H.; Alasaari, J.; Puttonen, S.; Härmä, M.; Viitasalo, K.; Lahtinen, A.; Lindström, J.; Toivola, A.; Sulkava, R.; et al. Common Genetic Variation Near Melatonin Receptor 1A Gene Linked to Job-Related Exhaustion in Shift Workers. Sleep 2017, 40, zsw011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schormair, B.; Zhao, C.; Bell, S.; Tilch, E.; Salminen, A.V.; Pütz, B.; Dauvilliers, Y.; Stefani, A.; Högl, B.; Poewe, W.; et al. Identification of Novel Risk Loci for Restless Legs Syndrome in Genome-Wide Association Studies in Individuals of European Ancestry: A Meta-Analysis. Lancet Neurol. 2017, 16, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Hill, W.D.; Davies, N.M.; Ritchie, S.J.; Skene, N.G.; Bryois, J.; Bell, S.; Di Angelantonio, E.; Roberts, D.J.; Xueyi, S.; Davies, G.; et al. Genome-Wide Analysis Identifies Molecular Systems and 149 Genetic Loci Associated with Income. Nat. Commun. 2019, 10, 5741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helgeland, H.; Sodeland, M.; Zoric, N.; Torgersen, J.S.; Grammes, F.; von Lintig, J.; Moen, T.; Kjøglum, S.; Lien, S.; Våge, D.I. Genomic and Functional Gene Studies Suggest a Key Role of Beta-Carotene Oxygenase 1 like (Bco1l) Gene in Salmon Flesh Color. Sci. Rep. 2019, 9, 20061. [Google Scholar] [CrossRef] [Green Version]
- Timmins, I.R.; Zaccardi, F.; Nelson, C.P.; Franks, P.W.; Yates, T.; Dudbridge, F. Genome-Wide Association Study of Self-Reported Walking Pace Suggests Beneficial Effects of Brisk Walking on Health and Survival. Commun. Biol. 2020, 3, 634. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.; Brown, A.M.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A Brief History of Human Disease Genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer's Disease in Late Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.S.; Pericakvance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of Apolipoprotein E Allele ϵ4 with Late-Onset Familial and Sporadic Alzheimer’s Disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.-C. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef] [Green Version]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a Breast Cancer Susceptibility Gene, BRCA2, to Chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef]
- Risch, N.; Merikangas, K. The Future of Genetic Studies of Complex Human Diseases. Science 1996, 273, 1516–1517. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Schork, N.J. Genetic Dissection of Complex Traits. Science 1994, 265, 2037–2048. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, R.A.; Belmont, J.W.; Hardenbol, P.; Willis, T.D.; Yu, F.; Yang, H.; Ch’ang, L.-Y.; Huang, W.; Liu, B.; Shen, Y.; et al. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, K.; Ohnishi, Y.; Iida, A.; Sekine, A.; Yamada, R.; Tsunoda, T.; Sato, H.; Sato, H.; Hori, M.; Nakamura, Y.; et al. Functional SNPs in the Lymphotoxin-A Gene that are Associated with Susceptibility to Myocardial Infarction. Nat. Genet. 2002, 32, 650–654. [Google Scholar] [CrossRef]
- Kennedy, G.C.; Matsuzaki, H.; Dong, S.; Liu, W.-M.; Huang, J.; Liu, G.; Su, X.; Cao, M.; Chen, W.; Zhang, J.; et al. Large-Scale Genotyping of Complex DNA. Nat. Biotechnol. 2003, 21, 1233–1237. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S. The HapMap and Genome-Wide Association Studies in Diagnosis and Therapy. Annu. Rev. Med. 2009, 60, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Plagnol, V.; Hu, T.T.; Toomajian, C.; Clark, R.M.; Ossowski, S.; Ecker, J.; Weigel, D.; Nordborg, M. Recombination and Linkage Disequilibrium in Arabidopsis thaliana. Nat. Genet. 2007, 39, 1151–1155. [Google Scholar] [CrossRef]
- Yang, H.; Ding, Y.; Hutchins, L.N.; Szatkiewicz, J.; Bell, A.T.; Paigen, B.J.; Graber, J.; De Villena, F.P.-M.; Churchill, G.A. A Customized and Versatile High-Density Genotyping Array for the Mouse. Nat. Methods 2009, 6, 663–666. [Google Scholar] [CrossRef]
- Barson, N.J.; Aykanat, T.; Hindar, K.; Baranski, M.; Bolstad, G.H.; Fiske, P.; Jacq, C.; Jensen, A.J.; Johnston, S.; Karlsson, S.; et al. Sex-Dependent Dominance at a Single Locus Maintains Variation in Age at Maturity in Salmon. Nature 2015, 528, 405–408. [Google Scholar] [CrossRef]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.-X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome Evolution across 1011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, A.; Sterken, M.G.; De Bos, J.U.; Van Creij, J.; Kamble, R.; Snoek, B.; Kammenga, J.E.; Houtkooper, R.H. Natural Genetic Variation in C. Elegansidentified Genomic Loci Controlling Metabolite Levels. Genome Res. 2018, 28, 1296–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, T.; Hicks, M.; Yu, H.-C.; Biggs, W.H.; Kirkness, E.F.; Menni, C.; Zierer, J.; Small, K.S.; Mangino, M.; Messier, H.; et al. Whole-Genome Sequencing Identifies Common-to-Rare Variants Associated with Human Blood Metabolites. Nat. Genet. 2017, 49, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Weng, L.-C.; Roselli, C.; Lin, H.; Haggerty, C.; Shoemaker, M.B.; Barnard, J.; Arking, D.E.; Chasman, D.I.; Albert, C.; et al. Association Between Titin Loss-of-Function Variants and Early-Onset Atrial Fibrillation. JAMA 2018, 320, 2354–2364. [Google Scholar] [CrossRef]
- Chia, R.; Center, T.A.G.; Sabir, M.S.; Bandres-Ciga, S.; Saez-Atienzar, S.; Reynolds, R.H.; Gustavsson, E.; Walton, R.L.; Ahmed, S.; Viollet, C.; et al. Genome Sequencing Analysis Identifies New Loci Associated with Lewy Body Dementia and Provides Insights into its Genetic Architecture. Nat. Genet. 2021, 53, 294–303. [Google Scholar] [CrossRef]
- Zhang, M.-Y.; Xue, C.; Hu, H.; Li, J.; Xue, Y.; Wang, R.; Fan, J.; Zou, C.; Tao, S.; Qin, M.; et al. Genome-Wide Association Studies Provide Insights into the Genetic Determination of Fruit Traits of Pear. Nat. Commun. 2021, 12, 1144. [Google Scholar] [CrossRef]
- Li, J.H.; Mazur, C.A.; Berisa, T.; Pickrell, J.K. Low-Pass Sequencing Increases the Power of GWAS and Decreases Measurement Error of Polygenic Risk Scores Compared to Genotyping Arrays. Genome Res. 2021, 31, 529–537. [Google Scholar] [CrossRef]
- Martin, A.R.; Atkinson, E.G.; Chapman, S.B.; Stevenson, A.; Stroud, R.E.; Abebe, T.; Akena, D.; Alemayehu, M.; Ashaba, F.K.; Atwoli, L.; et al. Low-Coverage Sequencing Cost-Effectively Detects Known and Novel Variation in Underrepresented Populations. Am. J. Hum. Genet. 2021, 108, 656–668. [Google Scholar] [CrossRef]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.-Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement Factor H Polymorphism in Age-Related Macular Degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Allen, H.L.; Estrada, K.; Lettre, G.; Berndt, S.I.; Weedon, M.N.; Rivadeneira, F.; Willer, C.J.; Jackson, A.U.; Vedantam, S.; Raychaudhuri, S.; et al. Hundreds of Variants Clustered in Genomic Loci and Biological Pathways Affect Human Height. Nature 2010, 467, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Corvin, A.; Sullivan, P.F. What Next in Schizophrenia Genetics for the Psychiatric Genomics Consortium? Schizophr. Bull. 2016, 42, 538–541. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, A.; Go, M.J.; Zhang, W.; Below, E.J.; Gaulton, K.J.; Ferreira, T.; Horikoshi, M.; Johnson, A.D.; Ng, M.C.Y.; Prokopenko, I.; et al. Genome-Wide Trans-Ancestry Meta-Analysis Provides Insight into the Genetic Architecture of Type 2 Diabetes Susceptibility. Nat. Genet. 2014, 46, 234–244. [Google Scholar] [CrossRef]
- The International Parkinson Disease Genomics Consortium (IPDGC). Ten Years of the International Parkinson Disease Genomics Consortium: Progress and Next Steps. J. Park. Dis. 2020, 10, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Evangelou, E.; Program, T.M.V.; Warren, H.R.; Mosen-Ansorena, D.; Mifsud, B.; Pazoki, R.; Gao, H.; Ntritsos, G.; Dimou, N.; Cabrera, C.P.; et al. Genetic Analysis of over 1 Million People Identifies 535 New Loci Associated with Blood Pressure Traits. Nat. Genet. 2018, 50, 1412–1425. [Google Scholar] [CrossRef] [Green Version]
- Klarin, D.; Damrauer, S.; Cho, K.; Sun, Y.V.; Teslovich, T.M.; Honerlaw, J.; Gagnon, D.R.; Duvall, S.L.; Li, J.; Peloso, G.M.; et al. Genetics of Blood Lipids among ~300,000 Multi-Ethnic Participants of the Million Veteran Program. Nat. Genet. 2018, 50, 1514–1523. [Google Scholar] [CrossRef]
- Chen, V.L.; Du, X.; Chen, Y.; Kuppa, A.; Handelman, S.K.; Vohnoutka, R.B.; Peyser, P.A.; Palmer, N.D.; Bielak, L.F.; Halligan, B.; et al. Genome-Wide Association Study of Serum Liver Enzymes Implicates Diverse Metabolic and Liver Pathology. Nat. Commun. 2021, 12, 816. [Google Scholar] [CrossRef]
- Ferreira, M.A.; Collaborators, E.; Gamazon, E.R.; Al-Ejeh, F.; Aittomäki, K.; Andrulis, I.L.; Anton-Culver, H.; Arason, A.; Arndt, V.; Aronson, K.J.; et al. Genome-Wide Association and Transcriptome Studies Identify Target Genes and Risk Loci for Breast Cancer. Nat. Commun. 2019, 10, 1741. [Google Scholar] [CrossRef] [Green Version]
- Wuttke, M.; Li, Y.; Li, M.; Sieber, K.B.; Feitosa, M.F.; Gorski, M.; Tin, A.; Wang, L.; Chu, A.Y.; Hoppmann, A.; et al. A Catalog of Genetic Loci associated with Kidney Function from Analyses of a Million Individuals. Nat. Genet. 2019, 51, 957–972. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.A.; Kemp, J.P.; Youlten, S.E.; Laurent, L.; Logan, J.G.; Chai, R.C.; Vulpescu, N.A.; Forgetta, V.; Kleinman, A.; Mohanty, S.T.; et al. An Atlas of Genetic Influences on Osteoporosis in Humans and Mice. Nat. Genet. 2019, 51, 258–266. [Google Scholar] [CrossRef]
- Foo, J.N.; Chew, E.G.Y.; Chung, S.J.; Peng, R.; Blauwendraat, C.; Nalls, M.A.; Mok, K.Y.; Satake, W.; Toda, T.; Chao, Y.; et al. Identification of Risk Loci for Parkinson Disease in Asians and Comparison of Risk Between Asians and Europeans. JAMA Neurol. 2020, 77, 746–754. [Google Scholar] [CrossRef]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 New Risk Loci for Type 2 Diabetes and Related Vascular Outcomes among 1.4 Million Participants in a Multi-Ancestry Meta-Analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef]
- Choquet, H.; Melles, R.B.; Anand, D.; Yin, J.; Cuellar-Partida, G.; Wang, W.; Hoffmann, T.J.; Nair, K.S.; Hysi, P.G.; Lachke, S.A.; et al. A Large Multiethnic GWAS Meta-Analysis of Cataract Identifies New Risk Loci and Sex-Specific Effects. Nat. Commun. 2021, 12, 3595. [Google Scholar] [CrossRef]
- Shungin, D.; Haworth, S.; Divaris, K.; Agler, C.; Kamatani, Y.; Lee, M.K.; Grinde, K.; Hindy, G.; Alaraudanjoki, V.; Pesonen, P.; et al. Genome-Wide Analysis of Dental Caries and Periodontitis Combining Clinical and Self-Reported Data. Nat. Commun. 2019, 10, 2773. [Google Scholar] [CrossRef] [Green Version]
- Yengo, L.; Sidorenko, J.; Kemper, E.K.; Zheng, Z.; Wood, A.R.; Weedon, M.; Frayling, T.; Hirschhorn, J.; Yang, J.; Visscher, P.M.; et al. Meta-Analysis of Genome-Wide Association Studies for Height and Body Mass Index In ∼700,000 Individuals of European Ancestry. Hum. Mol. Genet. 2018, 27, 3641–3649. [Google Scholar] [CrossRef]
- Wright, K.M.; Rand, K.A.; Kermany, A.; Noto, K.; Curtis, D.; Garrigan, D.; Slinkov, D.; Dorfman, I.; Granka, J.M.; Byrnes, J.; et al. A Prospective Analysis of Genetic Variants Associated with Human Lifespan. G3 Genes Genomes Genet. 2019, 9, 2863–2878. [Google Scholar] [CrossRef] [Green Version]
- Cuellar-Partida, G.; Tung, J.Y.; Eriksson, N.; Albrecht, E.; Aliev, F.; Andreassen, O.A.; Barroso, I.; Beckmann, J.S.; Boks, M.P.; Boomsma, D.I.; et al. Genome-Wide Association Study Identifies 48 Common Genetic Variants Associated with Handedness. Nat. Hum. Behav. 2021, 5, 59–70. [Google Scholar] [CrossRef]
- Pulit, S.L.; Stoneman, C.; Morris, A.P.; Wood, A.R.; Glastonbury, C.A.; Tyrrell, J.; Yengo, L.; Ferreira, T.; Marouli, E.; Ji, Y.; et al. Meta-Analysis of Genome-Wide Association Studies for Body Fat Distribution in 694 649 Individuals of European Ancestry. Hum. Mol. Genet. 2019, 28, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Kranzler, H.R.; Zhou, H.; Kember, R.L.; Smith, R.V.; Justice, A.C.; Damrauer, S.; Tsao, P.S.; Klarin, D.; Baras, A.; Reid, J.; et al. Genome-Wide Association Study of Alcohol Consumption and Use Disorder in 274,424 Individuals from Multiple Populations. Nat. Commun. 2019, 10, 1499. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Jiang, Y.; Wedow, R.; Li, Y.; Brazel, D.M.; Chen, F.; Datta, G.; Davila-Velderrain, J.; McGuire, D.; Tian, C.; et al. Association Studies of up to 1.2 Million Individuals Yield New Insights into the Genetic Etiology of Tobacco and Alcohol Use. Nat. Genet. 2019, 51, 237–244. [Google Scholar] [CrossRef]
- Jones, S.E.; Lane, J.M.; Wood, A.R.; Van Hees, V.T.; Tyrrell, J.; Beaumont, R.N.; Jeffries, A.R.; Dashti, H.S.; Hillsdon, M.; Ruth, K.S.; et al. Genome-Wide Association Analyses of Chronotype in 697,828 Individuals Provides Insights into Circadian Rhythms. Nat. Commun. 2019, 10, 343. [Google Scholar] [CrossRef] [Green Version]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-Wide Meta-Analysis of Depression Identifies 102 Independent Variants and Highlights the Importance of the Prefrontal Brain Regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnér, R.K.; Biroli, P.; Kong, E.; Meddens, S.F.W.; Wedow, R.; Fontana, M.A.; Lebreton, M.; Tino, S.P.; Abdellaoui, A.; Hammerschlag, A.R.; et al. Genome-Wide Association Analyses of Risk Tolerance and Risky Behaviors in over 1 Million Individuals Identify Hundreds of Loci and Shared Genetic Influences. Nat. Genet. 2019, 51, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; de Leeuw, C.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.I.; et al. Genome-Wide Association Meta-Analysis in 269,867 Individuals Identifies New Genetic and Functional Links to Intelligence. Nat. Genet. 2018, 50, 912–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselmans, B.M.L.; Jansen, R.; Ip, H.F.; Van Dongen, J.; Abdellaoui, A.; van de Weijer, M.; Bao, Y.; Smart, M.; Kumari, M.; Willemsen, G.; et al. Multivariate Genome-Wide Analyses of the Well-Being Spectrum. Nat. Genet. 2019, 51, 445–451. [Google Scholar] [CrossRef]
- Lo, M.-T.; Hinds, A.D.; Tung, J.Y.; Franz, C.; Fan, C.-C.; Wang, Y.; Smeland, O.B.; Schork, A.; Holland, D.; Kauppi, K.; et al. Genome-Wide Analyses for Personality Traits Identify Six Genomic Loci and Show Correlations with Psychiatric Disorders. Nat. Genet. 2017, 49, 152–156. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Wedow, R.; Okbay, A.; Kong, E.; Maghzian, O.; Zacher, M.; Nguyen-Viet, T.A.; Bowers, P.; Sidorenko, J.; Linnér, R.K.; et al. Gene Discovery and Polygenic Prediction from a Genome-Wide Association Study of Educational Attainment in 1.1 Million Individuals. Nat. Genet. 2018, 50, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Karlsson Linnér, R.; Mallard, T.T.; Barr, P.B.; Sanchez-Roige, S.; Madole, J.W.; Driver, M.N.; Poore, H.E.; de Vlaming, R.; Grotzinger, A.D.; Tielbeek, J.J.; et al. Multivariate Analysis of 1.5 Million People Identifies Genetic Associations with Traits Related to Self-Regulation and Addiction. Nat. Neurosci. 2021, 24, 1367–1376. [Google Scholar] [CrossRef]
- Atwell, S.; Huang, Y.S.; Vilhjalmsson, B.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.; Hu, T.T.; et al. Genome-wide Association Study of 107 Phenotypes in Arabidopsis Thaliana Inbred Lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef]
- Alonso-Blanco, C.; Andrade, J.; Becker, C.; Bemm, F.; Bergelson, J.; Borgwardt, K.M.; Cao, J.; Chae, E.; Dezwaan, T.M.; Ding, W.; et al. 1,135 Genomes Reveal the Global Pattern of Polymorphism in Arabidopsis thaliana. Cell 2016, 166, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.P.; Ramu, P.; Deshpande, S.P.; Hash, C.T.; Shah, T.; Upadhyaya, H.D.; Riera-Lizarazu, O.; Brown, P.J.; Acharya, C.B.; Mitchell, S.E.; et al. Population Genomic and Genome-Wide Association Studies of Agroclimatic Traits in Sorghum. Proc. Natl. Acad. Sci. USA 2013, 110, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Massouras, A.; Inoue, Y.; Peiffer, J.; Ràmia, M.; Tarone, A.; Turlapati, L.; Zichner, T.; Zhu, D.; Lyman, R.F.; et al. Natural Variation in Genome Architecture among 205 Drosophila melanogaster Genetic Reference Panel lines. Genome Res. 2014, 24, 1193–1208. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic Variation in 3010 Diverse Accessions of Asian Cultivated Rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Bloom, J.S.; Kotenko, I.; Sadhu, M.; Treusch, S.; Albert, F.; Kruglyak, L. Genetic Interactions Contribute Less than Additive Effects to Quantitative Trait Variation in Yeast. Nat. Commun. 2015, 6, 8712. [Google Scholar] [CrossRef] [Green Version]
- Burke, D.T.; Kozloff, K.M.; Chen, S.; West, J.L.; Wilkowski, J.M.; Goldstein, S.A.; Miller, R.A.; Galecki, A.T. Dissection of Complex Adult Traits in a Mouse Synthetic Population. Genome Res. 2012, 22, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.; Wang, X.; Zhou, T.; Jin, Y.; Tan, S.; Jiang, C.; Geng, X.; Li, N.; Shi, H.; Zeng, Q.; et al. Genome-Wide Association Study Reveals Multiple Novel QTL Associated with Low Oxygen Tolerance in Hybrid Catfish. Mar. Biotechnol. 2017, 19, 379–390. [Google Scholar] [CrossRef]
- Thelwall, M.; Munafo, M.; Mas-Bleda, A.; Stuart, E.; Makita, M.; Weigert, V.; Keene, C.; Khan, N.; Drax, K.; Kousha, K. Is Useful Research Data Usually shared? An Investigation of Genome-Wide Association Study Summary Statistics. PLoS ONE 2020, 15, e0229578. [Google Scholar] [CrossRef] [Green Version]
- Buniello, A. Why We Need More Freely Available Cancer GWAS Summary Statistics. Available online: https://blog.opentargets.org/open-sharing-of-cancer-summary-statistics/ (accessed on 16 July 2021).
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of Published Genome-Wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Togninalli, M.; Seren, A.; Freudenthal, A.J.; Monroe, J.G.; Meng, D.; Nordborg, M.; Weigel, D.; Borgwardt, K.M.; Korte, A.; Grimm, D.G. AraPheno and the AraGWAS Catalog 2020: A Major Database Update Including RNA-Seq and Knockout Mutation Data for Arabidopsis thaliana. Nucleic Acids Res. 2020, 48, D1063–D1068. [Google Scholar] [CrossRef]
- Collins, A.L.; Kim, Y.; Sklar, P.; O'Donovan, M.; Sullivan, P.F. International Schizophrenia Consortium Hypothesis-Driven Candidate Genes for Schizophrenia Compared to Genome-Wide Association Results. Psychol. Med. 2012, 42, 607–616. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Geschwind, D.H. Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders. Cell 2019, 177, 162–183. [Google Scholar] [CrossRef] [Green Version]
- Willoughby, E.A.; Love, A.; McGue, M.; Iacono, W.G.; Quigley, J.; Lee, J.J. Free Will, Determinism, and Intuitive Judgments About the Heritability of Behavior. Behav. Genet. 2019, 49, 136–153. [Google Scholar] [CrossRef] [Green Version]
- Kruuk, L.E.B.; Clutton-Brock, T.H.; Slate, J.; Pemberton, J.M.; Brotherstone, S.; Guinness, F.E. Heritability of Fitness in a Wild Mammal Population. Proc. Natl. Acad. Sci. USA 2000, 97, 698–703. [Google Scholar] [CrossRef] [Green Version]
- Vink, J.M.; Willemsen, G.; Boomsma, D.I. Heritability of Smoking Initiation and Nicotine Dependence. Behav. Genet. 2005, 35, 397–406. [Google Scholar] [CrossRef]
- Byrne, B.; Coventry, W.L.; Olson, R.K.; Samuelsson, S.; Corley, R.; Willcutt, E.G.; Wadsworth, S.; DeFries, J.C. Genetic and Environmental Influences on Aspects of Literacy and Language in Early Childhood: Continuity and Change from Preschool to Grade 2. J. Neurolinguist. 2009, 22, 219–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernandez, J.; Prati, D.; Baselli, G.; Asselta, R.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic Mechanisms of Critical Illness in COVID-19. Nature 2020, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Ganna, A.; COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 1626. [Google Scholar] [CrossRef]
- Wasser, S.K.; Clark, W.J.; Drori, O.; Kisamo, E.S.; Mailand, C.; Mutayoba, B.; Stephens, M. Combating the Illegal Trade in African Elephant Ivory with DNA Forensics. Conserv. Biol. 2008, 22, 1065–1071. [Google Scholar] [CrossRef]
- Novembre, J.; Johnson, T.; Bryc, K.; Kutalik, Z.; Boyko, A.R.; Auton, A.; Indap, A.; King, K.S.; Bergmann, S.; Nelson, M.R.; et al. Genes Mirror Geography within Europe. Nature 2008, 456, 98–101. [Google Scholar] [CrossRef] [Green Version]
- The 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A Reference Panel of 64,976 Haplotypes for Genotype Imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef] [Green Version]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Patterson, N.; Daly, M.J.; Price1, A.L.; Neale, B.M. LD Score Regression Distinguishes Confounding from Polygenicity in Genome-Wide Association Studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Gazal, S.; Loh, P.-R.; Finucane, H.K.; Ganna, A.; Schoech, A.; Sunyaev, S.; Price, A.L. Functional Architecture of Low-Frequency Variants Highlights Strength of Negative Selection across Coding and Non-Coding Annotations. Nat. Genet. 2018, 50, 1600–1607. [Google Scholar] [CrossRef]
- O'Connor, L.J.; Schoech, A.P.; Hormozdiari, F.; Gazal, S.; Patterson, N.; Price, A.L. Extreme Polygenicity of Complex Traits Is Explained by Negative Selection. Am. J. Hum. Genet. 2019, 105, 456–476. [Google Scholar] [CrossRef] [Green Version]
- Bloom, J.S.; Boocock, J.; Treusch, S.; Sadhu, M.J.; Day, L.; Oates-Barker, H.; Kruglyak, L. Rare Variants Contribute Disproportionately to Quantitative Trait Variation in Yeast. eLife 2019, 8, e49212. [Google Scholar] [CrossRef]
- Weiner, D.J.; Wigdor, E.M.; Ripke, S.; Walters, R.K.; Kosmicki, J.A.; Grove, J.; Samocha, K.E.; Goldstein, J.I.; Okbay, A.; Bybjerg-Grauholm, J.; et al. Polygenic Transmission Disequilibrium Confirms that Common and Rare Variation Act Additively to Create Risk for Autism Spectrum Disorders. Nat. Genet. 2017, 49, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Akbari, P.; Gilani, A.; Sosina, O.; Kosmicki, J.A.; Khrimian, L.; Fang, Y.-Y.; Persaud, T.; Garcia, V.; Sun, D.; Li, A.; et al. Sequencing of 640,000 Exomes identifies GPR75 Variants Associated with Protection from Obesity. Science 2021, 373, eabf8683. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-Wide Polygenic Scores for Common Diseases Identify Individuals with Risk Equivalent to Monogenic Mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Sebastiani, P.; Solovieff, N.; Hartley, S.W.; Milton, J.N.; Riva, A.; Dworkis, D.A.; Melista, E.; Klings, E.; Garrett, M.E.; Telen, M.J.; et al. Genetic Modifiers of the Severity of Sickle Cell Anemia Identified through a Genome-Wide Association Study. Am. J. Hematol. 2010, 85, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Wright, F.A.; Strug, L.J.; Doshi, V.K.; Commander, C.; Blackman, S.; Sun, L.; Berthiaume, Y.; Cutler, D.M.; Cojocaru, A.; Collaco, J.M.; et al. Genome-Wide Association and Linkage Identify Modifier Loci of Lung Disease Severity in Cystic Fibrosis at 11p13 and 20q13.2. Nat. Genet. 2011, 43, 539–546. [Google Scholar] [CrossRef]
- Navarini, A.A.; Simpson, M.A.; Weale, M.; Knight, J.; Carlavan, I.; Reiniche, P.; Burden, D.A.; Layton, A.; Bataille, V.; Allen, M.; et al. Genome-wide association study identifies three novel susceptibility loci for severe Acne vulgaris. Nat. Commun. 2014, 5, 4020. [Google Scholar] [CrossRef] [Green Version]
- Moss, D.J.H.; Pardiñas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; Holmans, P.; Jones, L.; et al. Identification of Genetic Variants Associated with Huntington’s Disease Progression: A Genome-Wide Association Study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef]
- Wei, W.; Hemani, G.; Haley, C. Detecting Epistasis in Human Complex Traits. Nat. Rev. Genet. 2014, 15, 722–733. [Google Scholar] [CrossRef]
- Chatelain, C.; Durand, G.; Thuillier, V.; Auge, F. Performance of Epistasis Detection Methods in Semi-Simulated GWAS. BMC Bioinform. 2018, 19, 231. [Google Scholar] [CrossRef]
- Fisher Sir, R.A.; Fisher, R.A. The Genetical Theory of Natural Selection: A Complete Variorum Edition; OUP: Oxford, UK, 1999; ISBN 9780198504405. [Google Scholar]
- Crow, J.F. On Epistasis: Why it is Unimportant in Polygenic Directional Selection. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1241–1244. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, M.; Besnier, F.; Siegel, P.B.; Carlborg, O. Replication and Explorations of High-Order Epistasis Using a Large Advanced Intercross Line Pedigree. PLoS Genet. 2011, 7, e1002180. [Google Scholar] [CrossRef] [Green Version]
- Hayward, J.J.; Castelhano, M.; Oliveira, K.C.; Corey, E.; Balkman, C.; Baxter, T.L.; Casal, M.L.; Center, S.A.; Fang, M.; Garrison, S.J.; et al. Complex Disease and Phenotype Mapping in the Domestic Dog. Nat. Commun. 2016, 7, 10460. [Google Scholar] [CrossRef]
- Gusareva, E.S.; Carrasquillo, M.M.; Bellenguez, C.; Cuyvers, E.; Colon, S.; Graff-Radford, N.R.; Petersen, R.C.; Dickson, D.W.; John, J.M.M.; Bessonov, K.; et al. Genome-Wide Association Interaction Analysis for Alzheimer's Disease. Neurobiol. Aging 2014, 35, 2436–2443. [Google Scholar] [CrossRef] [Green Version]
- Sinnott-Armstrong, N.; Naqvi, S.; Rivas, M.; Pritchard, J.K. GWAS of Three Molecular Traits Highlights Core Genes and Pathways Alongside a Highly Polygenic Background. eLife 2021, 10, e58615. [Google Scholar] [CrossRef]
- Fournier, T.; Saada, O.A.; Hou, J.; Peter, J.; Caudal, E.; Schacherer, J. Extensive Impact of Low-Frequency Variants on the Phenotypic Landscape at Population-Scale. eLife 2019, 8, e49258. [Google Scholar] [CrossRef]
- Mäki-Tanila, A.; Hill, W.G. Influence of Gene Interaction on Complex Trait Variation with Multilocus Models. Genetics 2014, 198, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marigorta, U.M.; Rodríguez, J.A.; Gibson, G.; Navarro, A. Replicability and Prediction: Lessons and Challenges from GWAS. Trends Genet. 2018, 34, 504–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, J.W.; Ioannidis, J.P.A. Reproducibility in the UK Biobank of Genome-Wide Significant Signals Discovered in Earlier Genome-Wide Association Studies. Sci. Rep. 2021, 11, 18625. [Google Scholar] [CrossRef] [PubMed]
- Marigorta, U.M.; Navarro, A. High Trans-Ethnic Replicability of GWAS Results Implies Common Causal Variants. PLoS Genet. 2013, 9, e1003566. [Google Scholar] [CrossRef] [Green Version]
- N'Diaye, A.; Chen, G.K.; Palmer, C.D.; Ge, B.; Tayo, B.; Mathias, R.A.; Ding, J.; Nalls, M.A.; Adeyemo, A.; Adoue, V.; et al. Identification, Replication, and Fine-Mapping of Loci Associated with Adult Height in Individuals of African Ancestry. PLoS Genet. 2011, 7, e1002298. [Google Scholar] [CrossRef]
- Coram, M.; Duan, Q.; Hoffmann, T.J.; Thornton, T.; Knowles, J.W.; Johnson, N.A.; Ochs-Balcom, H.M.; Donlon, T.A.; Martin, L.W.; Eaton, C.; et al. Genome-wide Characterization of Shared and Distinct Genetic Components that Influence Blood Lipid Levels in Ethnically Diverse Human Populations. Am. J. Hum. Genet. 2013, 92, 904–916. [Google Scholar] [CrossRef] [Green Version]
- Adeyemo, A.; Tekola-Ayele, F.; Doumatey, A.P.; Bentley, A.R.; Chen, G.; Huang, H.; Zhou, J.; Shriner, D.; Fasanmade, O.; Okafor, G.; et al. Evaluation of Genome Wide Association Study Associated Type 2 Diabetes Susceptibility Loci in Sub Saharan Africans. Front. Genet. 2015, 6, 335. [Google Scholar] [CrossRef] [Green Version]
- Spracklen, C.N.; Horikoshi, M.; Kim, Y.J.; Lin, K.; Bragg, F.; Moon, S.; Suzuki, K.; Tam, C.H.T.; Tabara, Y.; Kwak, S.-H.; et al. Identification of Type 2 Diabetes Loci in 433,540 East Asian Individuals. Nat. Cell Biol. 2020, 582, 240–245. [Google Scholar] [CrossRef]
- Tedja, M.S.; Wojciechowski, R.; Hysi, P.G.; Eriksson, N.; Furlotte, N.A.; Verhoeven, V.J.M.; Iglesias, A.I.; Meester-Smoor, M.A.; Tompson, S.W.; Fan, Q.; et al. Genome-Wide Association Meta-Analysis Highlights Light-Induced Signaling as a Driver for Refractive Error. Nat. Genet. 2018, 50, 834–848. [Google Scholar] [CrossRef]
- Lam, M.; Chen, C.-Y.; Li, Z.; Martin, A.R.; Bryois, J.; Ma, X.; Gaspar, H.; Ikeda, M.; Benyamin, B.; Brown, B.C.; et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat. Genet. 2019, 51, 1670–1678. [Google Scholar] [CrossRef]
- Liu, J.Z.; Van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.; Jostins, L.; Shah, T.; et al. Association Analyses Identify 38 Susceptibility Loci for Inflammatory Bowel Disease and Highlight Shared Genetic Risk across Populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Li, Y.R.; Keating, B.J. Trans-Ethnic Genome-Wide Association Studies: Advantages and Challenges of Mapping in Diverse Populations. Genome Med. 2014, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Schaid, D.J.; Chen, W.; Larson, N.B. From Genome-Wide Associations to Candidate Causal Variants by Statistical Fine-Mapping. Nat. Rev. Genet. 2018, 19, 491–504. [Google Scholar] [CrossRef]
- Wood, A.R.; The Electronic Medical Records and Genomics (eMERGE) Consortium; Esko, T.; Yang, J.; Vedantam, S.; Pers, T.H.; Gustafsson, S.; Chu, A.Y.; Estrada, K.; Luan, J.; et al. Defining the Role of Common Variation in the Genomic and Biological Architecture of Adult Human Height. Nat. Genet. 2014, 46, 1173–1186. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, A.C.; Daetwyler, H.D.; Chamberlain, A.J.; Ponce, C.H.; Sargolzaei, M.; Schenkel, F.S.; Sahana, G.; Govignon-Gion, A.; Boitard, S.; Dolezal, M.; et al. Meta-Analysis of Genome-Wide Association Studies for Cattle Stature Identifies Common Genes that Regulate Body Size in Mammals. Nat. Genet. 2018, 50, 362–367. [Google Scholar] [CrossRef]
- Makvandi-Nejad, S.; Hoffman, G.E.; Allen, J.; Chu, E.; Gu, E.; Chandler, A.M.; Loredo, A.I.; Bellone, R.R.; Mezey, J.G.; Brooks, S.; et al. Four Loci Explain 83% of Size Variation in the Horse. PLoS ONE 2012, 7, e39929. [Google Scholar] [CrossRef] [Green Version]
- Samaha, G.; Wade, C.M.; Beatty, J.; Lyons, L.A.; Fleeman, L.M.; Haase, B. Mapping the Genetic Basis of Diabetes Mellitus in the Australian Burmese Cat (Felis catus). Sci. Rep. 2020, 10, 19194. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host–Microbe Interactions have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Marouli, E.; Graff, M.; Medina-Gomez, C.; Lo, K.S.; Wood, A.R.; Kjaer, T.R.; Fine, R.S.; Lu, Y.; Schurmann, C.; Highland, H.M.; et al. Rare and Low-Frequency Coding Variants alter Human Adult Height. Nature 2017, 542, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; de Lange, K.M.; Jostins, L.; Moutsianas, L.; Randall, J.; Kennedy, A.N.; Lamb, A.C.; McCarthy, S.; Ahmad, T.; Edwards, C.; et al. Exploring the Genetic Architecture of Inflammatory Bowel Disease by Whole-Genome Sequencing identifies Association at ADCY7. Nat. Genet. 2017, 49, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Flannick, J.; Mercader, J.M.; Fuchsberger, C.; Udler, M.S.; Mahajan, A.; Wessel, J.; Teslovich, T.M.; Caulkins, L.; Koesterer, R.; Barajas-Olmos, F.; et al. Exome Sequencing of 20,791 Cases of Type 2 Diabetes and 24,440 Controls. Nature 2019, 570, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Al Eissa, M.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J.; et al. Exome Sequencing Identifies Rare Coding Variants in 10 Genes Which Confer Substantial Risk for Schizophrenia. medRxiv 2020. [Google Scholar] [CrossRef]
- Wang, Q.; Dhindsa, R.S.; Carss, K.; Harper, A.R.; Nag, A.; Tachmazidou, I.; Vitsios, D.; Deevi, S.V.V.; Mackay, A.; Muthas, D.; et al. Rare Variant Contribution to Human Disease in 281,104 UK Biobank Exomes. Nature 2021, 597, 527–532. [Google Scholar] [CrossRef]
- Blair, D.R.; Lyttle, C.S.; Mortensen, J.M.; Bearden, C.F.; Jensen, A.B.; Khiabanian, H.; Melamed, R.; Rabadan, R.; Bernstam, E.V.; Brunak, S.; et al. A Nondegenerate Code of Deleterious Variants in Mendelian Loci Contributes to Complex Disease Risk. Cell 2013, 155, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Freund, M.K.; Burch, K.S.; Shi, H.; Mancuso, N.; Kichaev, G.; Garske, K.M.; Pan, D.Z.; Miao, Z.; Mohlke, K.L.; Laakso, M.; et al. Phenotype-Specific Enrichment of Mendelian Disorder Genes near GWAS Regions across 62 Complex Traits. Am. J. Hum. Genet. 2018, 103, 535–552. [Google Scholar] [CrossRef] [Green Version]
- O’Seaghdha, C.M.; Wu, H.; Yang, Q.; Kapur, K.; Guessous, I.; Zuber, A.M.; Köttgen, A.; Stoudmann, C.; Teumer, A.; Kutalik, Z.; et al. Meta-Analysis of Genome-Wide Association Studies Identifies Six New Loci for Serum Calcium Concentrations. PLoS Genet. 2013, 9, e1003796. [Google Scholar] [CrossRef] [Green Version]
- Shelton, J.F.; Shastri, A.J.; Aslibekyan, S.; Auton, A. The 23andMe COVID-19 Team the UGT2A1/UGT2A2 Locus is Associated with COVID-19-Related Anosmia. bioRxiv 2021. [Google Scholar] [CrossRef]
- Nicolae, D.; Gamazon, E.; Zhang, W.; Duan, S.; Dolan, M.E.; Cox, N.J. Trait-Associated SNPs Are More Likely to be eQTLs: Annotation to Enhance Discovery from GWAS. PLoS Genet. 2010, 6, e1000888. [Google Scholar] [CrossRef]
- Nica, A.C.; Montgomery, S.; Dimas, A.S.; Stranger, B.; Beazley, C.; Barroso, I.; Dermitzakis, E.T. Candidate Causal Regulatory Effects by Integration of Expression QTLs with Complex Trait Genetic Associations. PLoS Genet. 2010, 6, e1000895. [Google Scholar] [CrossRef] [Green Version]
- Boix, C.A.; James, B.T.; Park, Y.P.; Meuleman, W.; Kellis, M. Regulatory Genomic Circuitry of Human Disease Loci by Integrative Epigenomics. Nature 2021, 590, 300–307. [Google Scholar] [CrossRef]
- Lee, P.H.; O’Dushlaine, C.; Thomas, B.; Purcell, S.M. INRICH: Interval-based Enrichment Analysis for Genome-Wide Association Studies. Bioinformatics 2012, 28, 1797–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pers, T.H.; Karjalainen, J.M.; Chan, Y.; Westra, H.-J.; Wood, A.R.; Yang, J.; Lui, J.C.; Vedantam, S.; Gustafsson, S.; Esko, T.; et al. Biological Interpretation of Genome-Wide Association Studies Using Predicted Gene Functions. Nat. Commun. 2015, 6, 5890. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.; Lee, S.H.; Trynka, G.; Finucane, H.; Vilhjalmsson, B.; Xu, H.; Zang, C.; Ripke, S.; Bulik-Sullivan, B.; Stahl, E.; et al. Partitioning Heritability of Regulatory and Cell-Type-Specific Variants across 11 Common Diseases. Am. J. Hum. Genet. 2014, 95, 535–552. [Google Scholar] [CrossRef] [Green Version]
- Finucane, H.K.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.-R.; Anttila, V.; Xu, H.; Zang, C.; Farh, K.; Ripke, S.; et al. Partitioning Heritability by Functional Annotation Using Genome-Wide Association Summary Statistics. Nat. Genet. 2015, 47, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Huang, Y.; Zhao, S.; Yao, Y.; Zhang, Y.; Qu, J.; Wu, N.; Su, J. Integrative Genomics Analysis reveals a 21q22.11 Locus Contributing Risk to COVID-19. Hum. Mol. Genet. 2021, 30, 1247–1258. [Google Scholar] [CrossRef]
- Elvsåshagen, T.; Shadrin, A.; Frei, O.; van der Meer, D.; Bahrami, S.; Kumar, V.J.; Smeland, O.; Westlye, L.T.; Andreassen, O.A.; Kaufmann, T. The Genetic Architecture of the Human Thalamus and its Overlap with Ten Common Brain Disorders. Nat. Commun. 2021, 12, 2909. [Google Scholar] [CrossRef]
- Tsetsos, F.; Yu, D.; Sul, J.H.; Huang, A.Y.; Illmann, C.; Osiecki, L.; Darrow, S.M.; Hirschtritt, M.E.; Greenberg, E.; Muller-Vahlet, K.R.; et al. Synaptic Processes and Immune-Related Pathways Implicated in Tourette Syndrome. Transl. Psychiatry 2021, 11, 56. [Google Scholar] [CrossRef]
- Hormozdiari, F.; Gazal, S.; Van De Geijn, B.; Finucane, H.K.; Ju, C.J.-T.; Loh, P.-R.; Schoech, A.; Reshef, Y.; Liu, X.; O’Connor, L.; et al. Leveraging Molecular Quantitative Trait Loci to Understand the Genetic Architecture of Diseases and Complex Traits. Nat. Genet. 2018, 50, 1041–1047. [Google Scholar] [CrossRef]
- Finucane, H.K.; Reshef, Y.A.; Anttila, V.; Slowikowski, K.; Gusev, A.; Byrnes, A.; Gazal, S.; Loh, P.-R.; Lareau, C.; Shoresh, N.; et al. Heritability Enrichment of Specifically Expressed Genes Identifies Disease-Relevant Tissues and Cell Types. Nat. Genet. 2018, 50, 621–629. [Google Scholar] [CrossRef]
- Fromer, M.; Roussos, P.; Sieberts, S.K.; Johnson, J.; Kavanagh, D.H.; Perumal, T.M.; Ruderfer, D.M.; Oh, E.C.; Topol, A.; Shah, H.R.; et al. Gene Expression Elucidates Functional Impact of Polygenic Risk for Schizophrenia. Nat. Neurosci. 2016, 19, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium; Ripke, S.; Walters, J.T.R.; O’Donovan, M.C. Schizophrenia Working Group of the Psychiatric Genomics Consortium; Ripke, S.; Walters, J.T.R.; O’Donovan, M.C. Mapping Genomic Loci Prioritises Genes and Implicates Synaptic Biology in Schizophrenia. medRxiv 2020. [Google Scholar] [CrossRef]
- Landi, M.T.; Bishop, D.T.; MacGregor, S.; Machiela, M.J.; Stratigos, A.J.; Ghiorzo, P.; Brossard, M.; Calista, D.; Choi, J.; Fargnoli, M.C.; et al. Genome-Wide Association Meta-Analyses Combining Multiple Risk Phenotypes Provide Insights into the Genetic Architecture of Cutaneous Melanoma Susceptibility. Nat. Genet. 2020, 52, 494–504. [Google Scholar] [CrossRef]
- Sey, N.Y.A.; Hu, B.; Mah, W.; Fauni, H.; McAfee, J.C.; Rajarajan, P.; Brennand, K.J.; Akbarian, S.; Won, H. A Computational Tool (H-MAGMA) for Improved Prediction of Brain-Disorder Risk Genes by Incorporating Brain Chromatin Interaction Profiles. Nat. Neurosci. 2020, 23, 583–593. [Google Scholar] [CrossRef]
- Matoba, N.; Liang, D.; Sun, H.; Aygün, N.; McAfee, J.C.; Davis, J.E.; Raffield, L.M.; Qian, H.; Piven, J.; Li, Y.; et al. Common Genetic Risk Variants Identified in the SPARK Cohort Support DDHD2 as a Candidate Risk Gene for Autism. Transl. Psychiatry 2020, 10, 265. [Google Scholar] [CrossRef]
- Gamazon, E.R.; GTEx Consortium; Wheeler, H.; Shah, K.P.; Mozaffari, S.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; Nicolae, D.; et al. A Gene-Based Association Method for Mapping Traits Using Reference Transcriptome Data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; De Geus, E.J.C.; Boomsma, I.D.; Wright, A.F.; et al. Integrative approaches for Large-Scale Transcriptome-Wide Association Studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Barbeira, A.N.; GTEx Consortium; Dickinson, S.P.; Bonazzola, R.; Zheng, J.; Wheeler, H.E.; Torres, J.M.; Torstenson, E.S.; Shah, K.P.; Garcia, T.; et al. Exploring the Phenotypic Consequences of Tissue Specific Gene Expression Variation Inferred from GWAS Summary Statistics. Nat. Commun. 2018, 9, 1825. [Google Scholar] [CrossRef]
- Wainberg, M.; Sinnott-Armstrong, N.; Mancuso, N.; Barbeira, A.N.; Knowles, D.A.; Golan, D.; Ermel, R.; Ruusalepp, A.; Quertermous, T.; Hao, K.; et al. Opportunities and Challenges for Transcriptome-Wide Association Studies. Nat. Genet. 2019, 51, 592–599. [Google Scholar] [CrossRef]
- Kim-Hellmuth, S.; Aguet, F.; Oliva, M.; Muñoz-Aguirre, M.; Kasela, S.; Wucher, V.; Castel, S.E.; Hamel, A.R.; Viñuela, A.; Roberts, A.L.; et al. Cell Type–Specific Genetic Regulation of Gene Expression across Human Tissues. Science 2020, 369, eaaz8528. [Google Scholar] [CrossRef]
- Mancuso, N.; Shi, H.; Goddard, P.; Kichaev, G.; Gusev, A.; Pasaniuc, B. Integrating Gene Expression with Summary Association Statistics to Identify Genes Associated with 30 Complex Traits. Am. J. Hum. Genet. 2017, 100, 473–487. [Google Scholar] [CrossRef]
- Wang, X.W.H.; Wang, H.; Liu, S.; Ferjani, A.; Li, J.; Yan, J.; Yang, J.L.X.; Qin, F. Genetic Variation in ZmVPP1 contributes to Drought Tolerance in Maize Seedlings. Nat. Genet. 2016, 48, 1233–1241. [Google Scholar] [CrossRef]
- Plenge, R.M.; Scolnick, E.M.; Altshuler, D. Validating Therapeutic Targets through Human Genetics. Nat. Rev. Drug Discov. 2013, 12, 581–594. [Google Scholar] [CrossRef]
- Diogo, D.; Bastarache, L.; Liao, K.P.; Graham, R.R.; Fulton, R.S.; Greenberg, J.D.; Eyre, S.; Bowes, J.; Cui, J.; Lee, A.; et al. TYK2 Protein-Coding Variants Protect against Rheumatoid Arthritis and Autoimmunity, with No Evidence of Major Pleiotropic Effects on Non-Autoimmune Complex Traits. PLoS ONE 2015, 10, e0122271. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.R.; Cheng, L.; Gillooly, K.M.; Strnad, J.; Zupa-Fernandez, A.; Catlett, I.M.; Zhang, Y.; Heimrich, E.M.; McIntyre, K.W.; Cunningham, M.D.; et al. Autoimmune Pathways in Mice and Humans are Blocked by Pharmacological Stabilization of the TYK2 Pseudokinase Domain. Sci. Transl. Med. 2019, 11, eaaw1736. [Google Scholar] [CrossRef]
- Suarez-Gestal, M.; Calaza, M.; Endreffy, E.; Pullmann, R.; Ordi-Ros, J.; Sebastiani, G.D.; Ruzickova, S.; Santos, M.J.; Papasteriades, C.; Marchini, M.; et al. Replication of Recently Identified Systemic Lupus Erythematosus Genetic Associations: A Case–Control Study. Arthritis Res. Ther. 2009, 11, R69. [Google Scholar] [CrossRef] [Green Version]
- Wallace, C.; Smyth, D.J.; Maisuria-Armer, M.; Walker, N.M.; Todd, A.J.A.; Clayton, D.G. The Imprinted DLK1-MEG3 Gene Region on Chromosome 14q32.2 Alters Susceptibility to Type 1 Diabetes. Nat. Genet. 2010, 42, 68–71. [Google Scholar] [CrossRef] [Green Version]
- Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A Genome-Wide Association Study Identifies New Psoriasis Susceptibility Loci and an Interaction between HLA-C and ERAP1. Nat. Genet. 2010, 42, 985–990. [CrossRef] [Green Version]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-Wide Meta-Analysis increases to 71 the Number of Confirmed Crohn's Disease Susceptibility Loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [Green Version]
- Maher, B. Personal Genomes: The Case of the Missing Heritability. Nature 2008, 456, 18–21. [Google Scholar] [CrossRef]
- Gibson, G. Hints of Hidden Heritability in GWAS. Nat. Genet. 2010, 42, 558–560. [Google Scholar] [CrossRef] [PubMed]
- The International Schizophrenia Consortium; Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P. Common Polygenic Variation Contributes to Risk of Schizophrenia and Bipolar Disorder. Nature 2009, 460, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Genetic Risk and Outcome in Psychosis (GROUP); Stefansson, H.; Ophoff, R.A.; Steinberg, S.; Andreassen, O.A.; Cichon, S.; Rujescu, D.; Werge, T.; Pietiläinen, O.P.H.; Mors, O.; et al. Common Variants Conferring Risk of Schizophrenia. Nature 2009, 460, 744–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Levinson, D.F.; Duan, J.; Sanders, A.R.; Zheng, Y.; Pe’Er, I.; Dudbridge, F.; Holmans, P.; Whittemore, A.S.; Mowry, B.J.; et al. Common Variants on Chromosome 6p22.1 are Associated with Schizophrenia. Nature 2009, 460, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Goddard, M.; Visscher, P. Prediction of Individual Genetic Risk to Disease from Genome-Wide Association Studies. Genome Res. 2007, 17, 1520–1528. [Google Scholar] [CrossRef] [Green Version]
- Privé, F.; Arbel, J.; Vilhjálmsson, B.J. LDpred2: Better, Faster, Stronger. bioRxiv 2020. [Google Scholar] [CrossRef]
- Visscher, P.M.; Hill, W.G.; Wray, N.R. Heritability in the Genomics Era—Concepts and Misconceptions. Nat. Rev. Genet. 2008, 9, 255–266. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Wang, W.; Bradfield, J.; Li, J.; Cardinale, C.; Frackelton, E.; Kim, C.; Mentch, F.; Van Steen, K.; Visscher, P.M.; et al. Large Sample Size, Wide Variant Spectrum, and Advanced Machine-Learning Technique Boost Risk Prediction for Inflammatory Bowel Disease. Am. J. Hum. Genet. 2013, 92, 1008–1012. [Google Scholar] [CrossRef] [Green Version]
- Romagnoni, A.; International Inflammatory Bowel Disease Genetics Consortium (IIBDGC); Jégou, S.; Van Steen, K.; Wain, L.; Hugot, J.-P. Comparative Performances of Machine Learning Methods for Classifying Crohn Disease Patients Using Genome-Wide Genotyping Data. Sci. Rep. 2019, 9, 1035. [Google Scholar] [CrossRef]
- Mieth, B.; Rozier, A.; Rodriguez, J.A.; Höhne, M.M.C.; Görnitz, N.; Müller, K.-R. DeepCOMBI: Explainable Artificial Intelligence for the Analysis and Discovery in Genome-Wide Association Studies. NAR Genom. Bioinform. 2021, 3, lqab065. [Google Scholar] [CrossRef]
- Wang, H.; Bennett, D.A.; De Jager, P.L.; Zhang, Q.-Y.; Zhang, H.-Y. Genome-Wide Epistasis Analysis for Alzheimer’s Disease and Implications for Genetic Risk Prediction. Alzheimer's Res. Ther. 2021, 13, 55. [Google Scholar] [CrossRef]
- Fahed, A.C.; Wang, M.; Homburger, J.R.; Patel, A.P.; Bick, A.G.; Neben, C.L.; Lai, C.; Brockman, D.; Philippakis, A.; Ellinor, P.T.; et al. Polygenic Background Modifies Penetrance of Monogenic Variants for Tier 1 Genomic Conditions. Nat. Commun. 2020, 11, 3635. [Google Scholar] [CrossRef]
- Aragam, K.G.; Dobbyn, A.; Judy, R.; Chaffin, M.; Chaudhary, K.; Hindy, G.; Cagan, A.; Finneran, P.; Weng, L.-C.; Loos, R.J.; et al. Limitations of Contemporary Guidelines for Managing Patients at High Genetic Risk of Coronary Artery Disease. J. Am. Coll. Cardiol. 2020, 75, 2769–2780. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; McGuffog, L.; Barrowdale, D.; Lee, A.; Soucy, P.; Dennis, J.; Domchek, S.M.; Robson, M.; Spurdle, A.B.; Ramus, S.; et al. Evaluation of Polygenic Risk Scores for Breast and Ovarian Cancer Risk Prediction in BRCA1 and BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 2017, 109, djw302. [Google Scholar] [CrossRef] [Green Version]
- Barnes, D.R.; Rookus, M.A.; McGuffog, L.; Leslie, G.; Mooij, T.M.; Dennis, J.; Mavaddat, N.; Adlard, J.; Ahmed, M.; Aittomäki, K.; et al. Polygenic Risk Scores and Breast and Epithelial Ovarian Cancer Risks for Carriers of BRCA1 and BRCA2 Pathogenic Variants. Genet. Med. 2020, 22, 1653–1666. [Google Scholar] [CrossRef]
- Lecarpentier, J.; Silvestri, V.; Kuchenbaecker, K.B.; Barrowdale, D.; Dennis, J.; McGuffog, L.; Soucy, P.; Leslie, G.; Rizzolo, P.; Navazio, A.S.; et al. Prediction of Breast and Prostate Cancer Risks in Male BRCA1 and BRCA2 Mutation Carriers Using Polygenic Risk Scores. J. Clin. Oncol. 2017, 35, 2240–2250. [Google Scholar] [CrossRef]
- Han, X.; Qassim, A.; An, J.; Marshall, H.; Zhou, T.; Ong, J.-S.; Hassall, M.M.; Hysi, P.G.; Foster, P.J.; Khaw, P.T.; et al. Genome-Wide Association Analysis of 95 549 Individuals Identifies Novel Loci and Genes Influencing Optic Disc Morphology. Hum. Mol. Genet. 2019, 28, 3680–3690. [Google Scholar] [CrossRef]
- Liu, X.; Song, Z.; Li, Y.; Yao, Y.; Fang, M.; Bai, C.; An, P.; Chen, H.; Chen, Z.; Tang, B.; et al. Integrated Genetic Analyses Revealed Novel Human Longevity Loci and Reduced Risks of Multiple Diseases in a Cohort Study of 15,651 Chinese Individuals. Aging Cell 2021, 20, e13323. [Google Scholar] [CrossRef]
- Zhang, Y.D.; Breast Cancer Association Consortium (BCAC); Hurson, A.N.; Zhang, H.; Choudhury, P.P.; Easton, D.F.; Milne, R.L.; Simard, J.; Hall, P.; Michailidou, K.; et al. Assessment of Polygenic Architecture and Risk Prediction Based on Common Variants across Fourteen Cancers. Nat. Commun. 2020, 11, 3353. [Google Scholar] [CrossRef]
- Escott-Price, V.; Myers, A.J.; Huentelman, M.; Hardy, J. Polygenic Risk Score Analysis of Pathologically confirmed Alzheimer Disease. Ann. Neurol. 2017, 82, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Escott-Price, V. Genes, Pathways and Risk Prediction in Alzheimer's Disease. Hum. Mol. Genet. 2019, 28, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Han, Y.; Teeple, E.; Shankara, S.; Sadeghi, M.; Zhu, C.; Liu, D.; FinnGen; Wang, C.; Frau, F.; Klinger, K.W.; et al. Genome-Wide Polygenic Risk Score Identifies Individuals at Elevated Parkinson’s Disease Risk. medRxiv 2020. [Google Scholar] [CrossRef]
- Allegrini, A.G.; Selzam, S.; Rimfeld, K.; von Stumm, S.; Pingault, J.B.; Plomin, R. Genomic Prediction of Cognitive Traits in Childhood and Adolescence. Mol. Psychiatry 2019, 24, 819–827. [Google Scholar] [CrossRef]
- Von Stumm, S.; Smith-Woolley, E.; Ayorech, Z.; McMillan, A.; Rimfeld, K.; Dale, P.S.; Plomin, R. Predicting Educational Achievement from Genomic Measures and Socioeconomic Status. Dev. Sci. 2019, 23, e12925. [Google Scholar] [CrossRef] [Green Version]
- Morris, T.T.; Davies, N.M.; Smith, G.D. Can Education be Personalised using Pupils’ Genetic Data? eLife 2020, 9, e49962. [Google Scholar] [CrossRef]
- Smith-Woolley, E.; Pingault, J.-B.; Selzam, S.; Rimfeld, K.; Krapohl, E.; Von Stumm, S.; Asbury, K.; Dale, P.S.; Young, T.; Allen, R.; et al. Differences in Exam Performance Between Pupils Attending Selective and Non-Selective Schools Mirror the Genetic Differences between Them. npj Sci. Learn. 2018, 3, 3. [Google Scholar] [CrossRef]
- Richardson, K.; Jones, M.C. Why Genome-Wide Associations with Cognitive Ability Measures are Probably Spurious. New Ideas Psychol. 2019, 55, 35–41. [Google Scholar] [CrossRef]
- Cheesman, R.; Hunjan, A.; Coleman, J.; Ahmadzadeh, Y.; Plomin, R.; McAdams, T.; Eley, T.C.; Breen, G. Comparison of Adopted and Nonadopted Individuals Reveals Gene–Environment Interplay for Education in the UK Biobank. Psychol. Sci. 2020, 31, 582–591. [Google Scholar] [CrossRef]
- Murphy, K.C.; Jones, L.A.; Owen, M.J. High Rates of Schizophrenia in Adults with Velo-Cardio-Facial Syndrome. Arch. Gen. Psychiatry 1999, 56, 940–945. [Google Scholar] [CrossRef] [Green Version]
- Zinkstok, J.; Van Amelsvoort, T. Neuropsychological Profile and Neuroimaging in Patients with 22Q11.2 Deletion Syndrome: A Review Keywords. Child Neuropsychol. 2005, 11, 21–37. [Google Scholar] [CrossRef]
- Davies, R.W.; Fiksinski, A.M.; Breetvelt, E.J.; Williams, N.M.; Hooper, S.R.; Monfeuga, T.; Bassett, A.S.; Owen, M.J.; Gur, R.E.; Morrow, B.E.; et al. Using Common Genetic Variation to Examine Phenotypic Expression and Risk Prediction in 22q11.2 Deletion Syndrome. Nat. Med. 2020, 26, 1912–1918. [Google Scholar] [CrossRef]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical Use of Current Polygenic Risk Scores May Exacerbate Health Disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef]
- Majara, L.; Kalungi, A.; Koen, N.; Zar, H.; Stein, D.J.; Kinyanda, E.; Atkinson, E.G.; Martin, A.R. Low Generalizability of Polygenic Scores in African Populations due to Genetic and Environmental Diversity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bigdeli, T.B.; Consortium on the Genetics of Schizophrenia (COGS) Investigators; Genovese, G.; Georgakopoulos, P.; Meyers, J.L.; Peterson, R.; Iyegbe, C.O.; Medeiros, H.; Valderrama, J.; Achtyes, E.D.; et al. Contributions of Common Genetic Variants to Risk of Schizophrenia among Individuals of African and Latino Ancestry. Mol. Psychiatry 2019, 25, 2455–2467. [Google Scholar] [CrossRef] [Green Version]
- Cross-Disorder Group of the Psychiatric Genomics Consortium Genetic relationship between Five Psychiatric Disorders Estimated from Genome-Wide SNPs. Nat. Genet. 2013, 45, 984–994. [CrossRef] [Green Version]
- Van Rheenen, W.; Peyrot, W.J.; Schork, A.J.; Lee, S.H.; Wray, N.R. Genetic Correlations of Polygenic Disease Traits: From Theory to Practice. Nat. Rev. Genet. 2019, 20, 567–581. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Finucane, H.K.; Anttila, V.; Gusev, A.; Day, F.R.; Loh, P.-R.; Duncan, E.L.; Perry, J.R.; Patterson, N.; Robinson, E.; et al. An Atlas of Genetic Correlations across Human Diseases and Traits. Nat. Genet. 2015, 47, 1236–1241. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Erzurumluoglu, A.M.; Elsworth, B.L.; Kemp, J.P.; Howe, L.; Haycock, P.C.; Hemani, G.; Tansey, K.; Laurin, C.; Pourcain, B.S.; et al. LD Hub: A Centralized Database and Web Interface to Perform LD Score Regression that Maximizes the Potential of Summary Level GWAS Data for SNP Heritability and Genetic Correlation Analysis. Bioinformatics 2017, 33, 272–279. [Google Scholar] [CrossRef]
- Watanabe, K.; Stringer, S.; Frei, O.; Mirkov, M.U.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A Global Overview of Pleiotropy and Genetic Architecture in Complex Traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef]
- Gao, J.; Davis, L.K.; Hart, A.B.; Sanchez-Roige, S.; Han, L.; Cacioppo, J.T.; Palmer, A.A. Genome-Wide Association Study of Loneliness Demonstrates a Role for Common Variation. Neuropsychopharmacology 2017, 42, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Bone, W.P.; Program, T.V.M.V.; Siewert, K.M.; Jha, A.; Klarin, D.; Damrauer, S.M.; Chang, K.-M.; Tsao, P.S.; Assimes, T.L.; Ritchie, M.D.; et al. Multi-Trait Association Studies Discover Pleiotropic Loci Between Alzheimer’s Disease and Cardiometabolic Traits. Alzheimer's Res. Ther. 2021, 13, 34. [Google Scholar] [CrossRef]
- Xicoy, H.; Klemann, C.J.; De Witte, W.; Martens, M.B.; Martens, G.J.; Poelmans, G. Shared Genetic Etiology between Parkinson’s Disease and Blood Levels of Specific Lipids. npj Park. Dis. 2021, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Denny, J.; Bastarache, L.; Ritchie, M.D.; Carroll, R.J.; Zink, R.; Mosley, J.; Field, J.R.; Pulley, J.M.; Ramirez, A.H.; Bowton, E.; et al. Systematic Comparison of Phenome-Wide Association Study of Electronic Medical Record Data and Genome-Wide Association Study Data. Nat. Biotechnol. 2013, 31, 1102–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, T.G.; Harrison, S.; Hemani, G.; Smith, G.D. An Atlas of Polygenic Risk Score Associations to Highlight Putative Causal Relationships across the Human Phenome. eLife 2019, 8, e43657. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.R.; Denny, J.C.; Roden, D.M.; Van Driest, S.L. Genome-wide and Phenome-wide Approaches to Understand Variable Drug Actions in Electronic Health Records. Clin. Transl. Sci. 2018, 11, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Luo, H.; Huang, X.; Wei, C.; Di, J.; Tian, Y.; Fu, X.; Li, B.; Liu, G.E.; Fang, L.; et al. Integration of a Single-Step Genome-Wide Association Study with a Multi-Tissue Transcriptome Analysis Provides Novel Insights into the Genetic Basis of Wool and Weight Traits in Sheep. Genet. Sel. Evol. 2021, 53, 56. [Google Scholar] [CrossRef]
- Evans, W.N.; Ringel, J.S. Can Higher Cigarette Taxes Improve Birth Outcomes? J. Public Econ. 1999, 72, 135–154. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.D.; Ebrahim, S. ‘Mendelian Randomization’: Can Genetic Epidemiology Contribute to Understanding Environmental Determinants of Disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Smith, G.D.; Harbord, R.M.; Lewis, S.J. Alcohol Intake and Blood Pressure: A Systematic Review Implementing a Mendelian Randomization Approach. PLoS Med. 2008, 5, e52. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Shin, S.-Y.; Won, S.; Relton, C.L.; Smith, G.D.; Shin, M.-J. Alcohol Intake and Cardiovascular Risk Factors: A Mendelian Randomisation Study. Sci. Rep. 2015, 5, 18422. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium; Howard, D.M.; Adams, M.J.; Hill, W.D.; Clarke, T.-K.; Deary, I.J.; Whalley, H.C.; McIntosh, A.M. A Phenome-Wide Association and Mendelian Randomisation Study of Polygenic Risk for Depression in UK Biobank. Nat. Commun. 2020, 11, 230. [Google Scholar] [CrossRef]
- Cao, L.; Li, Z.Q.; Shi, Y.Y.; Liu, Y. Telomere Length and Type 2 Diabetes: Mendelian Randomization Study and Polygenic Risk Score Analysis. Yi Chuan 2020, 42, 882–888. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; O'Reilly, P.; Marcora, E.; Goate, A.M.; Farrer, L.A.; Haines, J.L.; Mayeux, R.; Naj, A.C.; Pericak-Vance, M.A.; et al. Causal Associations Between Modifiable Risk Factors and the Alzheimer's Phenome. Ann. Neurol. 2020, 89, 54–65. [Google Scholar] [CrossRef]
- Higgins, H.; Mason, A.M.; Larsson, S.C.; Gill, D.; Langenberg, C.; Burgess, S. Estimating the Population Benefits of Blood Pressure Lowering: A Wide-Angled Mendelian Randomization Study in UK Biobank. J. Am. Hear. Assoc. 2021, 10, e021098. [Google Scholar] [CrossRef]
- Pierce, B.L.; VanderWeele, T.J. The Effect of Non-Differential Measurement Error on Bias, Precision and Power in Mendelian Randomization Studies. Int. J. Epidemiol. 2012, 41, 1383–1393. [Google Scholar] [CrossRef]
- Bowden, J.; Del Greco, M.F.; Minelli, C.; Smith, G.D.; Sheehan, N.; Thompson, J. A Framework for the Investigation of Pleiotropy in Two-Sample Summary Data Mendelian Randomization. Stat. Med. 2017, 36, 1783–1802. [Google Scholar] [CrossRef] [Green Version]
- Bowden, J.; Smith, G.D.; Burgess, S. Mendelian Randomization with Invalid Instruments: Effect Estimation and Bias Detection through Egger Regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.; Knoblauch, N.; Marcus, J.H.; Stephens, M.; He, X. Mendelian Randomization Accounting for Correlated and Uncorrelated Pleiotropic Effects Using Genome-Wide Summary Statistics. Nat. Genet. 2020, 52, 740–747. [Google Scholar] [CrossRef]
- Hu, X.; Zhao, J.; Lin, Z.; Wang, Y.; Peng, H.; Zhao, H.; Wan, X.; Yang, C. MR-APSS: A Unified Approach to Mendelian Randomization Accounting for Pleiotropy and Sample Structure Using Genome-Wide Summary Statistics. bioRxiv 2021. [Google Scholar] [CrossRef]
- Xue, H.; Shen, X.; Pan, W. Constrained Maximum Likelihood-Based Mendelian Randomization Robust to Both Correlated and Uncorrelated Pleiotropic Effects. Am. J. Hum. Genet. 2021, 108, 1251–1269. [Google Scholar] [CrossRef]
- Sekar, A.; Adolfsson, R.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; et al. Schizophrenia Risk from Complex Variation of Complement Component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Smemo, S.; Tena, J.J.; Kim, K.-H.; Gamazon, E.; Sakabe, N.J.; Gómez-Marín, C.; Aneas, I.; Credidio, F.L.; Sobreira, D.R.; Wasserman, N.F.; et al. Obesity-Associated Variants within FTO form Long-Range Functional Connections with IRX3. Nature 2014, 507, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Claussnitzer, M.; Dankel, S.N.; Kim, K.-H.; Quon, G.; Meuleman, W.; Haugen, C.; Glunk, V.; Sousa, I.S.; Beaudry, J.L.; Puviindran, V.; et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 2015, 373, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Liang, X.; Ma, Z.; Hu, H.; Liu, H.; Miao, Z.; Linkermann, A.; Hellwege, J.N.; Voight, B.F.; Susztak, K. A Single Genetic Locus Controls Both Expression of DPEP1/CHMP1A and Kidney Disease Development via Ferroptosis. Nat. Commun. 2021, 12, 5078. [Google Scholar] [CrossRef]
- Kichaev, G.; Pasaniuc, B. Leveraging Functional-Annotation Data in Trans-ethnic Fine-Mapping Studies. Am. J. Hum. Genet. 2015, 97, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Sinnott-Armstrong, N.; Sousa, I.S.; Laber, S.; Rendina-Ruedy, E.; Dankel, S.E.N.; Ferreira, T.; Mellgren, G.; Karasik, D.; Rivas, M.; Pritchard, J.; et al. A Regulatory Variant at 3q21.1 Confers an Increased Pleiotropic Risk for Hyperglycemia and Altered Bone Mineral Density. Cell Metab. 2021, 33, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Guan, Y.; Ma, Z.; Wu, J.; Liu, H.; Qiu, C.; Vitale, S.; Miao, Z.; Seasock, M.J.; Palmer, M.; et al. Mapping the Genetic Architecture of Human Traits to Cell Types in the Kidney Identifies Mechanisms of Disease and Potential Treatments. Nat. Genet. 2021, 53, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Stanzick, K.J.; Li, Y.; Schlosser, P.; Gorski, M.; Wuttke, M.; Thomas, L.F.; Rasheed, H.; Rowan, B.X.; Graham, S.E.; Vanderweff, B.R.; et al. Discovery and Prioritization of Variants and Genes for Kidney Function in >1.2 million Individuals. Nat. Commun. 2021, 12, 4350. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Frésard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-Cell Epigenomic Analyses Implicate Candidate Causal Variants at Inherited Risk Loci for Alzheimer’s and Parkinson’s Diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef]
- Kupari, J.; Usoskin, D.; Parisien, M.; Lou, D.; Hu, Y.; Fatt, M.; Lönnerberg, P.; Spångberg, M.; Eriksson, B.; Barkas, N.; et al. Single Cell Transcriptomics of Primate Sensory Neurons Identifies Cell Types Associated with Chronic Pain. Nat. Commun. 2021, 12, 1510. [Google Scholar] [CrossRef]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic Studies of Body Mass Index Yield New Insights for Obesity Biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Calderon, D.; Bhaskar, A.; Knowles, D.A.; Golan, D.; Raj, T.; Fu, A.Q.; Pritchard, J.K. Inferring Relevant Cell Types for Complex Traits by Using Single-Cell Gene Expression. Am. J. Hum. Genet. 2017, 101, 686–699. [Google Scholar] [CrossRef] [Green Version]
- Porcu, E.; Rüeger, S.; Lepik, K.; Santoni, F.A.; Reymond, A.; Kutalik, Z.; eQTLGen Consortium. BIOS Consortium Mendelian Randomization Integrating GWAS and eQTL Data Reveals Genetic Determinants of Complex and Clinical Traits. Nat. Commun. 2019, 10, 3300. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.; Matoba, N.; Wilson, E.P.; Tapia, A.L.; Li, Y.; Ibrahim, J.G.; Stein, J.L.; Love, M.I. MRLocus: Identifying causal genes mediating a trait through Bayesian Estimation of Allelic Heterogeneity. PLoS Genet. 2021, 17, e1009455. [Google Scholar] [CrossRef]
- Pain, O.; Glanville, K.P.; Hagenaars, S.; Selzam, S.; Fürtjes, A.; Coleman, I.J.R.; Rimfeld, K.; Breen, G.; Folkersen, L.; Lewis, C.M. Imputed Gene Expression Risk Scores: A Functionally Informed Component of Polygenic Risk. Hum. Mol. Genet. 2021, 30, 727–738. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-Scale Cis- and Trans-Eqtl Analyses Identify Thousands of Genetic Loci and Polygenic Scores that Regulate Blood Gene Expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- Finkel, Y.; Gassull, A.M.; Goossens, D.; Laukens, D.; Lémann, M.; Libioulle, C.; O’Morain, C.; Reenaers, C.; Rutgeerts, P.; Tysk, C.; et al. Resequencing of Positional Candidates Identifies Low Frequency IL23R Coding Variants Protecting against Inflammatory Bowel Disease. Nat. Genet. 2011, 43, 43–47. [Google Scholar] [CrossRef]
- Rivas, M.A.; National Institute of Diabetes and Digestive Kidney Diseases Inflammatory Bowel Disease Genetics Consortium (NIDDK IBDGC); Beaudoin, M.; Gardet, A.; Stevens, C.; Sharma, Y.; Zhang, C.K.; Boucher, G.; Ripke, S.; Ellinghaus, D.; et al. Deep Resequencing of GWAS Loci Identifies Independent Rare Variants Associated with Inflammatory Bowel Disease. Nat. Genet. 2011, 43, 1066–1073. [Google Scholar] [CrossRef] [Green Version]
- Seddon, J.M.; Yu, Y.; Miller, E.C.; Reynolds, R.; Tan, P.L.; Gowrisankar, S.; Goldstein, J.; Triebwasser, M.; Anderson, E.H.; Zerbib, J.; et al. Rare Variants in CFI, C3 and C9 are Associated with High Risk of Advanced Age-Related Macular Degeneration. Nat. Genet. 2013, 45, 1366–1370. [Google Scholar] [CrossRef] [Green Version]
- Flannick, J.; Thorleifsson, G.; Beer, N.L.; Jacobs, S.B.R.; Grarup, N.; Burtt, N.P.; Mahajan, A.; Fuchsberger, C.; Atzmon, G.; Benediktsson, R.; et al. Loss-of-Function Mutations in SLC30A8 Protect against Type 2 Diabetes. Nat. Genet. 2014, 46, 357–363. [Google Scholar] [CrossRef]
- Diogo, D.; Kurreeman, F.; Stahl, E.A.; Liao, K.P.; Gupta, N.; Greenberg, J.D.; Rivas, M.A.; Hickey, B.; Flannick, J.; Thomson, B.; et al. Rare, Low-Frequency, and Common Variants in the Protein-Coding Sequence of Biological Candidate Genes from GWASs Contribute to Risk of Rheumatoid Arthritis. Am. J. Hum. Genet. 2013, 92, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Motegi, T.; Kochi, Y.; Matsuda, K.; Kubo, M.; Yamamoto, K.; Momozawa, Y. Identification of Rare Coding Variants in TYK2 Protective for Rheumatoid Arthritis in the Japanese Population and their Effects on Cytokine Signalling. Ann. Rheum. Dis. 2019, 78, 1062–1069. [Google Scholar] [CrossRef]
- Bergen, S.E.; Ploner, A.; Howrigan, D.; O’Donovan, M.C.; Smoller, J.W.; Sullivan, P.F.; Sebat, J.; Neale, B.; Kendler, K.S. CNV Analysis Group and the Schizophrenia Working Group of the Psychiatric Genomics Consortium Joint Contributions of Rare Copy Number Variants and Common SNPs to Risk for Schizophrenia. Am. J. Psychiatry 2019, 176, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, S.; Ninomiya, K.; Kushima, I.; Saito, T.; Shimasaki, A.; Sakusabe, T.; Momozawa, Y.; Kubo, M.; Kamatani, Y.; Ozaki, N.; et al. Polygenic Risk Scores in Schizophrenia with Clinically Significant Copy Number Variants. Psychiatry Clin. Neurosci. 2020, 74, 35–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, E.; GROUP Investigators; Han, J.; Morgan, J.; Carrera, N.; Escott-Price, V.; Pocklington, A.J.; Duffield, M.; Hall, L.S.; Legge, S.E.; et al. De Novo Mutations Identified by Exome Sequencing Implicate Rare Missense Variants in SLC6A1 in Schizophrenia. Nat. Neurosci. 2020, 23, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Yu, D.; Scharf, J.M.; Mathews, C.A.; McGrath, L.; Cook, E.; Lee, S.H.; Davis, L.K.; Gamazon, E.R. Contextualizing Genetic Risk Score for Disease Screening and Rare Variant Discovery. Nat. Commun. 2021, 12, 4418. [Google Scholar] [CrossRef]
- Dobrindt, K.; Zhang, H.; Das, D.; Abdollahi, S.; Prorok, T.; Ghosh, S.; Weintraub, S.; Genovese, G.; Powell, S.K.; Lund, A.; et al. Publicly Available hiPSC Lines with Extreme Polygenic Risk Scores for Modeling Schizophrenia. Complex Psychiatry 2020, 6, 68–82. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondratyev, N.V.; Alfimova, M.V.; Golov, A.K.; Golimbet, V.E. Bench Research Informed by GWAS Results. Cells 2021, 10, 3184. https://doi.org/10.3390/cells10113184
Kondratyev NV, Alfimova MV, Golov AK, Golimbet VE. Bench Research Informed by GWAS Results. Cells. 2021; 10(11):3184. https://doi.org/10.3390/cells10113184
Chicago/Turabian StyleKondratyev, Nikolay V., Margarita V. Alfimova, Arkadiy K. Golov, and Vera E. Golimbet. 2021. "Bench Research Informed by GWAS Results" Cells 10, no. 11: 3184. https://doi.org/10.3390/cells10113184
APA StyleKondratyev, N. V., Alfimova, M. V., Golov, A. K., & Golimbet, V. E. (2021). Bench Research Informed by GWAS Results. Cells, 10(11), 3184. https://doi.org/10.3390/cells10113184