
Activation Mechanisms of the VPS34 Complexes


Abstract
1. Introduction
2. Basic Architectures of VPS34/Vps34 Complexes I and II
3. Activation Mechanisms of VPS34/Vps34 and Complexes I and II
3.1. Basic Architecture of the VPS34 Kinase Domain
3.2. Activation Mechanisms of VPS34/Vps34 Complexes I and II
4. Controlling the Intracellular PtdIns(3)P Concentration
5. Regulation of VPS34/Vps34 Complex Activity by Phosphorylation from Upstream Regulators, the ULK1 Complex and mTORC1
5.1. Complex I Regulation by Phosphorylation from the ULK1/Atg1 Complex
5.2. mTORC1–VPS34 and VPS34–mTORC1 Pathways
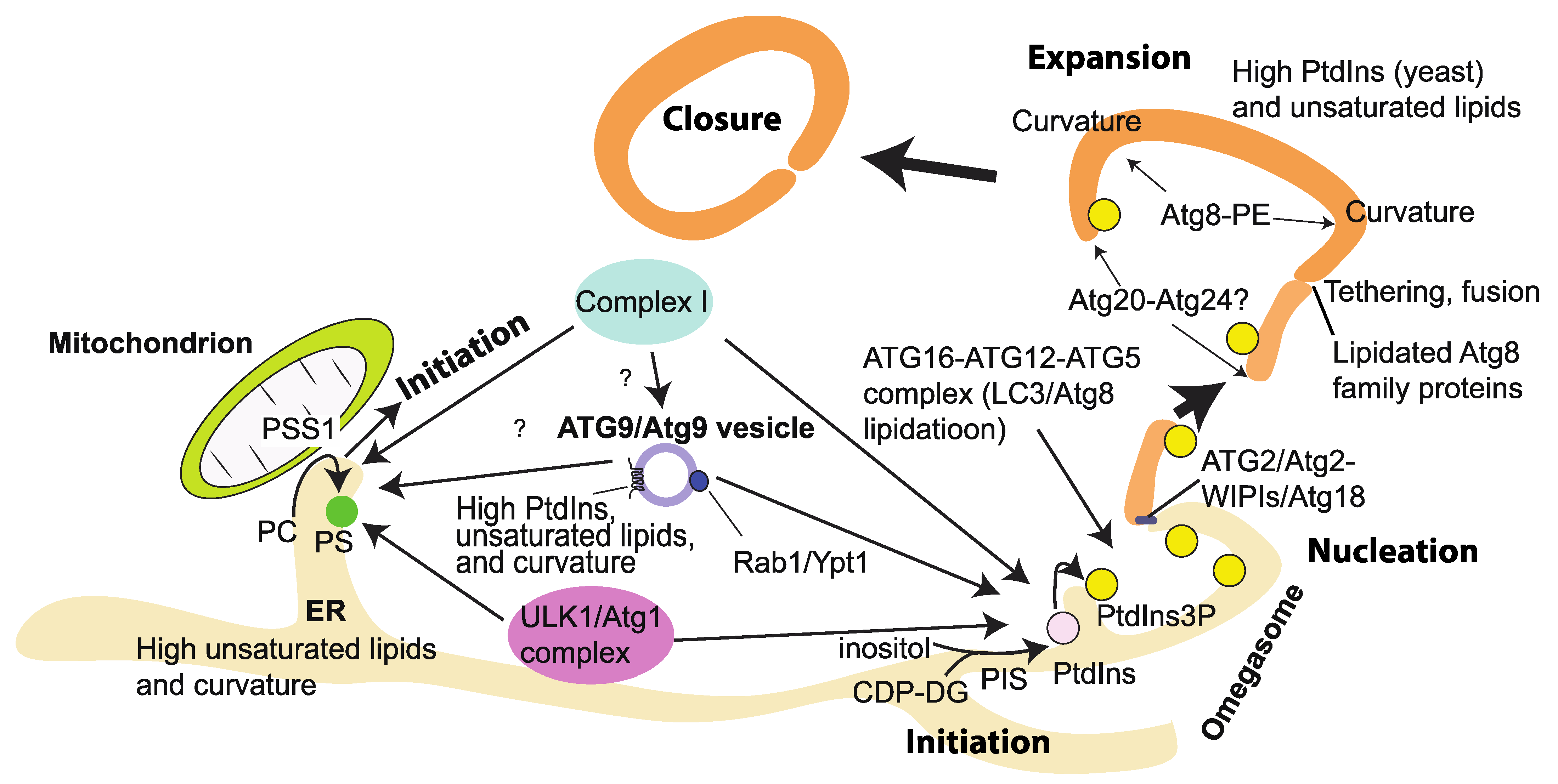
6. Genetic and Cell Biological Hierarchy of the Recruitment of Complex I at the Initiation Step of Autophagy
7. Lipid Environments That Influence the VPS34/Vps34 Complex Activities
7.1. Physicochemical Parameters That Affect the VPS34/Vps34 Activities
7.2. Lipid Packing of Acyl Chains
7.3. Membrane Curvature
7.4. Electrostatics from PS
7.5. The Use and Interpretation of High-Percentage PS In Vitro
7.6. Electrostatics from Phosphoinositides
7.7. Physicochemical Properties of Autophagosomal Membranes
7.8. Effects of Lipid Packing, Curvature, and Electrostatics on the Activity and Membrane Binding of VPS34 Complexes
7.9. Effects of Packing, Membrane Curvature, and Electrostatics on Membrane Tethering (MT) and Lipid Transfer (LT) of ATG2A/B/Atg2
7.10. Roles of Lipid Unpacking and Curvature on the Atg16/ATG16L Complex and the Lipidation of Atg8 Families
7.11. Phosphatidylinositol (PtdIns), the Sole Substrate for VPS34/Vps34
7.12. Membrane Properties in Health and Disease
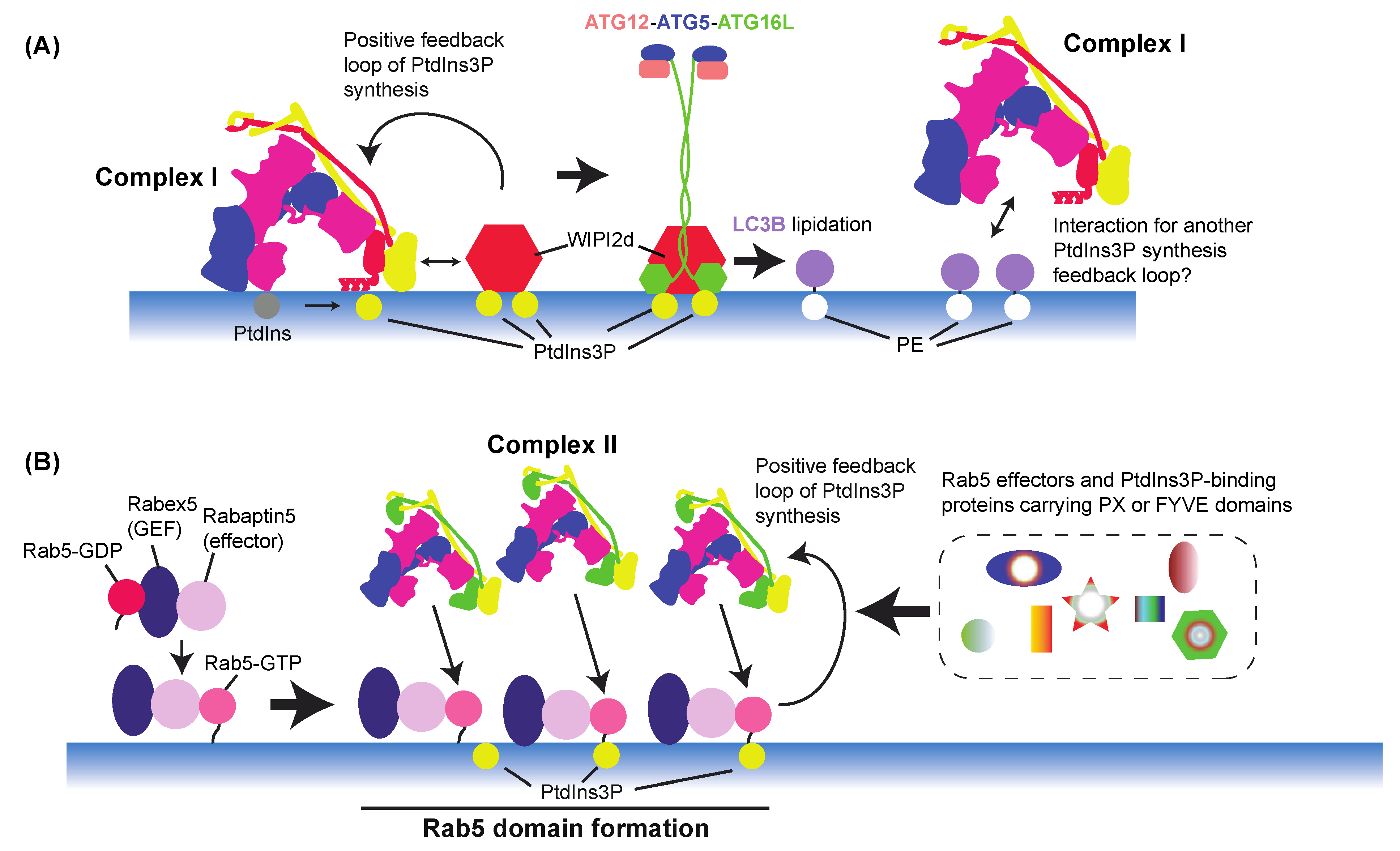
8. VPS34 Activation Regulation by Feedback Mechanisms and Downstream Effectors
8.1. Autophagy
8.2. Endocytic Pathway
8.3. PtdIns(3)P-Binding Proteins in Endocytic Pathways
9. Regulation of VPS34 Activation by Binding Proteins
10. Concluding Remarks and Future Perspectives
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Batrouni, A.G.; Baskin, J.M. A MAP for PI3K activation on endosomes. Nat. Cell Biol. 2020, 22, 1292–1294. [Google Scholar] [CrossRef] [PubMed]
- Thapa, N.; Chen, M.; Horn, H.T.; Choi, S.; Wen, T.; Anderson, R.A. Phosphatidylinositol-3-OH kinase signalling is spatially organized at endosomal compartments by microtubule-associated protein 4. Nat. Cell Biol. 2020, 22, 1357–1370. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef] [PubMed]
- Marat, A.L.; Wallroth, A.; Lo, W.T.; Muller, R.; Norata, G.D.; Falasca, M.; Schultz, C.; Haucke, V. mTORC1 activity repression by late endosomal phosphatidylinositol 3,4-bisphosphate. Science 2017, 356, 968–972. [Google Scholar] [CrossRef]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schoneberg, J.; Ullrich, A.; Lampe, A.; Muller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 2013, 499, 233–237. [Google Scholar] [CrossRef]
- Boukhalfa, A.; Nascimbeni, A.C.; Ramel, D.; Dupont, N.; Hirsch, E.; Gayral, S.; Laffargue, M.; Codogno, P.; Morel, E. PI3KC2alpha-dependent and VPS34-independent generation of PI3P controls primary cilium-mediated autophagy in response to shear stress. Nat. Commun. 2020, 11, 294. [Google Scholar] [CrossRef]
- Devereaux, K.; Dall’Armi, C.; Alcazar-Roman, A.; Ogasawara, Y.; Zhou, X.; Wang, F.; Yamamoto, A.; De Camilli, P.; Di Paolo, G. Regulation of mammalian autophagy by class II and III PI 3-kinases through PI3P synthesis. PLoS ONE 2013, 8, e76405. [Google Scholar] [CrossRef]
- Franco, I.; Gulluni, F.; Campa, C.C.; Costa, C.; Margaria, J.P.; Ciraolo, E.; Martini, M.; Monteyne, D.; De Luca, E.; Germena, G.; et al. PI3K class II alpha controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev. Cell 2014, 28, 647–658. [Google Scholar] [CrossRef]
- Schu, P.V.; Takegawa, K.; Fry, M.J.; Stack, J.H.; Waterfield, M.D.; Emr, S.D. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science 1993, 260, 88–91. [Google Scholar] [CrossRef]
- Welters, P.; Takegawa, K.; Emr, S.D.; Chrispeels, M.J. AtVPS34, a phosphatidylinositol 3-kinase of Arabidopsis thaliana, is an essential protein with homology to a calcium-dependent lipid binding domain. Proc. Natl. Acad. Sci. USA 1994, 91, 11398–11402. [Google Scholar] [CrossRef] [PubMed]
- Banta, L.M.; Robinson, J.S.; Klionsky, D.J.; Emr, S.D. Organelle assembly in yeast: Characterization of yeast mutants defective in vacuolar biogenesis and protein sorting. J. Cell Biol. 1988, 107, 1369–1383. [Google Scholar] [CrossRef] [PubMed]
- Herman, P.K.; Emr, S.D. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae. Mol. Cell Biol. 1990, 10, 6742–6754. [Google Scholar] [CrossRef] [PubMed]
- Stack, J.H.; Herman, P.K.; Schu, P.V.; Emr, S.D. A membrane-associated complex containing the Vps15 protein kinase and the Vps34 PI 3-kinase is essential for protein sorting to the yeast lysosome-like vacuole. EMBO J. 1993, 12, 2195–2204. [Google Scholar] [CrossRef]
- Stack, J.H.; DeWald, D.B.; Takegawa, K.; Emr, S.D. Vesicle-mediated protein transport: Regulatory interactions between the Vps15 protein kinase and the Vps34 PtdIns 3-kinase essential for protein sorting to the vacuole in yeast. J. Cell Biol. 1995, 129, 321–334. [Google Scholar] [CrossRef]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, W.; Chen, K.; Ding, X.; Chen, S.; Zhong, Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 19211–19216. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Rostislavleva, K.; Soler, N.; Ohashi, Y.; Zhang, L.; Pardon, E.; Burke, J.E.; Masson, G.R.; Johnson, C.; Steyaert, J.; Ktistakis, N.T.; et al. Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science 2015, 350, aac7365. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Tremel, S.; Masson, G.R.; McGinney, L.; Boulanger, J.; Rostislavleva, K.; Johnson, C.M.; Niewczas, I.; Clark, J.; Williams, R.L. Membrane characteristics tune activities of endosomal and autophagic human VPS34 complexes. eLife 2020, 9, e58281. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, S.; Carlson, L.A.; Stjepanovic, G.; Young, L.N.; Kim, D.J.; Grob, P.; Stanley, R.E.; Nogales, E.; Hurley, J.H. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. eLife 2014, 3, e05115. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Nassiri, A.; Zhong, Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc. Natl. Acad. Sci. USA 2011, 108, 7769–7774. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Thapa, N.; Liao, Y.; Choi, S.; Anderson, R.A. PtdIns(4,5)P2 signaling regulates ATG14 and autophagy. Proc. Natl. Acad. Sci. USA 2016, 113, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Liu, J.J.; Li, Y.; Huang, Y.; Ta, N.; Chen, Y.; Fu, H.; Ye, M.D.; Ding, Y.; Huang, W.; et al. Cryo-EM structure and biochemical analysis reveal the basis of the functional difference between human PI3KC3-C1 and -C2. Cell Res. 2017, 27, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Young, L.N.; Morris, K.L.; von Bulow, S.; Schoneberg, J.; Yamamoto-Imoto, H.; Oe, Y.; Yamamoto, K.; Nakamura, S.; Stjepanovic, G.; et al. Bidirectional Control of Autophagy by BECN1 BARA Domain Dynamics. Mol. Cell 2019, 73, 339–353.e6. [Google Scholar] [CrossRef]
- Huang, W.; Choi, W.; Hu, W.; Mi, N.; Guo, Q.; Ma, M.; Liu, M.; Tian, Y.; Lu, P.; Wang, F.L.; et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012, 22, 473–489. [Google Scholar] [CrossRef]
- Tremel, S.; Ohashi, Y.; Morado, D.R.; Bertram, J.; Perisic, O.; Brandt, L.T.L.; von Wrisberg, M.K.; Chen, Z.A.; Maslen, S.L.; Kovtun, O.; et al. Structural basis for VPS34 kinase activation by Rab1 and Rab5 on membranes. Nat. Commun. 2021, 12, 1564. [Google Scholar] [CrossRef]
- Stjepanovic, G.; Baskaran, S.; Lin, M.G.; Hurley, J.H. Vps34 Kinase Domain Dynamics Regulate the Autophagic PI 3-Kinase Complex. Mol. Cell 2017, 67, 528–534.e3. [Google Scholar] [CrossRef]
- Stack, J.H.; Emr, S.D. Vps34p required for yeast vacuolar protein sorting is a multiple specificity kinase that exhibits both protein kinase and phosphatidylinositol-specific PI 3-kinase activities. J. Biol. Chem. 1994, 269, 31552–31562. [Google Scholar] [CrossRef]
- Ohashi, Y. Class III phosphatidylinositol 3-kinase complex I subunit NRBF2/Atg38-from cell and structural biology to health and disease. Autophagy 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Tremel, S.; Williams, R.L. VPS34 complexes from a structural perspective. J. Lipid. Res. 2019, 60, 229–241. [Google Scholar] [CrossRef]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef]
- Honda, A.; Harrington, E.; Cornella-Taracido, I.; Furet, P.; Knapp, M.S.; Glick, M.; Triantafellow, E.; Dowdle, W.E.; Wiedershain, D.; Maniara, W.; et al. Potent, Selective, and Orally Bioavailable Inhibitors of VPS34 Provide Chemical Tools to Modulate Autophagy in Vivo. ACS Med. Chem. Lett. 2016, 7, 72–76. [Google Scholar] [CrossRef]
- Pasquier, B.; El-Ahmad, Y.; Filoche-Romme, B.; Dureuil, C.; Fassy, F.; Abecassis, P.Y.; Mathieu, M.; Bertrand, T.; Benard, T.; Barriere, C.; et al. Discovery of (2S)-8-[(3R)-3-methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)- 3,4-dihydro-2H-pyrimido[1,2-a]pyrimidin-6-one: A novel potent and selective inhibitor of Vps34 for the treatment of solid tumors. J. Med. Chem. 2015, 58, 376–400. [Google Scholar] [CrossRef] [PubMed]
- Dyczynski, M.; Yu, Y.; Otrocka, M.; Parpal, S.; Braga, T.; Henley, A.B.; Zazzi, H.; Lerner, M.; Wennerberg, K.; Viklund, J.; et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 2018, 435, 32–43. [Google Scholar] [CrossRef]
- Miller, S.; Tavshanjian, B.; Oleksy, A.; Perisic, O.; Houseman, B.T.; Shokat, K.M.; Williams, R.L. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science 2010, 327, 1638–1642. [Google Scholar] [CrossRef]
- Bilanges, B.; Alliouachene, S.; Pearce, W.; Morelli, D.; Szabadkai, G.; Chung, Y.L.; Chicanne, G.; Valet, C.; Hill, J.M.; Voshol, P.J.; et al. Vps34 PI 3-kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat. Commun. 2017, 8, 1804. [Google Scholar] [CrossRef]
- Christoforidis, S.; Miaczynska, M.; Ashman, K.; Wilm, M.; Zhao, L.; Yip, S.C.; Waterfield, M.D.; Backer, J.M.; Zerial, M. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol. 1999, 1, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.T.; Panaretou, C.; Stenmark, H.; Miaczynska, M.; Backer, J.M. Role of Rab5 in the recruitment of hVps34/p150 to the early endosome. Traffic 2002, 3, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Buckles, T.C.; Ohashi, Y.; Tremel, S.; McLaughlin, S.H.; Pardon, E.; Steyaert, J.; Gordon, M.T.; Williams, R.L.; Falke, J.J. The G-Protein Rab5A Activates VPS34 Complex II, a Class III PI3K, by a Dual Regulatory Mechanism. Biophys. J. 2020, 119, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Birkeland, H.C.; Stenmark, H. Protein targeting to endosomes and phagosomes via FYVE and PX domains. Curr Top. Microbiol. Immunol. 2004, 282, 89–115. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H.; Gillooly, D.J. Intracellular trafficking and turnover of phosphatidylinositol 3-phosphate. Semin. Cell Dev. Biol. 2001, 12, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Young, L.N.; Goerdeler, F.; Hurley, J.H. Structural pathway for allosteric activation of the autophagic PI 3-kinase complex I. Proc. Natl. Acad. Sci. USA 2019, 116, 21508–21513. [Google Scholar] [CrossRef]
- Lipatova, Z.; Belogortseva, N.; Zhang, X.Q.; Kim, J.; Taussig, D.; Segev, N. Regulation of selective autophagy onset by a Ypt/Rab GTPase module. Proc. Natl. Acad. Sci. USA 2012, 109, 6981–6986. [Google Scholar] [CrossRef]
- Wang, J.; Menon, S.; Yamasaki, A.; Chou, H.T.; Walz, T.; Jiang, Y.; Ferro-Novick, S. Ypt1 recruits the Atg1 kinase to the preautophagosomal structure. Proc. Natl. Acad. Sci. USA 2013, 110, 9800–9805. [Google Scholar] [CrossRef]
- Lynch-Day, M.A.; Bhandari, D.; Menon, S.; Huang, J.; Cai, H.; Bartholomew, C.R.; Brumell, J.H.; Ferro-Novick, S.; Klionsky, D.J. Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 7811–7816. [Google Scholar] [CrossRef] [PubMed]
- Kakuta, S.; Yamamoto, H.; Negishi, L.; Kondo-Kakuta, C.; Hayashi, N.; Ohsumi, Y. Atg9 vesicles recruit vesicle-tethering proteins Trs85 and Ypt1 to the autophagoso.ome formation site. J. Biol. Chem. 2012, 287, 44261–44269. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Tamura, N.; Kono, N.; Shimanaka, Y.; Arai, H.; Yamamoto, H.; Mizushima, N. Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J. 2017, 36, 1719–1735. [Google Scholar] [CrossRef]
- Steinfeld, N.; Lahiri, V.; Morrison, A.; Metur, S.P.; Klionsky, D.J.; Weisman, L.S. Elevating PI3P drives select downstream membrane trafficking pathways. Mol. Biol Cell 2021, 32, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Soler, N.; Garcia Ortegon, M.; Zhang, L.; Kirsten, M.L.; Perisic, O.; Masson, G.R.; Burke, J.E.; Jakobi, A.J.; Apostolakis, A.A.; et al. Characterization of Atg38 and NRBF2, a fifth subunit of the autophagic Vps34/PIK3C3 complex. Autophagy 2016, 12, 2129–2144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Takatoh, J.; Wang, F. The mammalian class 3 PI3K (PIK3C3) is required for early embryogenesis and cell proliferation. PLoS ONE 2011, 6, e16358. [Google Scholar] [CrossRef] [PubMed]
- Nemazanyy, I.; Blaauw, B.; Paolini, C.; Caillaud, C.; Protasi, F.; Mueller, A.; Proikas-Cezanne, T.; Russell, R.C.; Guan, K.L.; Nishino, I.; et al. Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease. EMBO Mol. Med. 2013, 5, 870–890. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef]
- Cheng, J.; Fujita, A.; Yamamoto, H.; Tatematsu, T.; Kakuta, S.; Obara, K.; Ohsumi, Y.; Fujimoto, T. Yeast and mammalian autophagosomes exhibit distinct phosphatidylinositol 3-phosphate asymmetries. Nat. Commun. 2014, 5, 3207. [Google Scholar] [CrossRef] [PubMed]
- Cebollero, E.; van der Vaart, A.; Zhao, M.; Rieter, E.; Klionsky, D.J.; Helms, J.B.; Reggiori, F. Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr. Biol. 2012, 22, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Vergne, I.; Roberts, E.; Elmaoued, R.A.; Tosch, V.; Delgado, M.A.; Proikas-Cezanne, T.; Laporte, J.; Deretic, V. Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J. 2009, 28, 2244–2258. [Google Scholar] [CrossRef] [PubMed]
- Taguchi-Atarashi, N.; Hamasaki, M.; Matsunaga, K.; Omori, H.; Ktistakis, N.T.; Yoshimori, T.; Noda, T. Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic 2010, 11, 468–478. [Google Scholar] [CrossRef]
- Zou, J.; Zhang, C.; Marjanovic, J.; Kisseleva, M.V.; Majerus, P.W.; Wilson, M.P. Myotubularin-related protein (MTMR) 9 determines the enzymatic activity, substrate specificity, and role in autophagy of MTMR8. Proc. Natl. Acad. Sci. USA 2012, 109, 9539–9544. [Google Scholar] [CrossRef]
- Dowling, J.J.; Lawlor, M.W.; Das, S. X-Linked Myotubular Myopathy. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Buj-Bello, A.; Furling, D.; Tronchere, H.; Laporte, J.; Lerouge, T.; Butler-Browne, G.S.; Mandel, J.L. Muscle-specific alternative splicing of myotubularin-related 1 gene is impaired in DM1 muscle cells. Hum. Mol. Genet. 2002, 11, 2297–2307. [Google Scholar] [CrossRef]
- Laporte, J.; Hu, L.J.; Kretz, C.; Mandel, J.L.; Kioschis, P.; Coy, J.F.; Klauck, S.M.; Poustka, A.; Dahl, N. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat. Genet. 1996, 13, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Bolino, A.; Muglia, M.; Conforti, F.L.; LeGuern, E.; Salih, M.A.; Georgiou, D.M.; Christodoulou, K.; Hausmanowa-Petrusewicz, I.; Mandich, P.; Schenone, A.; et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat. Genet. 2000, 25, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Amoasii, L.; Hnia, K.; Chicanne, G.; Brech, A.; Cowling, B.S.; Muller, M.M.; Schwab, Y.; Koebel, P.; Ferry, A.; Payrastre, B.; et al. Myotubularin and PtdIns3P remodel the sarcoplasmic reticulum in muscle in vivo. J. Cell Sci. 2013, 126, 1806–1819. [Google Scholar] [CrossRef]
- Ketel, K.; Krauss, M.; Nicot, A.S.; Puchkov, D.; Wieffer, M.; Muller, R.; Subramanian, D.; Schultz, C.; Laporte, J.; Haucke, V. A phosphoinositide conversion mechanism for exit from endosomes. Nature 2016, 529, 408–412. [Google Scholar] [CrossRef]
- Al-Qusairi, L.; Prokic, I.; Amoasii, L.; Kretz, C.; Messaddeq, N.; Mandel, J.L.; Laporte, J. Lack of myotubularin (MTM1) leads to muscle hypotrophy through unbalanced regulation of the autophagy and ubiquitin-proteasome pathways. FASEB J. 2013, 27, 3384–3394. [Google Scholar] [CrossRef]
- Sabha, N.; Volpatti, J.R.; Gonorazky, H.; Reifler, A.; Davidson, A.E.; Li, X.; Eltayeb, N.M.; Dall’Armi, C.; Di Paolo, G.; Brooks, S.V.; et al. PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J. Clin. Investig. 2016, 126, 3613–3625. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.J.; Gubas, A.; Tooze, S.A. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018, 293, 5386–5395. [Google Scholar] [CrossRef] [PubMed]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Puente, C.; Hendrickson, R.C.; Jiang, X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J. Biol. Chem. 2016, 291, 6026–6035. [Google Scholar] [CrossRef]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef]
- Noda, N.N.; Fujioka, Y. Atg1 family kinases in autophagy initiation. Cell Mol. Life Sci. 2015, 72, 3083–3096. [Google Scholar] [CrossRef]
- Suzuki, K.; Kubota, Y.; Sekito, T.; Ohsumi, Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 2007, 12, 209–218. [Google Scholar] [CrossRef]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014, 127, 4089–4102. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.J.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.M.; Starker, C.; Nho, R.S.; et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Raucci, S.; Jaquenoud, M.; Hatakeyama, R.; Stumpe, M.; Rohr, R.; Reggiori, F.; De Virgilio, C.; Dengjel, J. Multilayered Control of Protein Turnover by TORC1 and Atg1. Cell Rep. 2019, 28, 3486–3496.e6. [Google Scholar] [CrossRef] [PubMed]
- Kamber, R.A.; Shoemaker, C.J.; Denic, V. Receptor-Bound Targets of Selective Autophagy Use a Scaffold Protein to Activate the Atg1 Kinase. Mol. Cell. 2015, 59, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell. 2015, 59, 285–297. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, S.; He, L.; Rong, Y.; Brier, L.W.; Sun, Q.; Liu, R.; Fan, W.; Chen, S.; Yue, Z.; et al. MTORC1-mediated NRBF2 phosphorylation functions as a switch for the class III PtdIns3K and autophagy. Autophagy 2017, 13, 592–607. [Google Scholar] [CrossRef]
- Mercer, T.J.; Ohashi, Y.; Boeing, S.; Jefferies, H.B.J.; De Tito, S.; Flynn, H.; Tremel, S.; Zhang, W.; Wirth, M.; Frith, D.; et al. Phosphoproteomic identification of ULK substrates reveals VPS15-dependent ULK/VPS34 interplay in the regulation of autophagy. EMBO J. 2021, 40, e105985. [Google Scholar] [CrossRef]
- Chan, E.Y.; Kir, S.; Tooze, S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef]
- Scott, R.C.; Juhasz, G.; Neufeld, T.P. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 2007, 17, 1–11. [Google Scholar] [CrossRef]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef]
- Lazarus, M.B.; Novotny, C.J.; Shokat, K.M. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem. Biol. 2015, 10, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yokom, A.L.; Wang, C.; Young, L.N.; Youle, R.J.; Hurley, J.H. ULK complex organization in autophagy by a C-shaped FIP200 N-terminal domain dimer. J. Cell Biol. 2020, 219, e201911047. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Fujioka, Y.; Suzuki, S.W.; Noshiro, D.; Suzuki, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Ando, T.; Noda, N.N.; et al. The Intrinsically Disordered Protein Atg13 Mediates Supramolecular Assembly of Autophagy Initiation Complexes. Dev. Cell 2016, 38, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, P.; Wollert, T. Reconstituting Autophagy Initiation from Purified Components. Methods Mol. Biol. 2019, 1880, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Perna, M.G.; Hofmann, B.; Beier, V.; Wollert, T. The Atg1-kinase complex tethers Atg9-vesicles to initiate autophagy. Nat. Commun. 2016, 7, 10338. [Google Scholar] [CrossRef]
- Jao, C.C.; Ragusa, M.J.; Stanley, R.E.; Hurley, J.H. A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 5486–5491. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.W.; Yamamoto, H.; Oikawa, Y.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Ohsumi, Y. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc. Natl. Acad. Sci. USA 2015, 112, 3350–3355. [Google Scholar] [CrossRef]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Ganley, I.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1·ATG13·FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Fujioka, Y.; Suzuki, S.W.; Yamamoto, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Akada, R.; Inagaki, F.; Ohsumi, Y.; Noda, N.N. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 2014, 21, 513–521. [Google Scholar] [CrossRef]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef]
- Jung, C.H.; Seo, M.; Otto, N.M.; Kim, D.H. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy 2011, 7, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.X.; Russell, R.C.; Guan, K.L. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 2013, 9, 1983–1995. [Google Scholar] [CrossRef]
- Young, L.N.; Cho, K.; Lawrence, R.; Zoncu, R.; Hurley, J.H. Dynamics and architecture of the NRBF2-containing phosphatidylinositol 3-kinase complex I of autophagy. Proc. Natl. Acad. Sci. USA 2016, 113, 8224–8229. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Jung, C.H.; Seo, M.; Kim, E.K.; Park, J.M.; Bae, S.S.; Kim, D.H. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell 2015, 57, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Munson, M.J.; Allen, G.F.; Toth, R.; Campbell, D.G.; Lucocq, J.M.; Ganley, I.G. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015, 34, 2272–2290. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Autophagic lysosome reformation. Exp. Cell Res. 2013, 319, 142–146. [Google Scholar] [CrossRef]
- Byfield, M.P.; Murray, J.T.; Backer, J.M. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J. Biol. Chem. 2005, 280, 33076–33082. [Google Scholar] [CrossRef]
- Hirsch, D.S.; Shen, Y.; Dokmanovic, M.; Yu, J.; Mohan, N.; Elzarrad, M.K.; Wu, W.J. Insulin activation of vacuolar protein sorting 34 mediates localized phosphatidylinositol 3-phosphate production at lamellipodia and activation of mTOR/S6K1. Cell Signal 2014, 26, 1258–1268. [Google Scholar] [CrossRef]
- Nobukuni, T.; Joaquin, M.; Roccio, M.; Dann, S.G.; Kim, S.Y.; Gulati, P.; Byfield, M.P.; Backer, J.M.; Natt, F.; Bos, J.L.; et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 14238–14243. [Google Scholar] [CrossRef]
- Hornberger, T.A.; Chu, W.K.; Mak, Y.W.; Hsiung, J.W.; Huang, S.A.; Chien, S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc. Natl. Acad. Sci. USA 2006, 103, 4741–4746. [Google Scholar] [CrossRef]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar] [CrossRef]
- Foster, D.A. Regulation of mTOR by phosphatidic acid? Cancer Res. 2007, 67, 1–4. [Google Scholar] [CrossRef]
- Yoon, M.S.; Du, G.; Backer, J.M.; Frohman, M.A.; Chen, J. Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J. Cell Biol. 2011, 195, 435–447. [Google Scholar] [CrossRef]
- Fetalvero, K.M.; Yu, Y.; Goetschkes, M.; Liang, G.; Valdez, R.A.; Gould, T.; Triantafellow, E.; Bergling, S.; Loureiro, J.; Eash, J.; et al. Defective autophagy and mTORC1 signaling in myotubularin null mice. Mol. Cell. Biol. 2013, 33, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Veverka, V.; Crabbe, T.; Bird, I.; Lennie, G.; Muskett, F.W.; Taylor, R.J.; Carr, M.D. Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor: Compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene 2008, 27, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Anandapadamanaban, M.; Masson, G.R.; Perisic, O.; Berndt, A.; Kaufman, J.; Johnson, C.M.; Santhanam, B.; Rogala, K.B.; Sabatini, D.M.; Williams, R.L. Architecture of human Rag GTPase heterodimers and their complex with mTORC1. Science 2019, 366, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.H.; Kim, D.H.; Kim, I.S.; Kim, J.H.; Lee, M.N.; Lee, H.J.; Kim, J.H.; Jang, S.K.; Suh, P.G.; Ryu, S.H. PLD2 forms a functional complex with mTOR/raptor to transduce mitogenic signals. Cell Signal. 2006, 18, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S.; Son, K.; Arauz, E.; Han, J.M.; Kim, S.; Chen, J. Leucyl-tRNA Synthetase Activates Vps34 in Amino Acid-Sensing mTORC1 Signaling. Cell Rep. 2016, 16, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Pedersen, N.M.; Wang, L.; Torgersen, M.L.; Stenmark, H.; Raiborg, C. PtdIns3P controls mTORC1 signaling through lysosomal positioning. J. Cell Biol. 2017, 216, 4217–4233. [Google Scholar] [CrossRef] [PubMed]
- Hodson, N.; Dent, J.R.; Song, Z.; O’Leary, M.F.; Nicholson, T.; Jones, S.W.; Murray, J.T.; Jeromson, S.; Hamilton, D.L.; Breen, L.; et al. Protein-carbohydrate ingestion alters Vps34 cellular localization independent of changes in kinase activity in human skeletal muscle. Exp. Physiol. 2020, 105, 2178–2189. [Google Scholar] [CrossRef]
- Jaber, N.; Dou, Z.; Chen, J.S.; Catanzaro, J.; Jiang, Y.P.; Ballou, L.M.; Selinger, E.; Ouyang, X.; Lin, R.Z.; Zhang, J.; et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. USA 2012, 109, 2003–2008. [Google Scholar] [CrossRef] [PubMed]
- Ravenhill, B.J.; Boyle, K.B.; von Muhlinen, N.; Ellison, C.J.; Masson, G.R.; Otten, E.G.; Foeglein, A.; Williams, R.; Randow, F. The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell. 2019, 74, 320–329.e6. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Koyama-Honda, I.; Mizushima, N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J. Cell Sci. 2012, 125, 1488–1499. [Google Scholar] [CrossRef]
- Karanasios, E.; Stapleton, E.; Manifava, M.; Kaizuka, T.; Mizushima, N.; Walker, S.A.; Ktistakis, N.T. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J. Cell Sci. 2013, 126, 5224–5238. [Google Scholar] [CrossRef]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lee, M.; Fairn, G.D. Phospholipid subcellular localization and dynamics. J. Biol. Chem. 2018, 293, 6230–6240. [Google Scholar] [CrossRef]
- Antonny, B.; Vanni, S.; Shindou, H.; Ferreira, T. From zero to six double bonds: Phospholipid unsaturation and organelle function. Trends Cell. Biol. 2015, 25, 427–436. [Google Scholar] [CrossRef]
- Vanni, S.; Riccardi, L.; Palermo, G.; De Vivo, M. Structure and Dynamics of the Acyl Chains in the Membrane Trafficking and Enzymatic Processing of Lipids. Acc Chem. Res. 2019, 52, 3087–3096. [Google Scholar] [CrossRef]
- Bigay, J.; Antonny, B. Curvature, lipid packing, and electrostatics of membrane organelles: Defining cellular territories in determining specificity. Dev. Cell 2012, 23, 886–895. [Google Scholar] [CrossRef]
- Rawicz, W.; Olbrich, K.C.; McIntosh, T.; Needham, D.; Evans, E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 2000, 79, 328–339. [Google Scholar] [CrossRef]
- Small, D.M. Lateral chain packing in lipids and membranes. J. Lipid. Res. 1984, 25, 1490–1500. [Google Scholar] [CrossRef]
- Paltauf, F. Ether lipids in biomembranes. Chem. Phys. Lipids 1994, 74, 101–139. [Google Scholar] [CrossRef]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Momsen, M.M.; Smaby, J.M.; Brockman, H.L.; Brown, R.E. Cholesterol decreases the interfacial elasticity and detergent solubility of sphingomyelins. Biochemistry 2001, 40, 5954–5963. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Momsen, M.M.; Brockman, H.L.; Brown, R.E. Sterol structure and sphingomyelin acyl chain length modulate lateral packing elasticity and detergent solubility in model membranes. Biophys. J. 2003, 85, 3788–3801. [Google Scholar] [CrossRef][Green Version]
- Bigay, J.; Casella, J.F.; Drin, G.; Mesmin, B.; Antonny, B. ArfGAP1 responds to membrane curvature through the folding of a lipid packing sensor motif. EMBO J. 2005, 24, 2244–2253. [Google Scholar] [CrossRef]
- Drin, G.; Casella, J.F.; Gautier, R.; Boehmer, T.; Schwartz, T.U.; Antonny, B. A general amphipathic alpha-helical motif for sensing membrane curvature. Nat. Struct. Mol. Biol. 2007, 14, 138–146. [Google Scholar] [CrossRef]
- van Hilten, N.; Stroh, K.S.; Risselada, H.J. Membrane Thinning Induces Sorting of Lipids and the Amphipathic Lipid Packing Sensor (ALPS) Protein Motif. Front. Physiol. 2020, 11, 250. [Google Scholar] [CrossRef]
- Koller, D.; Lohner, K. The role of spontaneous lipid curvature in the interaction of interfacially active peptides with membranes. Biochim. Biophys. Acta 2014, 1838, 2250–2259. [Google Scholar] [CrossRef]
- Cooke, I.R.; Deserno, M. Coupling between lipid shape and membrane curvature. Biophys. J. 2006, 91, 487–495. [Google Scholar] [CrossRef]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef]
- Lee, M.C.; Orci, L.; Hamamoto, S.; Futai, E.; Ravazzola, M.; Schekman, R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell 2005, 122, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.M.; Morello, V.; Bigay, J.; Gibson, K.; Verbavatz, J.M.; Antonny, B.; Jackson, C.L. alpha-Synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding. J. Cell Biol. 2011, 194, 89–103. [Google Scholar] [CrossRef]
- Martens, S.; Kozlov, M.M.; McMahon, H.T. How synaptotagmin promotes membrane fusion. Science 2007, 316, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Cezanne, A.; Lauer, J.; Solomatina, A.; Sbalzarini, I.F.; Zerial, M. A non-linear system patterns Rab5 GTPase on the membrane. eLife 2020, 9, 54434. [Google Scholar] [CrossRef]
- Arumugam, S.; Kaur, A. The Lipids of the Early Endosomes: Making Multimodality Work. Chembiochem 2017, 18, 1053–1060. [Google Scholar] [CrossRef]
- Cullen, P.J. Endosomal sorting and signalling: An emerging role for sorting nexins. Nat. Rev. Mol. Cell Biol. 2008, 9, 574–582. [Google Scholar] [CrossRef]
- Hanley, S.E.; Cooper, K.F. Sorting Nexins in Protein Homeostasis. Cells 2020, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Boucrot, E.; Ferreira, A.P.; Almeida-Souza, L.; Debard, S.; Vallis, Y.; Howard, G.; Bertot, L.; Sauvonnet, N.; McMahon, H.T. Endophilin marks and controls a clathrin-independent endocytic pathway. Nature 2015, 517, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Kjaerulff, O.; Brodin, L.; Jung, A. The structure and function of endophilin proteins. Cell Biochem. Biophys. 2011, 60, 137–154. [Google Scholar] [CrossRef]
- Bhatt, V.S.; Ashley, R.; Sundborger-Lunna, A. Amphipathic Motifs Regulate N-BAR Protein Endophilin B1 Auto-inhibition and Drive Membrane Remodeling. Structure 2021, 29, 61–69e63. [Google Scholar] [CrossRef]
- Rao, Y.; Ma, Q.; Vahedi-Faridi, A.; Sundborger, A.; Pechstein, A.; Puchkov, D.; Luo, L.; Shupliakov, O.; Saenger, W.; Haucke, V. Molecular basis for SH3 domain regulation of F-BAR-mediated membrane deformation. Proc. Natl. Acad. Sci. USA 2010, 107, 8213–8218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Navarro, M.V.; Peng, G.; Molinelli, E.; Goh, S.L.; Judson, B.L.; Rajashankar, K.R.; Sondermann, H. Molecular mechanism of membrane constriction and tubulation mediated by the F-BAR protein Pacsin/Syndapin. Proc. Natl. Acad. Sci. USA 2009, 106, 12700–12705. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Alimohamadi, H.; Bakka, B.; Trementozzi, A.N.; Day, K.J.; Fawzi, N.L.; Rangamani, P.; Stachowiak, J.C. Membrane bending by protein phase separation. Proc. Natl. Acad. Sci. USA 2021, 118, 5118. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Molecular and cell biology of phosphatidylserine and phosphatidylethanolamine metabolism. Prog Nucleic Acid Res. Mol. Biol. 2003, 75, 69–111. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.G.; Fairn, G.D. Distribution, dynamics and functional roles of phosphatidylserine within the cell. Cell Commun. Signal. 2019, 17, 126. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.X.; Akbar, M.; Kevala, K.; Kim, H.Y. Phosphatidylserine is a critical modulator for Akt activation. J. Cell Biol. 2011, 192, 979–992. [Google Scholar] [CrossRef]
- Zhou, Y.; Ariotti, N.; Rae, J.; Liang, H.; Tillu, V.; Tee, S.; Bastiani, M.; Bademosi, A.T.; Collins, B.M.; Meunier, F.A.; et al. Caveolin-1 and cavin1 act synergistically to generate a unique lipid environment in caveolae. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef] [PubMed]
- Orr, J.W.; Newton, A.C. Interaction of protein kinase C with phosphatidylserine. 2. Specificity and regulation. Biochemistry 1992, 31, 4667–4673. [Google Scholar] [CrossRef]
- Shenoy, S.; Shekhar, P.; Heinrich, F.; Daou, M.C.; Gericke, A.; Ross, A.H.; Losche, M. Membrane association of the PTEN tumor suppressor: Molecular details of the protein-membrane complex from SPR binding studies and neutron reflection. PLoS ONE 2012, 7, e32591. [Google Scholar] [CrossRef]
- Zhong, S.; Hsu, F.; Stefan, C.J.; Wu, X.; Patel, A.; Cosgrove, M.S.; Mao, Y. Allosteric activation of the phosphoinositide phosphatase Sac1 by anionic phospholipids. Biochemistry 2012, 51, 3170–3177. [Google Scholar] [CrossRef] [PubMed]
- Fairn, G.D.; Schieber, N.L.; Ariotti, N.; Murphy, S.; Kuerschner, L.; Webb, R.I.; Grinstein, S.; Parton, R.G. High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J. Cell Biol. 2011, 194, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Leventis, P.A.; Grinstein, S. The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 2010, 39, 407–427. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Yeung, T.; Gilbert, G.E.; Shi, J.; Silvius, J.; Kapus, A.; Grinstein, S. Membrane phosphatidylserine regulates surface charge and protein localization. Science 2008, 319, 210–213. [Google Scholar] [CrossRef]
- Del Vecchio, K.; Stahelin, R.V. Investigation of the phosphatidylserine binding properties of the lipid biosensor, Lactadherin C2 (LactC2), in different membrane environments. J. Bioenerg. Biomembr. 2018, 50, 1–10. [Google Scholar] [CrossRef]
- Yin, H.L.; Janmey, P.A. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol 2003, 65, 761–789. [Google Scholar] [CrossRef]
- Lemmon, M.A. Phosphoinositide recognition domains. Traffic 2003, 4, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Balla, T. Inositol-lipid binding motifs: Signal integrators through protein-lipid and protein-protein interactions. J. Cell Sci. 2005, 118, 2093–2104. [Google Scholar] [CrossRef]
- Di Paolo, G.; De Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. [Google Scholar] [CrossRef]
- Ktistakis, N.T. ER platforms mediating autophagosome generation. Biochim. Biophys Acta Mol. Cell Biol. Lipids 2020, 1865, 158433. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Karanasios, E.; Walker, S.A.; Okkenhaug, H.; Manifava, M.; Hummel, E.; Zimmermann, H.; Ahmed, Q.; Domart, M.C.; Collinson, L.; Ktistakis, N.T. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat. Commun. 2016, 7, 12420. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, M.S.; Munro, S. Cholesterol and the Golgi apparatus. Science 1993, 261, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, M.J.; Stanley, R.E.; Hurley, J.H. Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell 2012, 151, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.J.; Vance, J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 2000, 275, 34534–34540. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Sakai, Y.; Koyama-Honda, I.; Tachikawa, M.; Knorr, R.L.; Mizushima, N. Modeling Membrane Morphological Change during Autophagosome Formation. iScience 2020, 23, 101466. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Klionsky, D.J. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol. Biol. Cell 2005, 16, 1593–1605. [Google Scholar] [CrossRef]
- Hettema, E.H.; Lewis, M.J.; Black, M.W.; Pelham, H.R. Retromer and the sorting nexins Snx4/41/42 mediate distinct retrieval pathways from yeast endosomes. EMBO J. 2003, 22, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Popelka, H.; Damasio, A.; Hinshaw, J.E.; Klionsky, D.J.; Ragusa, M.J. Structure and function of yeast Atg20, a sorting nexin that facilitates autophagy induction. Proc. Natl. Acad. Sci. USA 2017, 114, E10112–E10121. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Alam, J.M.; Fukuda, T.; Kageyama, S.; Kirisako, H.; Ishii, Y.; Shimada, I.; Ohsumi, Y.; Komatsu, M.; Kanki, T.; et al. Membrane perturbation by lipidated Atg8 underlies autophagosome biogenesis. Nat. Struct. Mol. Biol. 2021, 28, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Shteyn, V.; Melia, T.J. Sensing Membrane Curvature in Macroautophagy. J. Mol. Biol. 2017, 429, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Kakuta, S.; Watanabe, T.M.; Kitamura, A.; Sekito, T.; Kondo-Kakuta, C.; Ichikawa, R.; Kinjo, M.; Ohsumi, Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 2012, 198, 219–233. [Google Scholar] [CrossRef]
- Sawa-Makarska, J.; Baumann, V.; Coudevylle, N.; von Bulow, S.; Nogellova, V.; Abert, C.; Schuschnig, M.; Graef, M.; Hummer, G.; Martens, S. Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science 2020, 369. [Google Scholar] [CrossRef]
- Kotani, T.; Kirisako, H.; Koizumi, M.; Ohsumi, Y.; Nakatogawa, H. The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation. Proc. Natl. Acad. Sci. USA 2018, 115, 10363–10368. [Google Scholar] [CrossRef]
- Gomez-Sanchez, R.; Rose, J.; Guimaraes, R.; Mari, M.; Papinski, D.; Rieter, E.; Geerts, W.J.; Hardenberg, R.; Kraft, C.; Ungermann, C.; et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J. Cell Biol. 2018, 217, 2743–2763. [Google Scholar] [CrossRef]
- Maeda, S.; Otomo, C.; Otomo, T. The autophagic membrane tether ATG2A transfers lipids between membranes. eLife 2019, 8. [Google Scholar] [CrossRef]
- Chowdhury, S.; Otomo, C.; Leitner, A.; Ohashi, K.; Aebersold, R.; Lander, G.C.; Otomo, T. Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. Proc. Natl. Acad. Sci. USA 2018, 115, E9792–E9801. [Google Scholar] [CrossRef]
- Osawa, T.; Ishii, Y.; Noda, N.N. Human ATG2B possesses a lipid transfer activity which is accelerated by negatively charged lipids and WIPI4. Genes Cells 2020, 25, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Fracchiolla, D.; Chang, C.; Hurley, J.H.; Martens, S. A PI3K-WIPI2 positive feedback loop allosterically activates LC3 lipidation in autophagy. J. Cell Biol. 2020, 219, 7. [Google Scholar] [CrossRef] [PubMed]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef]
- Landajuela, A.; Hervas, J.H.; Anton, Z.; Montes, L.R.; Gil, D.; Valle, M.; Rodriguez, J.F.; Goni, F.M.; Alonso, A. Lipid Geometry and Bilayer Curvature Modulate LC3/GABARAP-Mediated Model Autophagosomal Elongation. Biophys. J. 2016, 110, 411–422. [Google Scholar] [CrossRef]
- Taniguchi, S.; Toyoshima, M.; Takamatsu, T.; Mima, J. Curvature-sensitive trans-assembly of human Atg8-family proteins in autophagy-related membrane tethering. Protein Sci. 2020, 29, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Sekito, T.; Ohsumi, Y. Assortment of phosphatidylinositol 3-kinase complexes-Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol. Biol. Cell 2006, 17, 1527–1539. [Google Scholar] [CrossRef]
- Araki, Y.; Ku, W.C.; Akioka, M.; May, A.I.; Hayashi, Y.; Arisaka, F.; Ishihama, Y.; Ohsumi, Y. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J. Cell Biol. 2013, 203, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome formation in mammalian cells. Cell Struct. Funct. 2002, 27, 421–429. [Google Scholar] [CrossRef]
- Kobayashi, T.; Suzuki, K.; Ohsumi, Y. Autophagosome formation can be achieved in the absence of Atg18 by expressing engineered PAS-targeted Atg2. FEBS Lett. 2012, 586, 2473–2478. [Google Scholar] [CrossRef]
- Graef, M.; Friedman, J.R.; Graham, C.; Babu, M.; Nunnari, J. ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell 2013, 24, 2918–2931. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Akioka, M.; Kondo-Kakuta, C.; Yamamoto, H.; Ohsumi, Y. Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J. Cell Sci. 2013, 126, 2534–2544. [Google Scholar] [CrossRef]
- Valverde, D.P.; Yu, S.; Boggavarapu, V.; Kumar, N.; Lees, J.A.; Walz, T.; Reinisch, K.M.; Melia, T.J. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 2019, 218, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Kotani, T.; Kawaoka, T.; Hirata, E.; Suzuki, K.; Nakatogawa, H.; Ohsumi, Y.; Noda, N.N. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol. 2019, 26, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.X.; Li, Y.; Ding, Y.H.; Liu, J.J.; Zhang, M.J.; Dong, M.Q.; Wang, H.W.; Yu, L. Architecture of the ATG2B-WDR45 complex and an aromatic Y/HF motif crucial for complex formation. Autophagy 2017, 13, 1870–1883. [Google Scholar] [CrossRef]
- Obara, K.; Sekito, T.; Niimi, K.; Ohsumi, Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J. Biol. Chem. 2008, 283, 23972–23980. [Google Scholar] [CrossRef]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell. 2008, 19, 2092–2100. [Google Scholar] [CrossRef]
- Sakoh-Nakatogawa, M.; Kirisako, H.; Nakatogawa, H.; Ohsumi, Y. Localization of Atg3 to autophagy-related membranes and its enhancement by the Atg8-family interacting motif to promote expansion of the membranes. FEBS Lett. 2015, 589, 744–749. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, J.G.; Kim, Y.J.; Humpolickova, J.; Eisenreichova, A.; Sengupta, N.; Toth, D.J.; Boura, E.; Balla, T. Defining the subcellular distribution and metabolic channeling of phosphatidylinositol. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Zewe, J.P.; Miller, A.M.; Sangappa, S.; Wills, R.C.; Goulden, B.D.; Hammond, G.R.V. Probing the subcellular distribution of phosphatidylinositol reveals a surprising lack at the plasma membrane. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Blunsom, N.J.; Cockcroft, S. Phosphatidylinositol synthesis at the endoplasmic reticulum. Biochim. Biophys Acta Mol. Cell Biol. Lipids 2020, 1865, 158471. [Google Scholar] [CrossRef]
- Lofke, C.; Ischebeck, T.; Konig, S.; Freitag, S.; Heilmann, I. Alternative metabolic fates of phosphatidylinositol produced by phosphatidylinositol synthase isoforms in Arabidopsis thaliana. Biochem. J. 2008, 413, 115–124. [Google Scholar] [CrossRef]
- Schutter, M.; Giavalisco, P.; Brodesser, S.; Graef, M. Local Fatty Acid Channeling into Phospholipid Synthesis Drives Phagophore Expansion during Autophagy. Cell 2020, 180, 135–149.e14. [Google Scholar] [CrossRef]
- Laczko-Dobos, H.; Maddali, A.K.; Jipa, A.; Bhattacharjee, A.; Vegh, A.G.; Juhasz, G. Lipid profiles of autophagic structures isolated from wild type and Atg2 mutant Drosophila. Biochim. Biophys Acta Mol. Cell Biol. Lipids 2021, 1866, 158868. [Google Scholar] [CrossRef]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Kochl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef]
- Ohashi, Y.; Munro, S. Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol. Biol. Cell 2010, 21, 3998–4008. [Google Scholar] [CrossRef]
- Mari, M.; Griffith, J.; Rieter, E.; Krishnappa, L.; Klionsky, D.J.; Reggiori, F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J. Cell Biol. 2010, 190, 1005–1022. [Google Scholar] [CrossRef]
- Ohashi, Y.; Tremel, S.; Williams, R.L. Unsaturation, curvature and charge: Effects of membrane parameters on PIK3C3/VPS34-containing complexes. Autophagy 2021, 17, 823–825. [Google Scholar] [CrossRef]
- Raiborg, C.; Wenzel, E.M.; Stenmark, H. ER-endosome contact sites: Molecular compositions and functions. EMBO J. 2015, 34, 1848–1858. [Google Scholar] [CrossRef]
- Honscher, C.; Ungermann, C. A close-up view of membrane contact sites between the endoplasmic reticulum and the endolysosomal system: From yeast to man. Crit. Rev. Biochem Mol. Biol. 2014, 49, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.H.; Copic, A.; Levine, T.P. Advances on the Transfer of Lipids by Lipid Transfer Proteins. Trends Biochem. Sci. 2017, 42, 516–530. [Google Scholar] [CrossRef]
- Wallroth, A.; Haucke, V. Phosphoinositide conversion in endocytosis and the endolysosomal system. J. Biol. Chem. 2018, 293, 1526–1535. [Google Scholar] [CrossRef]
- Carrard, J.; Gallart-Ayala, H.; Infanger, D.; Teav, T.; Wagner, J.; Knaier, R.; Colledge, F.; Streese, L.; Konigstein, K.; Hinrichs, T.; et al. Metabolic View on Human Healthspan: A Lipidome-Wide Association Study. Metabolites 2021, 11, 287. [Google Scholar] [CrossRef]
- Staps, P.; Rizzo, W.B.; Vaz, F.M.; Bugiani, M.; Giera, M.; Heijs, B.; van Kampen, A.H.C.; Pras-Raves, M.L.; Breur, M.; Groen, A.; et al. Disturbed brain ether lipid metabolism and histology in Sjogren-Larsson syndrome. J. Inherit. Metab. Dis. 2020, 43, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Naguib, A.; Bencze, G.; Engle, D.D.; Chio, I.I.C.; Herzka, T.; Watrud, K.; Bencze, S.; Tuveson, D.A.; Pappin, D.J.; Trotman, L.C. p53 mutations change phosphatidylinositol acyl chain composition. Cell Rep. 2015, 10, 8–19. [Google Scholar] [CrossRef]
- Rueda-Rincon, N.; Bloch, K.; Derua, R.; Vyas, R.; Harms, A.; Hankemeier, T.; Khan, N.A.; Dehairs, J.; Bagadi, M.; Binda, M.M.; et al. p53 attenuates AKT signaling by modulating membrane phospholipid composition. Oncotarget 2015, 6, 21240–21254. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, A.; Narita, S.; Nakanishi, H.; Ishikawa, M.; Eguchi, S.; Kimura, H.; Takasuga, S.; Huang, M.; Inoue, T.; Sasaki, J.; et al. Increased fatty acyl saturation of phosphatidylinositol phosphates in prostate cancer progression. Sci. Rep. 2019, 9, 13257. [Google Scholar] [CrossRef]
- Balogh, G.; Peter, M.; Liebisch, G.; Horvath, I.; Torok, Z.; Nagy, E.; Maslyanko, A.; Benko, S.; Schmitz, G.; Harwood, J.L.; et al. Lipidomics reveals membrane lipid remodelling and release of potential lipid mediators during early stress responses in a murine melanoma cell line. Biochim. Biophys. Acta 2010, 1801, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Tooze, S.A.; Jefferies, H.B.; Kalie, E.; Longatti, A.; McAlpine, F.E.; McKnight, N.C.; Orsi, A.; Polson, H.E.; Razi, M.; Robinson, D.J.; et al. Trafficking and signaling in mammalian autophagy. IUBMB Life 2010, 62, 503–508. [Google Scholar] [CrossRef]
- Watanabe, Y.; Kobayashi, T.; Yamamoto, H.; Hoshida, H.; Akada, R.; Inagaki, F.; Ohsumi, Y.; Noda, N.N. Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J. Biol. Chem. 2012, 287, 31681–31690. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, S.; Ragusa, M.J.; Boura, E.; Hurley, J.H. Two-site recognition of phosphatidylinositol 3-phosphate by PROPPINs in autophagy. Mol. Cell 2012, 47, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Ren, J.; Zhang, Y.; Feng, W. Structural Conservation of the Two Phosphoinositide-Binding Sites in WIPI Proteins. J. Mol. Biol. 2019, 431, 1494–1505. [Google Scholar] [CrossRef]
- Ren, J.; Liang, R.; Wang, W.; Zhang, D.; Yu, L.; Feng, W. Multi-site-mediated entwining of the linear WIR-motif around WIPI beta-propellers for autophagy. Nat. Commun. 2020, 11, 2702. [Google Scholar] [CrossRef]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef]
- Birgisdottir, A.B.; Mouilleron, S.; Bhujabal, Z.; Wirth, M.; Sjottem, E.; Evjen, G.; Zhang, W.; Lee, R.; O’Reilly, N.; Tooze, S.A.; et al. Members of the autophagy class III phosphatidylinositol 3-kinase complex I interact with GABARAP and GABARAPL1 via LIR motifs. Autophagy 2019, 15, 1333–1355. [Google Scholar] [CrossRef] [PubMed]
- Redpath, G.M.I.; Betzler, V.M.; Rossatti, P.; Rossy, J. Membrane Heterogeneity Controls Cellular Endocytic Trafficking. Front. Cell Dev. Biol. 2020, 8, 757. [Google Scholar] [CrossRef] [PubMed]
- Iraburu, M.J.; Garner, T.; Montiel-Duarte, C. Revising Endosomal Trafficking under Insulin Receptor Activation. Int. J. Mol. Sci. 2021, 22, 6978. [Google Scholar] [CrossRef] [PubMed]
- Estadella, I.; Pedros-Gamez, O.; Colomer-Molera, M.; Bosch, M.; Sorkin, A.; Felipe, A. Endocytosis: A Turnover Mechanism Controlling Ion Channel Function. Cells 2020, 9, 1833. [Google Scholar] [CrossRef] [PubMed]
- Gillooly, D.J.; Morrow, I.C.; Lindsay, M.; Gould, R.; Bryant, N.J.; Gaullier, J.M.; Parton, R.G.; Stenmark, H. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 2000, 19, 4577–4588. [Google Scholar] [CrossRef]
- Lauer, J.; Segeletz, S.; Cezanne, A.; Guaitoli, G.; Raimondi, F.; Gentzel, M.; Alva, V.; Habeck, M.; Kalaidzidis, Y.; Ueffing, M.; et al. Auto-regulation of Rab5 GEF activity in Rabex5 by allosteric structural changes, catalytic core dynamics and ubiquitin binding. eLife 2019, 8. [Google Scholar] [CrossRef]
- Stenmark, H.; Vitale, G.; Ullrich, O.; Zerial, M. Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell 1995, 83, 423–432. [Google Scholar] [CrossRef]
- Edler, E.; Schulze, E.; Stein, M. Membrane localization and dynamics of geranylgeranylated Rab5 hypervariable region. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1335–1349. [Google Scholar] [CrossRef]
- Seet, L.F.; Hong, W. The Phox (PX) domain proteins and membrane traffic. Biochim. Biophys Acta 2006, 1761, 878–896. [Google Scholar] [CrossRef]
- Hayakawa, A.; Hayes, S.J.; Lawe, D.C.; Sudharshan, E.; Tuft, R.; Fogarty, K.; Lambright, D.; Corvera, S. Structural basis for endosomal targeting by FYVE domains. J. Biol. Chem. 2004, 279, 5958–5966. [Google Scholar] [CrossRef]
- Chandra, M.; Chin, Y.K.; Mas, C.; Feathers, J.R.; Paul, B.; Datta, S.; Chen, K.E.; Jia, X.; Yang, Z.; Norwood, S.J.; et al. Classification of the human phox homology (PX) domains based on their phosphoinositide binding specificities. Nat. Commun. 2019, 10, 1528. [Google Scholar] [CrossRef]
- Marat, A.L.; Haucke, V. Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. EMBO J. 2016, 35, 561–579. [Google Scholar] [CrossRef]
- Misra, S.; Miller, G.J.; Hurley, J.H. Recognizing phosphatidylinositol 3-phosphate. Cell 2001, 107, 559–562. [Google Scholar] [CrossRef]
- He, J.; Vora, M.; Haney, R.M.; Filonov, G.S.; Musselman, C.A.; Burd, C.G.; Kutateladze, A.G.; Verkhusha, V.V.; Stahelin, R.V.; Kutateladze, T.G. Membrane insertion of the FYVE domain is modulated by pH. Proteins 2009, 76, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Eyeson, R.; Cheever, M.L.; Geng, J.; Verkhusha, V.V.; Burd, C.; Overduin, M.; Kutateladze, T.G. Targeting of the FYVE domain to endosomal membranes is regulated by a histidine switch. Proc. Natl. Acad. Sci. USA 2005, 102, 13052–13057. [Google Scholar] [CrossRef]
- Gallon, M.; Cullen, P.J. Retromer and sorting nexins in endosomal sorting. Biochem Soc. Trans. 2015, 43, 33–47. [Google Scholar] [CrossRef]
- Chen, K.E.; Healy, M.D.; Collins, B.M. Towards a molecular understanding of endosomal trafficking by Retromer and Retriever. Traffic 2019, 20, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, L.K.; Arlt, H.; Kuhlee, A.; Raunser, S.; Ungermann, C. Retromer-driven membrane tubulation separates endosomal recycling from Rab7/Ypt7-dependent fusion. Mol. Biol. Cell 2017, 28, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.L.; Sato, T.K.; de Beer, T.; Kutateladze, T.G.; Emr, S.D.; Overduin, M. Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat. Cell Biol. 2001, 3, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Stahelin, R.V.; Ananthanarayanan, B.; Blatner, N.R.; Singh, S.; Bruzik, K.S.; Murray, D.; Cho, W. Mechanism of membrane binding of the phospholipase D1 PX domain. J. Biol. Chem. 2004, 279, 54918–54926. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.; Karathanassis, D.; Pacold, C.M.; Pacold, M.E.; Ellson, C.D.; Anderson, K.E.; Butler, P.J.; Lavenir, I.; Perisic, O.; Hawkins, P.T.; et al. The crystal structure of the PX domain from p40(phox) bound to phosphatidylinositol 3-phosphate. Mol. Cell 2001, 8, 829–839. [Google Scholar] [CrossRef]
- Bago, R.; Sommer, E.; Castel, P.; Crafter, C.; Bailey, F.P.; Shpiro, N.; Baselga, J.; Cross, D.; Eyers, P.A.; Alessi, D.R. The hVps34-SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. EMBO J. 2016, 35, 2263. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, M.A.; Pearson, R.B.; Hannan, R.D.; Sheppard, K.E. AKT-independent PI3-K signaling in cancer-emerging role for SGK3. Cancer Manag. Res. 2013, 5, 281–292. [Google Scholar] [CrossRef]
- Tessier, M.; Woodgett, J.R. Role of the Phox homology domain and phosphorylation in activation of serum and glucocorticoid-regulated kinase-3. J. Biol. Chem. 2006, 281, 23978–23989. [Google Scholar] [CrossRef]
- Pokorny, D.; Truebestein, L.; Fleming, K.D.; Burke, J.E.; Leonard, T.A. In vitro reconstitution of Sgk3 activation by phosphatidylinositol 3-phosphate. J. Biol. Chem. 2021, 297, 100919. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Lippe, R.; Christoforidis, S.; Gaullier, J.M.; Brech, A.; Callaghan, J.; Toh, B.H.; Murphy, C.; Zerial, M.; Stenmark, H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 1998, 394, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Christoforidis, S.; McBride, H.M.; Burgoyne, R.D.; Zerial, M. The Rab5 effector EEA1 is a core component of endosome docking. Nature 1999, 397, 621–625. [Google Scholar] [CrossRef]
- McBride, H.M.; Rybin, V.; Murphy, C.; Giner, A.; Teasdale, R.; Zerial, M. Oligomeric complexes link Rab5 effectors with NSF and drive membrane fusion via interactions between EEA1 and syntaxin 13. Cell 1999, 98, 377–386. [Google Scholar] [CrossRef]
- Char, R.; Pierre, P. The RUFYs, a Family of Effector Proteins Involved in Intracellular Trafficking and Cytoskeleton Dynamics. Front. Cell Dev. Biol. 2020, 8, 779. [Google Scholar] [CrossRef]
- Hayes, S.; Chawla, A.; Corvera, S. TGF beta receptor internalization into EEA1-enriched early endosomes: Role in signaling to Smad2. J. Cell Biol. 2002, 158, 1239–1249. [Google Scholar] [CrossRef]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef]
- Stenmark, H.; Aasland, R.; Driscoll, P.C. The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Lett 2002, 513, 77–84. [Google Scholar] [CrossRef]
- Hasegawa, J.; Strunk, B.S.; Weisman, L.S. PI5P and PI(3,5)P2: Minor, but Essential Phosphoinositides. Cell Struct. Funct. 2017, 42, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Futter, C.E.; Collinson, L.M.; Backer, J.M.; Hopkins, C.R. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J. Cell Biol. 2001, 155, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Borja, M.; Wubbolts, R.; Calafat, J.; Janssen, H.; Divecha, N.; Dusseljee, S.; Neefjes, J. Multivesicular body morphogenesis requires phosphatidyl-inositol 3-kinase activity. Curr Biol. 1999, 9, 55–58. [Google Scholar] [CrossRef]
- Martinez, J.; Malireddi, R.K.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, H.K.; Tabata, K.; Byck, J.M.; Hamasaki, M.; Farrell, D.P.; Anishchenko, I.; DiMaio, F.; Im, Y.J.; Yoshimori, T.; Hurley, J.H. Structural basis for autophagy inhibition by the human Rubicon-Rab7 complex. Proc. Natl. Acad. Sci. USA 2020, 117, 17003–17010. [Google Scholar] [CrossRef]
- Tabata, K.; Matsunaga, K.; Sakane, A.; Sasaki, T.; Noda, T.; Yoshimori, T. Rubicon and PLEKHM1 negatively regulate the endocytic/autophagic pathway via a novel Rab7-binding domain. Mol. Biol. Cell 2010, 21, 4162–4172. [Google Scholar] [CrossRef]
- Stein, M.P.; Feng, Y.; Cooper, K.L.; Welford, A.M.; Wandinger-Ness, A. Human VPS34 and p150 are Rab7 interacting partners. Traffic 2003, 4, 754–771. [Google Scholar] [CrossRef]
- Sun, Q.; Westphal, W.; Wong, K.N.; Tan, I.; Zhong, Q. Rubicon controls endosome maturation as a Rab7 effector. Proc. Natl. Acad. Sci. USA 2010, 107, 19338–19343. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef]
- Xu, D.Q.; Wang, Z.; Wang, C.Y.; Zhang, D.Y.; Wan, H.D.; Zhao, Z.L.; Gu, J.; Zhang, Y.X.; Li, Z.G.; Man, K.Y.; et al. PAQR3 controls autophagy by integrating AMPK signaling to enhance ATG14L-associated PI3K activity. EMBO J. 2016, 35, 496–514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, L.; Ireland, S.C.; Ahat, E.; Li, J.; Bekier, M.E., 2nd; Zhang, Z.; Wang, Y. GORASP2/GRASP55 collaborates with the PtdIns3K UVRAG complex to facilitate autophagosome-lysosome fusion. Autophagy 2019, 15, 1787–1800. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhang, Y.; Deng, T.; Gu, J.; Liu, J.; Yang, H.; Xu, Y.; Yan, Y.; Yang, F.; Zhang, H.; et al. PDPK1 regulates autophagosome biogenesis by binding to PIK3C3. Autophagy 2020, 17, 2166–2183. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Meyerkord, C.L.; Wang, H.G. Bif-1/endophilin B1: A candidate for crescent driving force in autophagy. Cell Death Differ. 2009, 16, 947–955. [Google Scholar] [CrossRef]
- Antonioli, M.; Albiero, F.; Nazio, F.; Vescovo, T.; Perdomo, A.B.; Corazzari, M.; Marsella, C.; Piselli, P.; Gretzmeier, C.; Dengjel, J.; et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev. Cell 2014, 31, 734–746. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zou, Z.; Sun, D.; Li, Y.; Sinha, S.C.; Yu, L.; Bennett, L.; Levine, B. GLIPR2 is a negative regulator of autophagy and the BECN1-ATG14-containing phosphatidylinositol 3-kinase complex. Autophagy 2020, 17, 2891–2904. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Wu, S.; He, Y.; Qiu, X.; Yang, W.; Liu, W.; Li, X.; Li, Y.; Shen, H.M.; Wang, R.; Yue, Z.; et al. Targeting the potent Beclin 1-UVRAG coiled-coil interaction with designed peptides enhances autophagy and endolysosomal trafficking. Proc. Natl. Acad. Sci. USA 2018, 115, E5669–E5678. [Google Scholar] [CrossRef]
- Pavlinov, I.; Salkovski, M.; Aldrich, L.N. Beclin 1-ATG14L Protein-Protein Interaction Inhibitor Selectively Inhibits Autophagy through Disruption of VPS34 Complex I. J. Am. Chem Soc. 2020, 142, 8174–8182. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein (Complex) | Packing | Curvature | Electrostatics | Reference |
|---|---|---|---|---|
| Atg1 complex binding to membranes | Not known | Binds more strongly to smaller vesicles | PS concentration did not affect membrane binding of Atg1 and Atg13, PtdIns(3)P increased Atg1 binding to membranes | [97,187] |
| ATG9/Atg9 vesicle property | High unsaturated lipids (yeast) | High curvature (30–60 nm) | <10% PS (yeast) | [81,196,197] |
| Human VPS34 complexes I and II | More active on GUVs composed of unsaturated lipids | More active on smaller vesicles, BATS domain binds to smaller vesicles more strongly | More active with higher PS; complex II interacts with high-PS-containing membranes more strongly | [24,26,45] |
| ATG2A/B/Atg2 membrane binding (MB), tethering (MT), and lipid transfer (LT) | Atg2 recognizes packing defects and binds to smaller vesicles. LT and MT by ATG2A–WIPI4 with DO (18:1–18:1) lipids are more efficient than PO (16:0–18:1) lipids | Stronger MB, MT, and LT on smaller vesicles (ATG2A/B/Atg2) | PS and PtdIns(3)P increase MB, MT, and LT of ATG2A and ATG2B, whereas they decrease MT and LT activity of Atg2 | [198,199,200,201,202] |
| LC3B/Atg8 lipidation | More efficient on GUVs with DO (18:1–18:1) lipids than those with PO (16:0–18:1) lipids (LC3B) | Possible with Atg12–Atg5 on SUVs, requires Atg16–Atg12–Atg5 on GUVs (Atg8) | Not known | [203,204] |
| Membrane tethering and fusion by GATE-16 and GABARAP | Not known | Smaller vesicles, CL and DAG facilitate fusion | Not known | [205] |
| Tethering comparison between LC3B and GATE-16 | Not known | LC3B > GATE-16 on 50 nm vesicles, GATE-16 > LC3B on 200–400 nm vesicles | Not known | [206] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohashi, Y. Activation Mechanisms of the VPS34 Complexes. Cells 2021, 10, 3124. https://doi.org/10.3390/cells10113124
Ohashi Y. Activation Mechanisms of the VPS34 Complexes. Cells. 2021; 10(11):3124. https://doi.org/10.3390/cells10113124
Chicago/Turabian StyleOhashi, Yohei. 2021. "Activation Mechanisms of the VPS34 Complexes" Cells 10, no. 11: 3124. https://doi.org/10.3390/cells10113124
APA StyleOhashi, Y. (2021). Activation Mechanisms of the VPS34 Complexes. Cells, 10(11), 3124. https://doi.org/10.3390/cells10113124