Abstract
Pulmonary arterial hypertension (PAH) sometimes co-exists with hereditary hemorrhagic telangiectasia (HHT). Despite being clinically diagnosable according to Curaçao criteria, HHT can be difficult to diagnose due to its clinically heterogenicity and highly overlapping with PAH. Genetic analysis of the associated genes ACVRL1, ENG, SMAD4 and GDF2 can help to confirm or discard the presumptive diagnosis. As part of the clinical routine and to establish a genetic diagnosis, we have analyzed a cohort of patients with PAH and overlapping HHT features through a customized Next Generation Sequencing (NGS) panel of 21 genes, designed and validated in-house. We detected a homozygous missense variant in GDF2 in a pediatric patient diagnosed with PAH associated with HHT and a missense variant along with a heterozygous deletion in another idiopathic PAH patient (compound heterozygous inheritance). In order to establish variant segregation, we analyzed all available family members. In both cases, parents were carriers for the variants, but neither was affected. Our results expand the clinical spectrum and the inheritance pattern associated with GDF2 pathogenic variants suggesting incomplete penetrance and/or variability of expressivity with a semi-dominant pattern of inheritance.
1. Introduction
Pulmonary arterial hypertension (PAH) is a severe disease defined as a persistently elevated mean pulmonary artery pressure (mPAP ≥ 20 mmHg) with a mean pulmonary arterial wedge pressure (PAWP) ≤ 15 mmHg and pulmonary vascular resistance (PVR) ≥ 3 UW [1]. PAH causes progressive right-sided heart failure leading to premature death if it remains untreated [1,2]. It is known that there is a genetic component that contributes to a predisposition to develop the disease. Pathogenic variants in the gene encoding the bone morphogenetic protein type 2 receptor (BMPR2), a receptor for the transforming growth factor-beta (TGF-β) superfamily, explain approximately 20–25% of idiopathic PAH (IPAH) cases and 60% of heritable PAH (HPAH) [3,4,5].
The etiology of PAH is highly heterogeneous, and it occasionally coexists with distinct clinical entities such as hereditary hemorrhagic telangiectasia (HHT). HHT is characterized by the presence of epistaxis, mucocutaneous telangiectasias and multiple arteriovenous malformations (AVMs) in the lung, gastrointestinal tract, liver, brain, nasal mucosa, spine, and conjunctiva [6]. The AVMs consist of the lack of intervening capillaries and result in direct connections between arteries and veins [6]. According to the Curaçao criteria [7,8], the diagnosis of HHT is established in a patient with at least three of the following clinical features: epistaxis, mucocutaneous telangiectases, visceral AVMs, and/or a family history of HHT. HHT might be sometimes difficult to be diagnosed because observable manifestations may be absent until adulthood [9]. Therefore, genetic testing is considered the gold standard in the pediatric population for patients with a family history of HHT and even when genetic test results are positive, it will confirm the diagnosis regardless of family history or symptoms.
Currently, all known genetic defects that cause HHT are found within the TGF-β signaling pathway. Identification of pathogenic variants in activin A receptor type I-like (ACVRL1 or ALK1), endoglin (ENG), growth differentiation factor 2 (GDF2, also known as BMP9 or SMAD9), or SMAD family member 4 (SMAD4) establish a definitely molecular diagnosis, even if the clinical features are inconclusive. On endothelial cells, BMPR2 and ACVRL1 participate in a complex signaling in which ENG acts as a co-receptor and SMAD1, SMAD4 and SMAD9 as signaling intermediaries [10,11,12,13]. These results provide further evidence of the importance of BMP signaling and its dysregulation in the pathogenesis of PAH and HHT. Pathogenic variants in ENG and ACVRL1, count 80% of the total number of patients with HHT [14,15,16,17]. Also, pathogenic variants in SMAD4 are responsible for a combined syndrome of juvenile polyposis with or without HHT [MIM# 174900, 175050], which occurs approximately in less than 2% of the HHT patients [18]. Finally, pathogenic variants in GDF2 have been described in PAH patients [19,20,21] and were shown to cause an HHT-like phenotype [22].
Genetic analysis of the associated genes is extremely useful to confirm the initial presumptive diagnosis, differential diagnosis with PAH, progression and management of the disease and family risks based on the results. In families where there is an affected member with a known disease-causing variant and for those family members who do not manifest symptoms of HHT, diagnostic genetic testing is a more cost-effective solution than conventional clinical screening and may benefit from early treatment, if necessary [23]. Therefore, in order to address the molecular diagnosis of PAH patients, we applied a custom NGS panel, which included genes ACVRL1, ENG, SMAD4 and GDF2 and 17 additional genes related to PAH [24]. In the present study, we have identified an homozygous missense pathogenic variant in GDF2 in a patient with PAH-HHT and a missense variant and a copy number variant (CNV) in a patient with IPAH. In both cases, their healthy parents were carriers of the same GDF2 variants in heterozygosis state. These results add more evidence of the semi-dominant inheritance with variable expression and incomplete penetrance model related to GDF2 variants. This also supports the co-exist variable clinical spectrum of PAH and HHT in individuals with GDF2 pathogenic variants.
2. Materials and Methods
This study was approved by the ethical committee for scientific research of each participant center and by the ethical committee of the La Paz University Hospital (CEIC-HULP PI-1210). All patients or parents of the children involved in this project signed a clinical consent form to accept their participation.
2.1. Cases Presentation
The index patients included in the study were selected from the Spanish pediatric PAH registry (REHIPED). Prior to genetic diagnosis, family history information was collected. DNA samples from the proband and parents were extracted. Both probands and first-degree relatives were enrolled in the study after informed written consent was obtained in each case. The most relevant clinical information of the two included patients is summarized in Table 1.

Table 1.
Clinical information. Main clinical characteristics of the patients and results obtained in the tests performed from diagnosis to the present time.
Patient 1 was evaluated for the first time in 2013, when he was five years old. He complained of shortness of breath on exertion and recurrent epistaxis. Peripheral oxygen saturation was normal at that time. A transthoracic echocardiography (TTE) revealed indirect data of significant pulmonary hypertension. During the first right heart catheterization (RHC), mean pulmonary artery pressure (mPAP) was 55 mmHg, pulmonary artery wedge pressure (PAWP) was 8 mmHg, and pulmonary vascular resistance (PVR) was 5.4 wood units (WU). The cardiac index (CI) was surprisingly high (8.7 L/min/m2), consistent with the initial diagnosis of PAH related with high cardiac output. Nevertheless, neither of the parents presented symptoms suggestive of the disease and there was no known relevant family history. Despite the patient being initially diagnosed with IPAH, further examinations revealed the presence of telangiectasias on the face and back, which led to the diagnosis being extended to “possible PAH associated with HHT” according to the Curaçao criteria [7,8]. Although fecal occult blood tests showed positive results, neither a computed tomography nor a colonoscopy demonstrated the presence of arteriovenous malformations in the gastrointestinal tract. Double initial oral therapy with tadalafil (40 mg/24 h) and bosentan (125 mg/12 h) was initiated from the diagnosis, maintaining the patient stable to date.
Patient 2 was also diagnosed in 2013 in the setting of chest pain evaluation when she was four years old. Similarly, the TTE showed data of significant pulmonary hypertension. Then, the RHC demonstrated a mPAP of 33 mmHg, a PAWP of 6 mmHg, a CI of 4.2 L/min/m2 and PVR of 6.5 WU. In this patient, neither the index case nor their first-degree relatives had any relevant signs or symptoms suggestive of HHT. At diagnosis, thoracic computed tomography was normal. Equally, blood samples had always been normal, without evidence of anemia during follow-up. Oxygen saturation at rest and during exercise was likewise unremarkable. Currently, the patient remains stable without any limitation in her daily life, estimating a functional class I at her twelve years of age, under double oral therapy with macitentan and tadalafil.
2.2. Next Generation Sequencing
Genetic analysis was performed in the probands and on their parents after obtaining DNA from peripheral blood. Samples were included into a customized Next Generation Sequencing (NGS) panel of 21 genes (HAP v1.2) designed in-house, which included: ACVRL1; GDF2; BMPR1B; BMPR2; CAV1; EIF2AK4; ENG; KCNA5; KCNK3; NOTCH3; SMAD1; SMAD4; SMAD5; SMAD9; TBX4; TOPBP1; SARS2; CPS1; ABCC8; CBLN2; MMACHC. This custom panel was designed using NimbleDesign tool (Roche, Indianapolis, IN, USA). Fragmentation and library preparation were performed with SeqCap EZ Choice Enrichment Kit (Roche, Indianapolis, IN, USA). Sequencing was performed with the Illumina MiSeq platform (Illumina, San Diego, CA, USA) following the manufacturer’s instructions [25].
Variant prioritization was performed according to a custom filtering algorithm considering quality of the base pair reads, variant allele frequency in pseudo control populations and pathogenicity predictors as previously described [25], and summarized in Figure 1. Finally, variants were classified according to the guidelines described by the American College of Medical Genetics and Genomics (ACMG) [26].
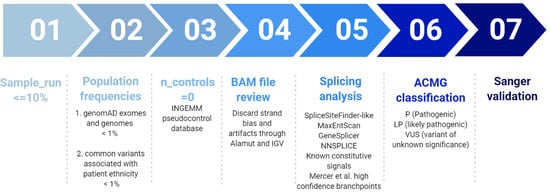
Figure 1.
Filtering pipeline for NGS panel HAP v1.2. Modified from Tenorio et al. 2020 [25].
Additionally, in order to carry out the analysis of CNVs, we used a custom script developed in-house called “LACONv” v1 (https://github.com/kibanez/LACONv, accessed on 21 June 2021) [27].
2.3. Segregation Analysis
In addition to the study of the probands, the analysis of all available first-degree relatives of the index cases was completed. Samples were obtained from the parents of the proband and siblings and the genetic segregation study was performed by sanger sequencing.
In patient 2, we performed a SNP array to confirm the deletion detected in the proband and her relatives. For this purpose, a SNP array study was carried out using the Infinium OmniExpressExome 8 v1.6 platform (Illumina). Image data were analyzed using the Chromosome Viewer tool contained in the Genome Studio package (Illumina, San Diego, CA, USA). In Chromosome Viewer, gene call scores < 0.15 at any locus were considered “no calls”. In addition, allele frequency analysis was applied for all SNPs. All genomic coodinates were established according to the 2009 human genome build 19 (GRCh37/NCBI build 37.1). Deletion sizes were plotted on the genome browser using the University os California at Santa Cruz Genome Browser (http://genome.ucsc.edu/, accessed on 25 June 2021).
In unaffected carriers, a complete and specific diagnostic follow-up was performed in order to rule out the disease.
3. Results
In patient 1, we identified a homozygous missense variant in exon 1 of GDF2 (Table 2) in the index patient (GDF2:NM_016204.4:c.328C > T:p.(Arg110Trp)). This variant causes a substitution of an arginine to tryptophan at amino acid position 110. The parents and brother were heterozygotes for the same variant, suggesting that the child inherited a copy from each parent (Figure 2A). In silico analyses showed that this variant was absent in several population databases, and the majority of the pathogenic prediction tools (dbNSFP) suggested a damaging effect for this variant. Therefore, this variant was classified as likely pathogenic variant (LP) by applying the ACMG criteria (Table 2).
Table 2.
Variants information.
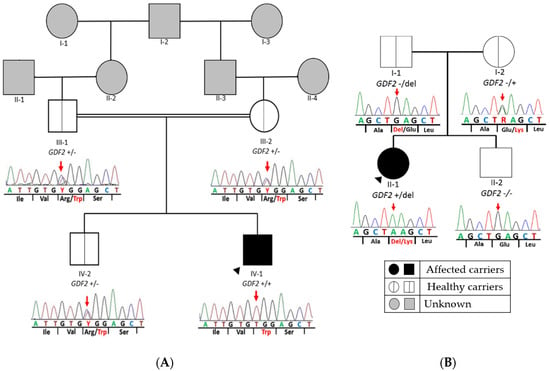
Figure 2.
Pedigree of both families with GDF2 variants. Segregation analysis of GDF2 in all available family members. The figure shows a fragment of the GDF2 sequence with corresponding amino acids below. (A) Patient 1: It represents the homozygous missense variant at nucleotide position c.328 (c.328C > T) in exon 1 of GDF2 (NM_016204.3), which causes the amino acid substitution arginine to tryptophane at peptide position 110 (p.Arg110Trp) in the index patient. In addition, it shows the same heterozygous missense variant found in healthy parents and brother. (B) Patient 2: The missense variant at nucleotide position c.445 in exon 2 of GDF2 (NM_ 016204.4), which causes the amino acid substitution glutamine to lysine at peptide position 149 in the index patient and mother and the deletion inherited from her father. Double line denotes consanguinity. Legend: +/+ homozygous for the alternative allele; +/− heterozygous for the alternative allele; del: deletion.
In patient 2, we detected a missense variant (GDF2:NM_016204.4:c.445G > A:p.(Glu149Lys)) in exon 2 of GDF2 (Table 2), absent in pseudo control population databases. Based on the results of the variant segregation analysis, we determine that this variant has been inherited from her mother. This patient also inherited a deletion from her father of a size of 4.24 Mb located on chromosome 10, in the region q11.22–q11.23, which includes 41 genes (AGAP4, FAM25E, PTPN20B, SYT15, GPRIN2, ANXA8L1, FAM25G, NPYAR, FAM25BP, AGAP10P, ANTXRL, ASAH2B, AGAP9, ANXA8, ZNF488, RBP3, GDF2, GDF10, FAM25C, FRMPD2, MAPK8, ARHGAP22, WDFYA, LRRC18, VSTM4, C10orf128, DRGX, ERCC6, CHAT, SLC18A3, OGDHL, PARG, AGAP7, TIM23, NOCOA4, MSMB, AGAP6, ASAH2), which included GDF2 (Figure 3).
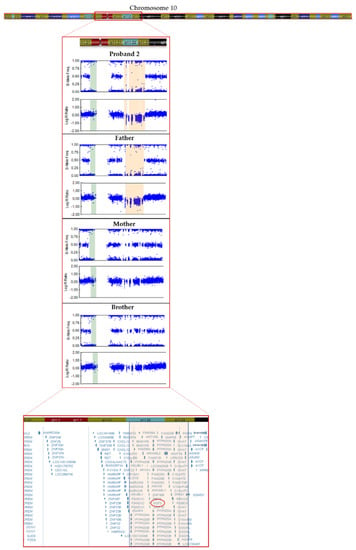
Figure 3.
Resolution of the SNP array carried out in proband 2 and family members showing the 4.2 Mb deletion in q11.22−q11.23 region of chromosome 10 (in orange).
4. Discussion
The recent advances in massive parallel sequencing have contributed to an improvement in the knowledge of the genetic basis of PAH. Although during many years it was thought that only the primary forms of PAH were associated with a genetic factor, several studies have demonstrated that associated forms such as PAH associated with HHT also had a genetic contribution [22]. As far as we know, HHT has been classically described as an autosomal-dominantly inherited vascular malformation syndrome since the detection of heterozygous variants in ACVRL1, ENG, SMAD4 and GDF2 [14,15,16,17].
Variants in GDF2 have been the most recent genetic discover related to PAH associated with HHT [22] and it is one of the genes with higher level of evidence of association with the development of PAH [28]. According to the analyses of the variant proteins performed by Wooderchak et al. (2013), pathogenic variants in GDF2 negatively affect protein processing and/or function; a bmp9-deficient zebrafish model demonstrated that BMP9, encoded by GDF2, is involved in angiogenesis [22]. These data confirm a genetic contribution of a vascular-anomaly syndrome that has phenotypic overlap with HHT [22]. This is directly related to the main biological processes dysregulated in PAH: proliferation, apoptosis and endothelial cell migration.
In the present study, we identified two missense variants in GDF2 and a CNV, which includes this gene in a PAH-HHT patient but also in an idiopathic PAH case. In patient 1, segregation analysis of the variant has allowed to confirm that both parents and older brother were heterozygous carriers of the same variant in GDF2. There is increasing evidence of homozygous variants in GDF2 associated with HHT. In 2016, Wang et al., detected a homozygous nonsense pathogenic variant in a Chinese boy with a suggestive diagnosis of HHT but the consanguineous parents were heterozygous and apparently unaffected carriers. However, it must be noted that the father and his paternal uncle had had epistaxis meanwhile the mother and his younger sister showed vascular lesions. Sanger sequencing revealed the same homozygous variant in his sister, but at the time of publication she did not show any symptoms of PAH [29].
As in the previous case, our consanguineous parents of the patient and brother were heterozygous carriers of the same GDF2 variant but they did not present any symptoms at the time of this study and there is no known family history. In addition, both consanguineous parents carry the same heterozygous variant, so we speculate that the variant was inherited in heterozygous form from the shared grandfather, through any of their parents. In other words, we speculate that there are at least three additional family members (I-2, II-2 and II-3 in Figure 2A) with the same heterozygous variant who had not developed the disease. Hypothetically, these results support the idea of an incomplete penetrance or variable expressivity may occur in heterozygous GDF2 carriers. Moreover, the older brother (IV-2 in Figure 2A), who is 16 years old, presented the same heterozygous variant, but he did not show any symptoms. This may be explained by the fact that epistaxis caused by telangiectasias in the nasal mucosa eventually develop in 95% of cases but only 50% of individuals report having nosebleeds before the age of 10 years [9] and, similarly, oral or dermal telangiectasias are not usually observed until the third decade of life [22,30]. As we have already commented, in many cases an early diagnosis of the disease is difficult, thus demonstrating the great importance of genetic screening.
As in several patients previously described, our patient presented with clinical features of GDF2-like disorder including epistaxis and dermal lesions described as telangiectases but whose location and appearance are atypical compared to HHT patients. Meanwhile patients with homozygous variants in GDF2 showed spider-like/linear telangiectases only on their chin and/or cheeks; patients with variants in ACVRL1, ENG and SMAD4 have them typically on the lips and oral cavity [22].
Not only have homozygous variants in GDF2 been detected in patients with HHT, but they have also been identified in patients with PAH [20,21,31,32]. Previous studies have measured BMP9 and pBMP10 plasma levels along with the serum-derived endothelial BMP activity in patients with homozygous nonsense GDF2 mutations, PAH patient and a patient with “HHT-like” phenotype. They confirmed that it results in reduced plasma BMP9 and pBMP10 levels, even in asymptomatic heterozygous parents [33]. Furthermore, the variant specifically detected in patient 1 in this study has been previously described in heterozygosis by Hodson et al. [32] in a female patient diagnosed at 46 years with PAH. She manifested a functional class 2 of the World Health Organization (WHO) scale, with a mPAP of 38 mmHg, PAWP 5 mmHg, PVR of 6.9 WU and a CO 4.8 L/min, with no family history [32]. Hodson et al. confirmed that this variant exhibited impaired BMP9 processing, secretion or stability, thus indicating that this variant is loss-of-function and it is likely the cause of PAH. Loss of function of BMP9 may reduce the expression of BMPR2 and ALK-1 and disrupt its interaction with ENG, both of which may contribute to PAH pathogenesis [32].
Likewise, in patient 2, we detected a missense variant of unknown significance inherited from the mother and a CNV, which includes GDF2 inherited from her father. The missense variant is absent in pseudo control population databases and has not been previously described. Although both parents were heterozygous for those variants, neither of them is affected. Strikingly, this proband is an idiopathic patient and she does not meet any of the Curaçao criteria. Likewise, although the CNV includes 40 genes more, the individual did not have reported comorbidities. Previous studies have reported other patients with similar deletions (4.29 Mb and 4.28 Mb) who exhibited earlier onset of disease and with no family history [32]. However, to the best of our knowledge, no segregation analysis of the variants was carried out. In view of all the above we suggest that the presence of one of the variants is not enough for the development of the disease but that the presence of a second hit is necessary. However, it would be necessary to extend this study with functional studies.
The analysis through our customized gene panel within the diagnostic routine has allowed us to describe a homozygous variant in GDF2 and a compound heterozygous of GDF2. Therefore, the design of our customized panel has proven to be effective for genetic diagnosis of conflicting cases of PAH and HHT or even with overlapping features. Moreover, it was possible to detect heterozygous variants in healthy relatives, which will help them to receive detailed follow-up of their progression with the aim of early diagnosis and/or prevention of the disease. Our work adds evidence to the notion that variants in the GDF2 may generate a greater predisposition to develop both idiopathic PAH and a “HHT-like” syndrome. Likewise, we described five healthy individuals with heterozygous variants, suggesting that there is a wider spectrum of inheritance patterns associated with GDF2 variants, an incomplete penetrance or even the need for the presence of a second mutational event in an individual for the development of the disease.
Finally, in view of the results obtained, the importance of genetic counselling for families is clear, which can be useful when planning future pregnancies or if there is family history of a condition.
Author Contributions
Conceptualization, J.T.-C. and P.L.; methodology, J.T.-C., N.G., P.L. and P.A.; software, J.T.-C., N.G. and P.A.; validation, J.T.-C., N.G., J.N. and P.A.; formal analysis, J.T.-C., N.G. and I.G., A.M.B.; investigation, P.A., A.C.-U. and N.O.; resources, A.C.-U. and N.O.; data curation, N.G., A.C.-U., I.G., J.T.-C.; writing—original draft preparation, N.G.; writing—review and editing, N.G. and I.G.; visualization, N.G.; supervision, J.T.-C., P.L. and P.E.-S.; project administration, P.L. and P.E.-S.; funding acquisition, P.E.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Instituto de Salud Carlos III, grant number FISPI18/01233, Annual supported by the Foundation against PAH (FCHP, https://www.fchp.es/, accessed on 9 September 2021) and FEDER Annual Grant (https://enfermedades-raras.org/index.php, accessed on 9 September 2021). Alejandro Cruz-Utrilla holds a research-training contract “Rio-Hostega” (CM20/00164) from the Spanish Ministry of Science and Innovation (Instituto de Salud Carlos III).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of Hospital Universitario La Paz (CEIC-HULP PI-1210).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patient(s) to publish this paper.
Acknowledgments
We would like to thank all patients and families for their collaboration and participation in this study and all the center participants centers in the PAH Consortium, including the REHAP registry.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar]
- Machado, R.D.; Eickelberg, O.; Elliott, C.G.; Geraci, M.W.; Hanaoka, M.; Loyd, J.E.; Newman, J.H.; Phillips, J.A.; Soubrier, F.; Trembath, R.C. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S32–S42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J. Familial primary pulmonary hypertension (Gene PPH1) is caused by mutations in the bone morphogenetic protein receptor–II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A.; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.; Bayrak-Toydemir, P.; Pyeritz, R.E. Hereditary hemorrhagic telangiectasia: An overview of diagnosis, management, and pathogenesis. Genet. Med. 2011, 13, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu Osler Weber syndrome). Am. J. Med. Genet. 2000, 91, 66–67. [Google Scholar] [CrossRef]
- Faughnan, M.E.; Palda, V.A.; Garcia-Tsao, G.; Geisthoff, U.W.; McDonald, J.; Proctor, D.D.; Spears, J.; Brown, D.H.; Buscarini, E.; Chesnutt, M.S. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J. Med. Genet. 2011, 48, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Porteous, M.; Reinhardt, D.; Gallione, C.; Holloway, S.; Umasunthar, T.; Lux, A.; McKinnon, W.; Marchuk, D.; Guttmacher, A. Hereditary haemorrhagic telangiectasia: A questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J. Med. Genet. 2003, 40, 585–590. [Google Scholar] [CrossRef]
- Shintani, M.; Yagi, H.; Nakayama, T.; Saji, T.; Matsuoka, R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J. Med. Genet. 2009, 46, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Nasim, M.T.; Ogo, T.; Ahmed, M.; Randall, R.; Chowdhury, H.M.; Snape, K.M.; Bradshaw, T.Y.; Southgate, L.; Lee, G.J.; Jackson, I. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum. Mutat. 2011, 32, 1385–1389. [Google Scholar] [CrossRef] [Green Version]
- Harrison, R.E.; Berger, R.; Haworth, S.G.; Tulloh, R.; Mache, C.J.; Morrell, N.W.; Aldred, M.A.; Trembath, R.C. Transforming growth factor-β receptor mutations and pulmonary arterial hypertension in childhood. Circulation 2005, 111, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Trembath, R.C.; Thomson, J.R.; Machado, R.D.; Morgan, N.V.; Atkinson, C.; Winship, I.; Simonneau, G.; Galie, N.; Loyd, J.E.; Humbert, M. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2001, 345, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, J.; Wooderchak-Donahue, W.; VanSant Webb, C.; Whitehead, K.; Stevenson, D.A.; Bayrak-Toydemir, P. Hereditary hemorrhagic telangiectasia: Genetics and molecular diagnostics in a new era. Front. Genet. 2015, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, M.; Ishiguro, T.; Yamada, O.; Morisaki, H.; Morisaki, T. Hereditary hemorrhagic telangiectasia in Japanese patients. J. Hum. Genet. 2014, 59, 37–41. [Google Scholar] [CrossRef]
- Olivieri, C.; Pagella, F.; Semino, L.; Lanzarini, L.; Valacca, C.; Pilotto, A.; Corno, S.; Scappaticci, S.; Manfredi, G.; Buscarini, E. Analysis of ENG and ACVRL1 genes in 137 HHT Italian families identifies 76 different mutations (24 novel). Comparison with other European studies. J. Hum. Genet. 2007, 52, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Llorente, L.; Gallardo-Vara, E.; Rossi, E.; Smadja, D.M.; Botella, L.M.; Bernabeu, C. Endoglin and alk1 as therapeutic targets for hereditary hemorrhagic telangiectasia. Expert Opin. Ther. Targets 2017, 21, 933–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, É.; Westermann, C.J.; Marchuk, D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef]
- Wang, X.; Lian, T.; Jiang, X.; Liu, S.; Li, S.; Jiang, R.; Wu, W.; Ye, J.; Cheng, C.; Du, Y. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801609–1801619. [Google Scholar] [CrossRef]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416–1432. [Google Scholar] [CrossRef] [Green Version]
- Eyries, M.; Montani, D.; Nadaud, S.; Girerd, B.; Levy, M.; Bourdin, A.; Trésorier, R.; Chaouat, A.; Cottin, V.; Sanfiorenzo, C. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur. Respir. J. 2019, 53, 1801371–1801383. [Google Scholar] [CrossRef] [Green Version]
- Wooderchak-Donahue, W.L.; McDonald, J.; O’Fallon, B.; Upton, P.D.; Li, W.; Roman, B.L.; Young, S.; Plant, P.; Fülöp, G.T.; Langa, C. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 2013, 93, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhardt, B.A.; Zayac, C.; Trerotola, S.O.; Asch, D.A.; Pyeritz, R.E. Cost savings through molecular diagnosis for hereditary hemorrhagic telangiectasia. Genet. Med. 2012, 14, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Castaño, J.A.T.; Hernández-Gonzalez, I.; Gallego, N.; Pérez-Olivares, C.; Ochoa Parra, N.; Arias, P.; Granda, E.; Acebo, G.G.; Lago-Docampo, M.; Palomino-Doza, J. Customized Massive Parallel Sequencing Panel for Diagnosis of Pulmonary Arterial Hypertension. Genes 2020, 11, 1158. [Google Scholar] [CrossRef]
- Tenorio, J.A.; Arias, P.; Hernandez, I.; Ochoa, N.; Granda, E.; Navas, P. Customized massive paralleled sequencing panel for diagnosis of Pulmonary Arterial Hypertension. Eur. J. Hum. Genet. 2019, 27, 1328–1342. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Hernandez-Gonzalez, I.; Tenorio, J.; Palomino-Doza, J.; Martinez Meñaca, A.; Morales Ruiz, R.; Lago-Docampo, M.; Valverde Gomez, M.; Gomez Roman, J.; Enguita Valls, A.B.; Perez-Olivares, C. Clinical heterogeneity of Pulmonary Arterial Hypertension associated with variants in TBX4. PLoS ONE 2020, 15, e0232216. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yang, J.; Tang, X.; Li, H.; Shen, Y.; Gu, W.; Zhao, S. Homozygous GDF2-Related Hereditary Hemorrhagic Telangiectasia in a Chinese Family. Pediatrics 2020, 146, e20191970. [Google Scholar] [CrossRef]
- Bayrak-Toydemir, P.; Mao, R.; Lewin, S.; McDonald, J. Hereditary hemorrhagic telangiectasia: An overview of diagnosis and management in the molecular era for clinicians. Genet. Med. 2004, 6, 175–191. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Fan, R.; Ji, R.; Zou, W.; Penny, D.J.; Varghese, N.P.; Fan, Y. Novel homozygous BMP9 nonsense mutation causes pulmonary arterial hypertension: A case report. BMC Pulm. Med. 2016, 16, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgson, J.; Swietlik, E.M.; Salmon, R.M.; Hadinnapola, C.; Nikolic, I.; Wharton, J.; Guo, J.; Liley, J.; Haimel, M.; Bleda, M. Characterization of GDF2 mutations and levels of BMP9 and BMP10 in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.; Ruiz Llorente, L.; McDonald, J.; Quarrell, O.; Ugonna, K.; Bentham, J.; Mason, R.; Martin, J.; Moore, D.; Bergstrom, K. Homozygous GDF2 nonsense mutations result in a loss of circulating BMP9 and BMP10 and are associated with either PAH or an “HHT like” syndrome in children. Mol. Genet. Genom. Med. 2021, e1685. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).