
Mechanisms Driving Palmitate-Mediated Neuronal Dysregulation in the Hypothalamus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Obesity and Palmitate Consumption
1.2. The Hypothalamus as a Central Homeostatic Regulator
1.3. Mechanisms and Biogenesis of microRNAs
2. Energy Homeostasis
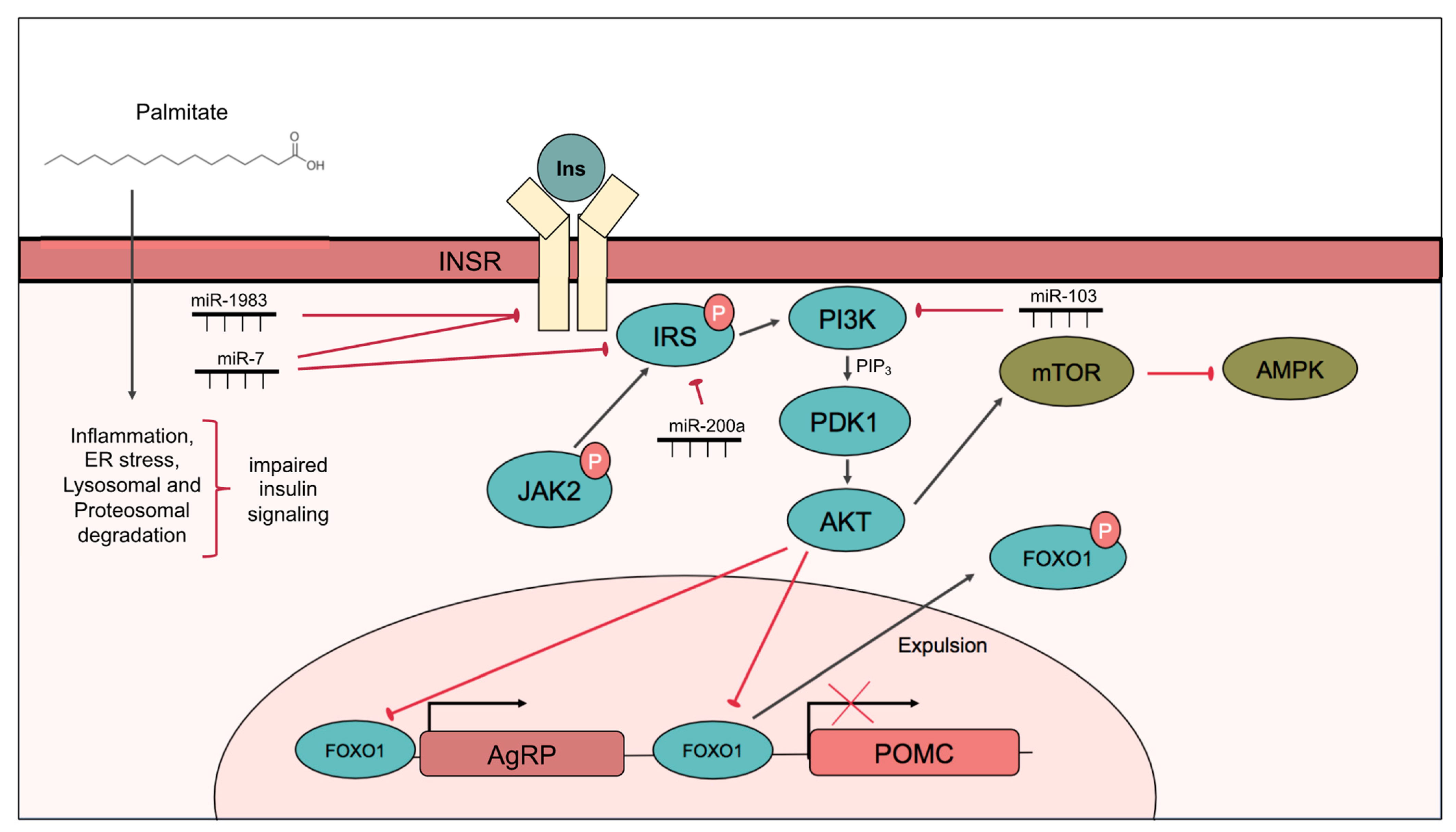
2.1. Hypothalamic Insulin Signaling Is Impaired by Palmitate
2.1.1. Insulin Signaling in the Hypothalamus
2.1.2. Hypothalamic Insulin Resistance
2.1.3. Induction of Central Insulin Resistance by Palmitate
2.1.4. Role of miRNAs in Hypothalamic Insulin Signaling and Resistance
2.2. Leptin Signaling Is Impaired by Palmitate
2.2.1. The Mechanisms of Hypothalamic Leptin Signaling
2.2.2. The Induction of Central Leptin Resistance by Palmitate
2.2.3. The Role of miRNAs in Leptin Signaling and Resistance
2.3. Feeding Neuropeptides
2.3.1. Palmitate-Induced Changes in Neuropeptide Expression
2.3.2. The Control of Feeding Neuropeptides by miRNAs
3. Reproduction
3.1. Hypothalamic-Pituitary-Gonadal Axis
3.2. The Integration of Energy Homeostasis in the Control of Reproduction
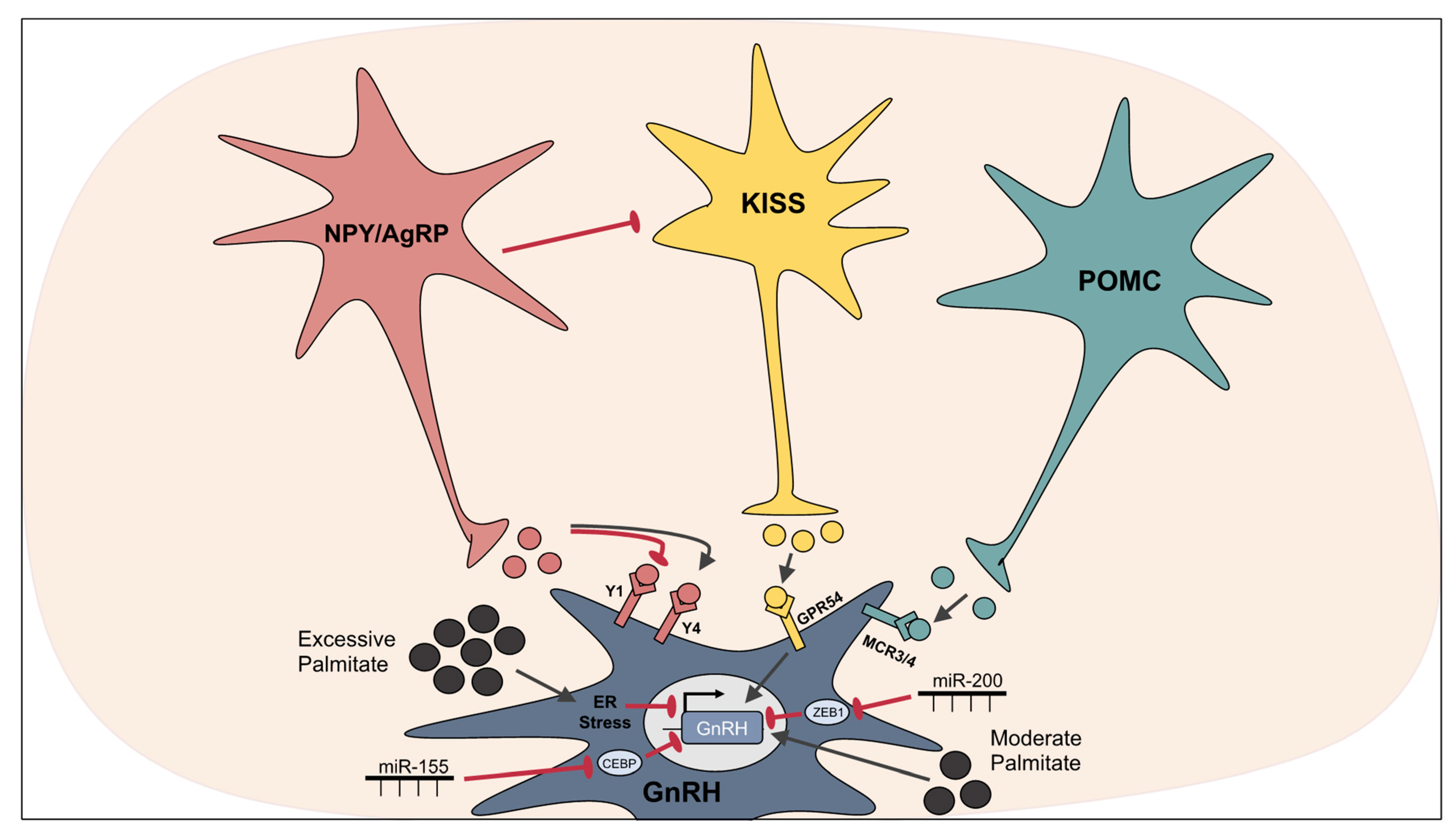
3.3. Palmitate-Mediated Disruption of Hypothalamic Reproductive Control
3.4. miRNAs Involved in the Hypothalamic Control of Reproduction
4. Circadian Rhythms
4.1. Control of Circadian Rhythms by the SCN and other Hypothalamic Nuclei
4.2. Palmitate-Induced Dysregulation of Circadian Rhythms in Hypothalamic Neurons
4.3. Role of miRNAs in Palmitate-Mediated Circadian Dysregulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| AGO | Argonaute |
| AgRP | Agouti related peptide |
| AICAR | Aminoimidazole carboxamide ribonucleotide |
| AMPK | AMP-activated protein kinase |
| Arc | Arcuate nucleus |
| ATF3 | Activating transcription factor 3 |
| AVPV | Anteroventral periventricular nucleus |
| BMAL1 | Brain and muscle ARNT-like 1 |
| BMI | Body mass index |
| CEBPß | CCAAT-enhancer binding protein beta |
| CLOCK | Circadian locomotor output cycles protein kaput |
| CNTF | Ciliary neurotrophic factor |
| CREB | cAMP response element binding protein |
| CRY(1-2) | Cryptochrome (1-2) |
| CSF | Cerebrospinal fluid |
| DHA | Docosahexaenoic acid |
| eGFP | Enhanced green fluorescent protein |
| ER ERK | Endoplasmic reticulum Extracellular signal-regulated kinase |
| FACS | Fluorescent activated cell sorting |
| FOXO1 | Forkhead box protein O1 |
| FSH GABA | Follicle stimulating hormone γ-aminobutyric acid |
| GnRH | Gonadotropin releasing hormone |
| GNRH-R | Gonadotropin releasing hormone receptor |
| HFD | High fat diet |
| HOMA-IR | Homeostatic model for insulin resistance |
| HPG | Hypothalamus-pituitary-gonadal |
| ICV | Intracerebroventricular |
| IL1-β | Interleukin 1 beta |
| IL6 | Interleukin 6 |
| InsR | Insulin receptor |
| IRS | Insulin receptor substrate |
| IRβ | Insulin receptor beta |
| JAK2 | Janus kinase 2 |
| JNK | cJun N-terminal kinases |
| KISS1 | Kisspeptin |
| KISS1r/GPR54 | Kisspeptin receptor |
| KLF4 | Kruppel-like factor 4 |
| LepR | Leptin receptor |
| LH | Luteinizing hormone |
| MAPK | Mitogen activated protein kinase |
| MC3R | Melanocortin 3 receptor |
| MC4R | Melanocortin 4 receptor |
| MEK | Mitogen-activated protein kinase kinase |
| MIF | Macrophage migration inhibitory factor |
| miRNA | microRNA |
| MKRN3 | Makorin RING-finger protein 3 |
| mRNA | Messenger RNA |
| NF𝜅B | Nuclear factor kappa B |
| NNAT | Neuronatin |
| NPY | Neuropeptide Y |
| OCT1 | Octamer transcription factor 1 |
| pCREB | Phosphorylated cAMP response element binding protein |
| PER (1-3) | Period (1-3) |
| PI3K | phosphoinositide 3-kinase |
| PIP3 | Phosphatidylinositol 3-phosphate |
| PKA | Protein Kinase A |
| PNX | Phoenexin |
| POMC | Proopiomelanocortin |
| PPAR | Peroxisome proliferator-activated receptor |
| pri-miRNA | Primary miRNA |
| PTP1B | Protein tyrosine phosphatase 1B |
| PVN | Paraventricular nucleus |
| Rev-erb | Nuclear receptor subfamily 1, group D, member 1 |
| RISC | RNA induced silencing complex |
| ROR | Retinoic acid-related orphan receptor |
| SCN | Suprachiasmatic nucleus |
| SOCS3 | Suppressor of cytokine signaling 3 |
| STAT3 | signal transducer and activator of transcription 3 |
| TLR4 | Toll like receptor 4 |
| TNFα | Tumour necrosis factor alpha |
| UTR | Untranslated region |
| WAT | White adipose tissue |
| WD | Western diet |
| ZEB1 | Zinc finger E-box binding homeobox 1 |
| α-MSH | α-melanocyte stimulating hormone |
References
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 5 October 2021).
- Bhupathiraju, S.N.; Hu, F.B. Epidemiology of obesity and diabetes and their cardiovascular complications. Circ. Res. 2016, 118, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Weihrauch-Bluher, S.; Schwarz, P.; Klusmann, J.H. Childhood obesity: Increased risk for cardiometabolic disease and cancer in adulthood. Metabolism 2019, 92, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, H.; Roser, M. Diet Compositions by Macronutrient. Daily Caloric Supply Derived from Carbohydrates, Protein and Fat, United States, 1961 to 2013. Available online: https://ourworldindata.org/diet-compositions (accessed on 5 October 2021).
- Rodriguez-Correa, E.; Gonzalez-Perez, I.; Clavel-Perez, P.I.; Contreras-Vargas, Y.; Carvajal, K. Biochemical and nutritional overview of diet-induced metabolic syndrome models in rats: What is the best choice? Nutr. Diabetes 2020, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic acid: Physiological role, metabolism and nutritional implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [PubMed]
- Martínez de Morentin, P.B.; Varela, L.; Fernø, J.; Nogueiras, R.; Diéguez, C.; López, M. Hypothalamic lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 2010, 1801, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Bhatt, N.; Cortassa, S.C. Mitochondrial and cellular mechanisms for managing lipid excess. Front. Physiol. 2014, 5, 282. [Google Scholar] [CrossRef]
- Tran, D.Q.; Ramos, E.H.; Belsham, D.D. Induction of Gnrh mRNA expression by the omega-3 polyunsaturated fatty acid docosahexaenoic acid and the saturated fatty acid palmitate in a GnRH-synthesizing neuronal cell model, mHypoA-GnRH/GFP. Mol. Cell. Endocrinol. 2016, 426, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef]
- McFadden, J.W.; Aja, S.; Li, Q.; Bandaru, V.V.; Kim, E.K.; Haughey, N.J.; Kuhajda, F.P.; Ronnett, G.V. Increasing fatty acid oxidation remodels the hypothalamic neurometabolome to mitigate stress and inflammation. PLoS ONE 2014, 9, e115642. [Google Scholar] [CrossRef]
- Melo, H.M.; Seixas da Silva, G.D.S.; Sant’Ana, M.R.; Teixeira, C.V.L.; Clarke, J.R.; Miya Coreixas, V.S.; de Melo, B.C.; Fortuna, J.T.S.; Forny-Germano, L.; Ledo, J.H.; et al. Palmitate Is increased in the cerebrospinal fluid of humans with obesity and induces memory impairment in mice via pro-inflammatory TNF-α. Cell. Rep. 2020, 30, 2180–2194. [Google Scholar] [CrossRef]
- Karmi, A.; Iozzo, P.; Viljanen, A.; Hirvonen, J.; Fielding, B.A.; Virtanen, K.; Oikonen, V.; Kemppainen, J.; Viljanen, T.; Guiducci, L.; et al. Increased brain fatty acid uptake in metabolic syndrome. Diabetes 2010, 59, 2171–2177. [Google Scholar] [CrossRef] [PubMed]
- Saper, C.B.; Lowell, B.B. The hypothalamus. Curr. Biol. 2014, 24, R1111–R1116. [Google Scholar] [CrossRef] [PubMed]
- Flament-Durand, J. The hypothalamus: Anatomy and functions. Acta Psychiatr. Belg. 1980, 80, 364–375. [Google Scholar] [PubMed]
- Wellhauser, L.; Gojska, N.M.; Belsham, D.D. Delineating the regulation of energy homeostasis using hypothalamic cell models. Front. Neuroendocrinol. 2015, 36, 130–149. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, P.S.; Nazarians-Armavil, A.; Tung, S.; Belsham, D.D. Immortalized neurons for the study of hypothalamic function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1030–R1052. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, P.S.; Loganathan, N.; McIlwraith, E.K.; Tran, A.; Belsham, D.D. (Eds.) Chapter 2—Hypothalamic Cell Models, 2nd ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 27–77. [Google Scholar]
- Ellwanger, D.C.; Büttner, F.A.; Mewes, H.W.; Stümpflen, V. The sufficient minimal set of miRNA seed types. Bioinformatics 2011, 27, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Pratt, A.J.; MacRae, I.J. The RNA-induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Ludwig, N.; Yerneni, S.S.; Razzo, B.M.; Whiteside, T.L. Exosomes from HNSCC Promote Angiogenesis through reprogramming of endothelial cells. Mol. Cancer Res. 2018, 16, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.A.; Ludwig, R.G.; Garcia-Martin, R.; Brandao, B.B.; Kahn, C.R. Extracellular miRNAs: From biomarkers to mediators of physiology and disease. Cell. Metab. 2019, 30, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Castaño, C.; Kalko, S.; Novials, A.; Párrizas, M. Obesity-associated exosomal miRNAs modulate glucose and lipid metabolism in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 12158–12163. [Google Scholar] [CrossRef] [PubMed]
- Deiuliis, J.A. MicroRNAs as regulators of metabolic disease: Pathophysiologic significance and emerging role as biomarkers and therapeutics. Int. J. Obes. 2016, 40, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose tissue macrophage-derived Exosomal miRNAs can modulate in vivo and in vitro insulin sensitivity. Cell 2017, 171, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Bernstein, E.; Kim, S.Y.; Carmell, M.A.; Murchison, E.P.; Alcorn, H.; Li, M.Z.; Mills, A.A.; Elledge, S.J.; Anderson, K.V.; Hannon, G.J. Dicer is essential for mouse development. Nat. Genet. 2003, 35, 215–217. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, J.; Yang, C.; Fan, P.; Balazs, L.; Jiao, Y.; Lu, M.; Gu, W.; Li, C.; Pfeffer, L.M.; et al. DiGeorge syndrome critical region 8 (DGCR8) protein-mediated microRNA biogenesis is essential for vascular smooth muscle cell development in mice. J. Biol. Chem. 2012, 287, 19018–19028. [Google Scholar] [CrossRef]
- Mang, G.M.; Pradervand, S.; Du, N.H.; Arpat, A.B.; Preitner, F.; Wigger, L.; Gatfield, D.; Franken, P. A neuron-specific deletion of the microRNA-processing enzyme DICER induces severe but transient obesity in mice. PLoS ONE 2015, 10, e0116760. [Google Scholar] [CrossRef]
- Greenman, Y.; Kuperman, Y.; Drori, Y.; Asa, S.L.; Navon, I.; Forkosh, O.; Gil, S.; Stern, N.; Chen, A. Postnatal ablation of POMC neurons induces an obese phenotype characterized by decreased food intake and enhanced anxiety-like behavior. Mol. Endocrinol. 2013, 27, 1091–1102. [Google Scholar] [CrossRef]
- Ouyang, S.; Tang, R.; Liu, Z.; Ma, F.; Li, Y.; Wu, J. Characterization and predicted role of microRNA expression profiles associated with early childhood obesity. Mol. Med. Rep. 2017, 16, 3799–3806. [Google Scholar] [CrossRef] [PubMed][Green Version]
- de Candia, P.; Spinetti, G.; Specchia, C.; Sangalli, E.; La Sala, L.; Uccellatore, A.; Lupini, S.; Genovese, S.; Matarese, G.; Ceriello, A. A unique plasma microRNA profile defines type 2 diabetes progression. PLoS ONE 2017, 12, e0188980. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Croce, C.M. miRNA profiling of cancer. Curr. Opin. Genet. Dev. 2013, 23, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Sangiao-Alvarellos, S.; Pena-Bello, L.; Manfredi-Lozano, M.; Tena-Sempere, M.; Cordido, F. Perturbation of hypothalamic microRNA expression patterns in male rats after metabolic distress: Impact of obesity and conditions of negative energy balance. Endocrinology 2014, 155, 1838–1850. [Google Scholar] [CrossRef]
- Benoit, C.; Doubi-Kadmiri, S.; Benigni, X.; Crepin, D.; Riffault, L.; Poizat, G.; Vacher, C.M.; Taouis, M.; Baroin-Tourancheau, A.; Amar, L. miRNA Long-term response to early metabolic environmental challenge in hypothalamic arcuate nucleus. Front. Mol. Neurosci. 2018, 11, 90. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Cansell, C.; Denis, R.G.; Delbes, A.S.; Castel, J.; Martinez, S.; Luquet, S. The hypothalamic arcuate nucleus and the control of peripheral substrates. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Obici, S.; Morgan, K.; Barzilai, N.; Feng, Z.; Rossetti, L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes 2001, 50, 2786–2791. [Google Scholar] [CrossRef]
- Darbre, P.D. Endocrine Disruptors and Obesity. Curr. Obes. Rep. 2017, 6, 18–27. [Google Scholar] [CrossRef]
- Ruud, J.; Steculorum, S.M.; Bruning, J.C. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat. Commun. 2017, 8, 15259. [Google Scholar] [CrossRef]
- Belsham, D.D.; Dalvi, P.S. Insulin signalling in hypothalamic neurones. J. Neuroendocrinol. 2020, 33, e12919. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Loh, K.; Zhang, L.; Brandon, A.; Wang, Q.; Begg, D.; Qi, Y.; Fu, M.; Kulkarni, R.; Teo, J.; Baldock, P.; et al. Insulin controls food intake and energy balance via NPY neurons. Mol. Metab. 2017, 6, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Hallschmid, M.; Benedict, C.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin reduces body fat in men but not in women. Diabetes 2004, 53, 3024–3029. [Google Scholar] [CrossRef]
- Konner, A.C.; Janoschek, R.; Plum, L.; Jordan, S.D.; Rother, E.; Ma, X.; Xu, C.; Enriori, P.; Hampel, B.; Barsh, G.S.; et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell. Metab. 2007, 5, 438–449. [Google Scholar] [CrossRef]
- Shin, A.C.; Filatova, N.; Lindtner, C.; Chi, T.; Degann, S.; Oberlin, D.; Buettner, C. Insulin receptor signaling in POMC, but not AgRP, neurons controls adipose tissue insulin action. Diabetes 2017, 66, 1560–1571. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhang, C.; Borgquist, A.; Nestor, C.C.; Smith, A.W.; Bosch, M.A.; Ku, S.; Wagner, E.J.; Ronnekleiv, O.K.; Kelly, M.J. Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell. Metab. 2014, 19, 682–693. [Google Scholar] [CrossRef]
- Mayer, C.M.; Belsham, D.D. Insulin directly regulates NPY and AgRP gene expression via the MAPK MEK/ERK signal transduction pathway in mHypoE-46 hypothalamic neurons. Mol. Cell. Endocrinol. 2009, 307, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Nazarians-Armavil, A.; Chalmers, J.A.; Lee, C.B.; Ye, W.; Belsham, D.D. Cellular insulin resistance disrupts hypothalamic mHypoA-POMC/GFP neuronal signaling pathways. J. Endocrinol. 2014, 220, 13–24. [Google Scholar] [CrossRef]
- Nazarians-Armavil, A.; Menchella, J.A.; Belsham, D.D. Cellular insulin resistance disrupts leptin-mediated control of neuronal signaling and transcription. Mol. Endocrinol. 2013, 27, 990–1003. [Google Scholar] [CrossRef]
- Mayer, C.M.; Belsham, D.D. Central insulin signaling is attenuated by long-term insulin exposure via insulin receptor substrate-1 serine phosphorylation, proteasomal degradation, and lysosomal insulin receptor degradation. Endocrinology 2010, 151, 75–84. [Google Scholar] [CrossRef]
- de Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.L.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef]
- Benoit, S.C.; Kemp, C.J.; Elias, C.F.; Abplanalp, W.; Herman, J.P.; Migrenne, S.; Lefevre, A.L.; Cruciani-Guglielmacci, C.; Magnan, C.; Yu, F.; et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellular localization in rodents. J. Clin. Investing. 2009, 119, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Schenten, D.; Konner, A.C.; Belgardt, B.F.; Mauer, J.; Okamura, T.; Wunderlich, F.T.; Medzhitov, R.; Bruning, J.C. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell. Metab. 2009, 10, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.M.; Belsham, D.D. Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: Rescue of resistance and apoptosis through adenosine 5′ monophosphate-activated protein kinase activation. Endocrinology 2010, 151, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Tse, E.K.; Belsham, D.D. Palmitate induces neuroinflammation, ER stress, and Pomc mRNA expression in hypothalamic mHypoA-POMC/GFP neurons through novel mechanisms that are prevented by oleate. Mol. Cell. Endocrinol. 2018, 472, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Clemenzi, M.N.; Wellhauser, L.; Aljghami, M.E.; Belsham, D.D. Tumour necrosis factor alpha induces neuroinflammation and insulin resistance in immortalised hypothalamic neurones through independent pathways. J. Neuroendocr. 2019, 31, e12678. [Google Scholar] [CrossRef]
- Amine, H.; Benomar, Y.; Taouis, M. Palmitic acid promotes resistin-induced insulin resistance and inflammation in SH-SY5Y human neuroblastoma. Sci. Rep. 2021, 11, 5427. [Google Scholar] [CrossRef]
- Wellhauser, L.; Belsham, D.D. Activation of the omega-3 fatty acid receptor GPR120 mediates anti-inflammatory actions in immortalized hypothalamic neurons. J. Neuroinflammation 2014, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Halseth, A.E.; Ensor, N.J.; White, T.A.; Ross, S.A.; Gulve, E.A. Acute and chronic treatment of ob/ob and db/db mice with AICAR decreases blood glucose concentrations. Biochem. Biophys. Res. Commun. 2002, 294, 798–805. [Google Scholar] [CrossRef]
- Buhl, E.S.; Jessen, N.; Pold, R.; Ledet, T.; Flyvbjerg, A.; Pedersen, S.B.; Pedersen, O.; Schmitz, O.; Lund, S. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes 2002, 51, 2199–2206. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Ong, P.S.; Wang, L.Z.; Dai, X.; Tseng, S.H.; Loo, S.J.; Sethi, G. Judicious toggling of mTOR activity to combat insulin resistance and cancer: Current evidence and perspectives. Front. Pharmacol. 2016, 7, 395. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef] [PubMed]
- Vinnikov, I.A.; Hajdukiewicz, K.; Reymann, J.; Beneke, J.; Czajkowski, R.; Roth, L.C.; Novak, M.; Roller, A.; Dorner, N.; Starkuviene, V.; et al. Hypothalamic miR-103 protects from hyperphagic obesity in mice. J. Neurosci. 2014, 34, 10659–10674. [Google Scholar] [CrossRef]
- Mu, J.; Yu, P.; Li, Q. microRNA-103 Contributes to progression of polycystic ovary syndrome through modulating the IRS1/PI3K/AKT signal axis. Arch. Med. Res. 2021, 52, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.D.; Toda, C.; Ramirez, C.M.; Fernandez-Hernando, C.; Diano, S. Hypothalamic ventromedial Lin28a enhances glucose metabolism in diet-induced obesity. Diabetes 2017, 66, 2102–2111. [Google Scholar] [CrossRef]
- Crepin, D.; Benomar, Y.; Riffault, L.; Amine, H.; Gertler, A.; Taouis, M. The over-expression of miR-200a in the hypothalamus of ob/ob mice is linked to leptin and insulin signaling impairment. Mol. Cell. Endocrinol. 2014, 384, 1–11. [Google Scholar] [CrossRef]
- Chalmers, J.A.; Dalvi, P.S.; Wellhauser, L.; Nazarians-Armavil, A.; Eversley, J.A.; Mohan, H.; Wheeler, M.B.; Belsham, D.D. Hypothalamic miR-1983 targets insulin receptor beta to induce insulin resistance, and the insulin-mediated increase in miR-1983 is blocked by metformin. Endocrinology 2021. in revision. [Google Scholar]
- Vaitheesvaran, B.; LeRoith, D.; Kurland, I.J. MKR mice have increased dynamic glucose disposal despite metabolic inflexibility, and hepatic and peripheral insulin insensitivity. Diabetologia 2010, 53, 2224–2232. [Google Scholar] [CrossRef]
- Amar, L.; Benoit, C.; Beaumont, G.; Vacher, C.M.; Crepin, D.; Taouis, M.; Baroin-Tourancheau, A. MicroRNA expression profiling of hypothalamic arcuate and paraventricular nuclei from single rats using Illumina sequencing technology. J. Neurosci. Methods 2012, 209, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-de Frutos, M.; Galan-Chilet, I.; Goedeke, L.; Kim, B.; Pardo-Marques, V.; Perez-Garcia, A.; Herrero, J.I.; Fernandez-Hernando, C.; Kim, J.; Ramirez, C.M. MicroRNA 7 impairs insulin signaling and regulates abeta levels through posttranscriptional regulation of the Insulin receptor substrate 2, insulin receptor, insulin-degrading enzyme, and liver X receptor pathway. Mol. Cell. Biol. 2019, 39, e00170-19. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, J.; Zhang, Z.; Zhang, R.; Pollock, A.; Sun, T. MicroRNA miR-7 and miR-17-92 in the arcuate nucleus of Mouse Hypothalamus regulate sex-specific diet-induced obesity. Mol. Neurobiol. 2019, 56, 7508–7521. [Google Scholar] [CrossRef] [PubMed]
- Romon, M.; Lebel, P.; Velly, C.; Marecaux, N.; Fruchart, J.C.; Dallongeville, J. Leptin response to carbohydrate or fat meal and association with subsequent satiety and energy intake. Am. J. Physiol. 1999, 277, E855–E861. [Google Scholar] [CrossRef] [PubMed]
- Ostlund, R.E.; Yang, J.W.; Klein, S.; Gingerich, R. Relation between plasma leptin concentration and body fat, gender, diet, age, and metabolic covariates. J. Clin. Endocrinol. Metab. 1996, 81, 3909–3913. [Google Scholar] [CrossRef]
- Clement, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998, 392, 398–401. [Google Scholar] [CrossRef]
- de Luca, C.; Kowalski, T.J.; Zhang, Y.; Elmquist, J.K.; Lee, C.; Kilimann, M.W.; Ludwig, T.; Liu, S.M.; Chua, S.C., Jr. Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J. Clin. Investig. 2005, 115, 3484–3493. [Google Scholar] [CrossRef]
- Havel, P.J.; Kasim-Karakas, S.; Mueller, W.; Johnson, P.R.; Gingerich, R.L.; Stern, J.S. Relationship of plasma leptin to plasma insulin and adiposity in normal weight and overweight women: Effects of dietary fat content and sustained weight loss. J. Clin. Endocrinol. Metab. 1996, 81, 4406–4413. [Google Scholar] [CrossRef][Green Version]
- Banks, W.A.; Farrell, C.L. Impaired transport of leptin across the blood-brain barrier in obesity is acquired and reversible. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E10–E15. [Google Scholar] [CrossRef]
- Zhao, S.; Zhu, Y.; Schultz, R.D.; Li, N.; He, Z.; Zhang, Z.; Caron, A.; Zhu, Q.; Sun, K.; Xiong, W.; et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell. Metab. 2019, 30, 706–719. [Google Scholar] [CrossRef]
- Donato, J., Jr.; Cravo, R.M.; Frazao, R.; Elias, C.F. Hypothalamic sites of leptin action linking metabolism and reproduction. Neuroendocrinology 2011, 93, 9–18. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Woods, S.C.; Weigle, D.S.; Campfield, L.A.; Burn, P.; Baskin, D.G. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes 1997, 46, 2119–2123. [Google Scholar] [CrossRef]
- Kim, Y.B.; Uotani, S.; Pierroz, D.D.; Flier, J.S.; Kahn, B.B. In vivo administration of leptin activates signal transduction directly in insulin-sensitive tissues: Overlapping but distinct pathways from insulin. Endocrinology 2000, 141, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bing, C.; Al-Barazanji, K.; Mossakowaska, D.E.; Wang, X.M.; McBay, D.L.; Neville, W.A.; Taddayon, M.; Pickavance, L.; Dryden, S.; et al. Interactions between leptin and hypothalamic neuropeptide Y neurons in the control of food intake and energy homeostasis in the rat. Diabetes 1997, 46, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Korner, J.; Savontaus, E.; Chua, S.C., Jr.; Leibel, R.L.; Wardlaw, S.L. Leptin regulation of Agrp and Npy mRNA in the rat hypothalamus. J. Neuroendocrinol. 2001, 13, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, Z.; Rui, L. Leptin stimulates both JAK2-dependent and JAK2-independent signaling pathways. J. Biol. Chem. 2008, 283, 28066–28073. [Google Scholar] [CrossRef]
- Gong, L.; Yao, F.; Hockman, K.; Heng, H.H.; Morton, G.J.; Takeda, K.; Akira, S.; Low, M.J.; Rubinstein, M.; MacKenzie, R.G. Signal transducer and activator of transcription-3 is required in hypothalamic agouti-related protein/neuropeptide Y neurons for normal energy homeostasis. Endocrinology 2008, 149, 3346–3354. [Google Scholar] [CrossRef]
- Ernst, M.B.; Wunderlich, C.M.; Hess, S.; Paehler, M.; Mesaros, A.; Koralov, S.B.; Kleinridders, A.; Husch, A.; Munzberg, H.; Hampel, B.; et al. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J. Neurosci. 2009, 29, 11582–11593. [Google Scholar] [CrossRef]
- Ma, W.; Fuentes, G.; Shi, X.; Verma, C.; Radda, G.K.; Han, W. FoxO1 negatively regulates leptin-induced POMC transcription through its direct interaction with STAT3. Biochem. J. 2015, 466, 291–298. [Google Scholar] [CrossRef]
- Lubis, A.R.; Widia, F.; Soegondo, S.; Setiawati, A. The role of SOCS-3 protein in leptin resistance and obesity. Acta Med. Indones. 2008, 40, 89–95. [Google Scholar]
- Carvalheira, J.B.; Torsoni, M.A.; Ueno, M.; Amaral, M.E.; Araujo, E.P.; Velloso, L.A.; Gontijo, J.A.; Saad, M.J. Cross-talk between the insulin and leptin signaling systems in rat hypothalamus. Obes. Res. 2005, 13, 48–57. [Google Scholar] [CrossRef]
- Dhillon, S.S.; McFadden, S.A.; Chalmers, J.A.; Centeno, M.L.; Kim, G.L.; Belsham, D.D. Cellular leptin resistance impairs the leptin-mediated suppression of neuropeptide Y secretion in hypothalamic neurons. Endocrinology 2011, 152, 4138–4147. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yu, Y.; Szabo, A.; Wu, Y.; Wang, H.; Camer, D.; Huang, X.F. Palmitic acid induces central leptin resistance and impairs hepatic glucose and lipid metabolism in male mice. J. Nutr. Biochem. 2015, 26, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Dhillon, H.; Yin, H.; Yoshimura, A.; Lowell, B.B.; Maratos-Flier, E.; Flier, J.S. Selective inactivation of Socs3 in SF1 neurons improves glucose homeostasis without affecting body weight. Endocrinology 2008, 149, 5654–5661. [Google Scholar] [CrossRef] [PubMed]
- Zabolotny, J.M.; Kim, Y.B.; Welsh, L.A.; Kershaw, E.E.; Neel, B.G.; Kahn, B.B. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J. Biol. Chem. 2008, 283, 14230–14241. [Google Scholar] [CrossRef]
- Hakim, F.; Wang, Y.; Carreras, A.; Hirotsu, C.; Zhang, J.; Peris, E.; Gozal, D. Chronic sleep fragmentation during the sleep period induces hypothalamic endoplasmic reticulum stress and PTP1b-mediated leptin resistance in male mice. Sleep 2015, 38, 31–40. [Google Scholar] [CrossRef]
- Ozcan, L.; Ergin, A.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell. Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef]
- Marwarha, G.; Claycombe, K.; Schommer, J.; Collins, D.; Ghribi, O. Palmitate-induced endoplasmic reticulum stress and subsequent C/EBPalpha homologous protein activation attenuates leptin and insulin-like growth factor 1 expression in the brain. Cell. Signal 2016, 28, 1789–1805. [Google Scholar] [CrossRef]
- Derghal, A.; Djelloul, M.; Azzarelli, M.; Degonon, S.; Tourniaire, F.; Landrier, J.F.; Mounien, L. MicroRNAs are involved in the hypothalamic leptin sensitivity. Epigenetics 2018, 13, 1127–1140. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, W.; He, Y.; Dawar, F.U.; Xiong, S.; Mei, J. Potential Contributions of miR-200a/-200b and their target gene-leptin to the sexual Size dimorphism in Yellow Catfish. Front. Physiol. 2017, 8, 970. [Google Scholar] [CrossRef]
- Collins, A.S.; McCoy, C.E.; Lloyd, A.T.; O’Farrelly, C.; Stevenson, N.J. miR-19a: An effective regulator of SOCS3 and enhancer of JAK-STAT signalling. PLoS ONE 2013, 8, e69090. [Google Scholar] [CrossRef] [PubMed]
- Derghal, A.; Astier, J.; Sicard, F.; Couturier, C.; Landrier, J.F.; Mounien, L. Leptin modulates the expression of miRNAs-targeting POMC mRNA by the JAK2-STAT3 and PI3K-Akt pathways. J. Clin. Med. 2019, 8, 2213. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.-Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and TNF-α on appetite-regulating NPY neurons. Int. J. Obes. 2017, 41, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Ramos, E.H.; Wong, B.C.; Belsham, D.D. Beneficial effects of metformin and/or salicylate on palmitate- or TNFalpha-induced neuroinflammatory marker and neuropeptide gene regulation in immortalized NPY/AgRP neurons. PLoS ONE 2016, 11, e0166973. [Google Scholar] [CrossRef]
- Fick, L.J.; Fick, G.H.; Belsham, D.D. Palmitate alters the rhythmic expression of molecular clock genes and orexigenic neuropeptide Y mRNA levels within immortalized, hypothalamic neurons. Biochem. Biophys. Res. Commun. 2011, 413, 414–419. [Google Scholar] [CrossRef]
- Jais, A.; Bruning, J.C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.; He, W.; Chen, J.T.C.; Wellhauser, L.; Hopperton, K.E.; Bazinet, R.P.; Belsham, D.D. Palmitate-mediated induction of neuropeptide Y expression occurs through intracellular metabolites and not direct exposure to proinflammatory cytokines. J. Neurochem. 2021, 159, 574–589. [Google Scholar] [CrossRef]
- Clemenzi, M.N.; Martchenko, A.; Loganathan, N.; Tse, E.K.; Brubaker, P.L.; Belsham, D.D. Analysis of Western diet, palmitate and BMAL1 regulation of neuropeptide Y expression in the murine hypothalamus and BMAL1 knockout cell models. Mol. Cell. Endocrinol. 2020, 507, 110773. [Google Scholar] [CrossRef]
- Park, S.; Oh, T.S.; Kim, S.; Kim, E.K. Palmitate-induced autophagy liberates monounsaturated fatty acids and increases Agrp expression in hypothalamic cells. Anim. Cells. Syst. 2019, 23, 384–391. [Google Scholar] [CrossRef]
- Titolo, D.; Mayer, C.M.; Dhillon, S.S.; Cai, F.; Belsham, D.D. Estrogen facilitates both phosphatidylinositol 3-kinase/Akt and ERK1/2 mitogen-activated protein kinase membrane signaling required for long-term neuropeptide Y transcriptional regulation in clonal, immortalized neurons. J. Neurosci. 2008, 28, 6473–6482. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-S.; Yang, S.-F.; Kuo, D.-Y. Intracerebral administration of protein kinase A or cAMP response element-binding protein antisense oligonucleotide can modulate amphetamine-mediated appetite suppression in free-moving rats. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E123–E131. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.M.; Cai, F.; Cui, H.; Gillespie, J.M.; MacMillan, M.; Belsham, D.D. Analysis of a repressor region in the human neuropeptide Y gene that binds Oct-1 and Pbx-1 in GT1-7 neurons. Biochem. Biophys. Res. Commun. 2003, 307, 847–854. [Google Scholar] [CrossRef]
- Loganathan, N.; Salehi, A.; Chalmers, J.A.; Belsham, D.D. Bisphenol A alters Bmal1, Per2, and Rev-Erba mRNA and requires Bmal1 to increase neuropeptide Y expression in hypothalamic neurons. Endocrinology 2019, 160, 181–192. [Google Scholar] [CrossRef]
- Friedrich, M.; Heimer, N.; Stoehr, C.; Steven, A.; Wach, S.; Taubert, H.; Hartmann, A.; Seliger, B. CREB1 is affected by the microRNAs miR-22-3p, miR-26a-5p, miR-27a-3p, and miR-221-3p and correlates with adverse clinicopathological features in renal cell carcinoma. Sci. Rep. 2020, 10, 6499. [Google Scholar] [CrossRef]
- Feng, Y.Y.; Zeng, D.Z.; Tong, Y.N.; Lu, X.X.; Dun, G.D.; Tang, B.; Zhang, Z.J.; Ye, X.L.; Li, Q.; Xie, J.P.; et al. Alteration of microRNA-4474/4717 expression and CREB-binding protein in human colorectal cancer tissues infected with Fusobacterium nucleatum. PLoS ONE 2019, 14, e0215088. [Google Scholar] [CrossRef] [PubMed]
- Curtis, A.M.; Fagundes, C.T.; Yang, G.; Palsson-McDermott, E.M.; Wochal, P.; McGettrick, A.F.; Foley, N.H.; Early, J.O.; Chen, L.; Zhang, H.; et al. Circadian control of innate immunity in macrophages by miR-155 targeting Bmal1. Proc. Nat. Acad. Sci. USA 2015, 112, 7231–7236. [Google Scholar] [CrossRef]
- Karkeni, E.; Astier, J.; Tourniaire, F.; El Abed, M.; Romier, B.; Gouranton, E.; Wan, L.; Borel, P.; Salles, J.; Walrand, S.; et al. Obesity-associated inflammation induces microRNA-155 expression in adipocytes and adipose tissue: Outcome on adipocyte function. J. Clin. Endocrinol. Metab. 2016, 101, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- McIlwraith, E.K.; Lieu, C.V.; Belsham, D.D. Bisphenol A induces miR-708-5p through an ER stress-mediated mechanism achieving its obesogenic effects by altering neuronatin and neuropeptide Y expression in hypothalamic neuronal models. Mol. Cell. Endocrinol. 2021, in press. [Google Scholar]
- Lin, H.H.; Bell, E.; Uwanogho, D.; Perfect, L.W.; Noristani, H.; Bates, T.J.; Snetkov, V.; Price, J.; Sun, Y.M. Neuronatin promotes neural lineage in ESCs via Ca(2+) signaling. Stem Cells 2010, 28, 1950–1960. [Google Scholar] [CrossRef] [PubMed]
- Cimino, I.; Rimmington, D.; Tung, Y.C.L.; Lawler, K.; Larraufie, P.; Kay, R.G.; Virtue, S.; Lam, B.Y.H.; Fagnocchi, L.; Ma, M.K.L.; et al. Murine neuronatin deficiency is associated with a hypervariable food intake and bimodal obesity. Sci. Rep. 2021, 11, 17571. [Google Scholar] [CrossRef]
- Kitamura, T.; Feng, Y.; Kitamura, Y.I.; Chua, S.C., Jr.; Xu, A.W.; Barsh, G.S.; Rossetti, L.; Accili, D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat. Med. 2006, 12, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Imbernon, M.; Sanchez-Rebordelo, E.; Gallego, R.; Gandara, M.; Lear, P.; Lopez, M.; Dieguez, C.; Nogueiras, R. Hypothalamic KLF4 mediates leptin’s effects on food intake via AgRP. Mol. Metab. 2014, 3, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, N.; McIlwraith, E.K.; Belsham, D.D. Bisphenol A Induces Agrp Gene Expression in Hypothalamic Neurons through a Mechanism Involving ATF3. Neuroendocrinology 2021, 111, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Liu, L.Z.; Addison, J.B.; Wonderlin, W.F.; Ivanov, A.V.; Ruppert, J.M. A KLF4-miRNA-206 autoregulatory feedback loop can promote or inhibit protein translation depending upon cell context. Mol. Cell. Biol. 2011, 31, 2513–2527. [Google Scholar] [CrossRef]
- Xu, N.; Papagiannakopoulos, T.; Pan, G.; Thomson, J.A.; Kosik, K.S. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell 2009, 137, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Vinod, M.; Patankar, J.V.; Sachdev, V.; Frank, S.; Graier, W.F.; Kratky, D.; Kostner, G.M. MiR-206 is expressed in pancreatic islets and regulates glucokinase activity. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E175–E185. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Sasaki, T.; Kobayashi, M.; Kikuchi, O.; Kim, H.J.; Yokota-Hashimoto, H.; Shimpuku, M.; Susanti, V.Y.; Ido-Kitamura, Y.; Kimura, K.; et al. Hypothalamic ATF3 is involved in regulating glucose and energy metabolism in mice. Diabetologia 2013, 56, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Choe, N.; Kwon, D.H.; Ryu, J.; Shin, S.; Cho, H.J.; Joung, H.; Eom, G.H.; Ahn, Y.; Park, W.J.; Nam, K.I.; et al. Targets ATF3 to Reduce Calcium Deposition in Vascular Smooth Muscle Cells. Mol. Ther. Nucleic Acids 2020, 22, 627–639. [Google Scholar] [CrossRef]
- Shi, B.; Yan, W.; Liu, G.; Guo, Y. MicroRNA-488 inhibits tongue squamous carcinoma cell invasion and EMT by directly targeting ATF3. Cell. Mol. Biol. Lett. 2018, 23, 28. [Google Scholar] [CrossRef]
- Li, S.; Yan, G.; Yue, M.; Wang, L. Extracellular vesicles-derived microRNA-222 promotes immune escape via interacting with ATF3 to regulate AKT1 transcription in colorectal cancer. BMC Cancer 2021, 21, 349. [Google Scholar] [CrossRef]
- Li, D.; Song, H.; Shuo, L.; Wang, L.; Xie, P.; Li, W.; Liu, J.; Tong, Y.; Zhang, C.Y.; Jiang, X.; et al. Gonadal white adipose tissue-derived exosomal MiR-222 promotes obesity-associated insulin resistance. Aging 2020, 12, 22719–22743. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Sexual and Reproductive Health: Infertility Is a Global Public Health Issue. Available online: https://www.who.int/reproductivehealth/topics/infertility/perspective/en/ (accessed on 20 September 2020).
- Silvestris, E.; de Pergola, G.; Rosania, R.; Loverro, G. Obesity as disruptor of the female fertility. Reprod. Biol. Endocrinol. 2018, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Rich-Edwards, J.W.; Goldman, M.B.; Willett, W.C.; Hunter, D.J.; Stampfer, M.J.; Colditz, G.A.; Manson, J.E. Adolescent body mass index and infertility caused by ovulatory disorder. Am. J. Obstet. Gynecol. 1994, 171, 171–177. [Google Scholar] [CrossRef]
- Sallmen, M.; Sandler, D.P.; Hoppin, J.A.; Blair, A.; Baird, D.D. Reduced fertility among overweight and obese men. Epidemiology 2006, 17, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Canada Clinical Practice Guidelines Expert Committee; Feig, D.S.; Berger, H.; Donovan, L.; Godbout, A.; Kader, T.; Keely, E.; Sanghera, R. Diabetes and Pregnancy. Can. J. Diabetes 2018, 42, S255–S282. [Google Scholar] [CrossRef]
- Katib, A. Mechanisms linking obesity to male infertility. Cent. European J. Urol. 2015, 68, 79–85. [Google Scholar] [CrossRef]
- Skorupskaite, K.; George, J.T.; Anderson, R.A. The kisspeptin-GnRH pathway in human reproductive health and disease. Hum. Reprod. Update 2014, 20, 485–500. [Google Scholar] [CrossRef]
- Lapatto, R.; Pallais, J.C.; Zhang, D.; Chan, Y.M.; Mahan, A.; Cerrato, F.; Le, W.W.; Hoffman, G.E.; Seminara, S.B. Kiss1-/- mice exhibit more variable hypogonadism than Gpr54-/- mice. Endocrinology 2007, 148, 4927–4936. [Google Scholar] [CrossRef] [PubMed]
- Seminara, S.B.; Messager, S.; Chatzidaki, E.E.; Thresher, R.R.; Acierno, J.S., Jr.; Shagoury, J.K.; Bo-Abbas, Y.; Kuohung, W.; Schwinof, K.M.; Hendrick, A.G.; et al. The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 2003, 349, 1614–1627. [Google Scholar] [CrossRef]
- Kirilov, M.; Clarkson, J.; Liu, X.; Roa, J.; Campos, P.; Porteous, R.; Schutz, G.; Herbison, A.E. Dependence of fertility on kisspeptin-Gpr54 signaling at the GnRH neuron. Nat. Commun. 2013, 4, 2492. [Google Scholar] [CrossRef]
- Ikegami, K.; Goto, T.; Nakamura, S.; Watanabe, Y.; Sugimoto, A.; Majarune, S.; Horihata, K.; Nagae, M.; Tomikawa, J.; Imamura, T.; et al. Conditional kisspeptin neuron-specific Kiss1 knockout with newly generated Kiss1-floxed and Kiss1-Cre mice replicates a hypogonadal phenotype of global Kiss1 knockout mice. J. Reprod. Dev. 2020, 66, 359–367. [Google Scholar] [CrossRef]
- De Bond, J.A.; Smith, J.T. Kisspeptin and energy balance in reproduction. Reproduction 2014, 147, R53–R63. [Google Scholar] [CrossRef] [PubMed]
- Messager, S.; Chatzidaki, E.E.; Ma, D.; Hendrick, A.G.; Zahn, D.; Dixon, J.; Thresher, R.R.; Malinge, I.; Lomet, D.; Carlton, M.B.; et al. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc. Nat. Acad. Sci. USA 2005, 102, 1761–1766. [Google Scholar] [CrossRef]
- Gottsch, M.L.; Cunningham, M.J.; Smith, J.T.; Popa, S.M.; Acohido, B.V.; Crowley, W.F.; Seminara, S.; Clifton, D.K.; Steiner, R.A. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology 2004, 145, 4073–4077. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Takatsu, Y.; Kumano, S.; Matsumoto, H.; Ohtaki, T. Peripheral administration of metastin induces marked gonadotropin release and ovulation in the rat. Biochem. Biophys. Res. Commun. 2004, 320, 383–388. [Google Scholar] [CrossRef]
- Yosten, G.L.; Lyu, R.M.; Hsueh, A.J.; Avsian-Kretchmer, O.; Chang, J.K.; Tullock, C.W.; Dun, S.L.; Dun, N.; Samson, W.K. A novel reproductive peptide, phoenixin. J. Neuroendocrinol. 2013, 25, 206–215. [Google Scholar] [CrossRef]
- Treen, A.K.; Luo, V.; Belsham, D.D. Phoenixin activates immortalized GnRH and kisspeptin neurons through the novel receptor GPR173. Mol. Endocrinol. 2016, 30, 872–888. [Google Scholar] [CrossRef] [PubMed]
- Elias, S.G.; van Noord, P.A.; Peeters, P.H.; den Tonkelaar, I.; Grobbee, D.E. Childhood exposure to the 1944–1945 Dutch famine and subsequent female reproductive function. Hum. Reprod. 2005, 20, 2483–2488. [Google Scholar] [CrossRef] [PubMed]
- Hessler, S.; Liu, X.; Herbison, A.E. Direct inhibition of arcuate kisspeptin neurones by neuropeptide Y in the male and female mouse. J. Neuroendocrinol. 2020, 32, e12849. [Google Scholar] [CrossRef]
- Roa, J.; Herbison, A.E. Direct regulation of GnRH neuron excitability by arcuate nucleus POMC and NPY neuron neuropeptides in female mice. Endocrinology 2012, 153, 5587–5599. [Google Scholar] [CrossRef] [PubMed]
- Padilla, S.L.; Qiu, J.; Nestor, C.C.; Zhang, C.; Smith, A.W.; Whiddon, B.B.; Ronnekleiv, O.K.; Kelly, M.J.; Palmiter, R.D. AgRP to Kiss1 neuron signaling links nutritional state and fertility. Proc. Nat. Acad. Sci. USA 2017, 114, 2413–2418. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Ohkura, S.; Sakurai, K.; Tsukamura, H.; Maeda, K.; Okamura, H. Activation of melanocortin receptors accelerates the gonadotropin-releasing hormone pulse generator activity in goats. Neurosci. Lett. 2005, 383, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Divall, S.A.; Williams, T.R.; Carver, S.E.; Koch, L.; Bruning, J.C.; Kahn, C.R.; Wondisford, F.; Radovick, S.; Wolfe, A. Divergent roles of growth factors in the GnRH regulation of puberty in mice. J. Clin. Investig. 2010, 120, 2900–2909. [Google Scholar] [CrossRef] [PubMed]
- Quennell, J.H.; Mulligan, A.C.; Tups, A.; Liu, X.; Phipps, S.J.; Kemp, C.J.; Herbison, A.E.; Grattan, D.R.; Anderson, G.M. Leptin indirectly regulates gonadotropin-releasing hormone neuronal function. Endocrinology 2009, 150, 2805–2812. [Google Scholar] [CrossRef]
- Quennell, J.H.; Howell, C.S.; Roa, J.; Augustine, R.A.; Grattan, D.R.; Anderson, G.M. Leptin deficiency and diet-induced obesity reduce hypothalamic kisspeptin expression in mice. Endocrinology 2011, 152, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.T.; Acohido, B.V.; Clifton, D.K.; Steiner, R.A. KiSS-1 neurones are direct targets for leptin in the ob/ob mouse. J. Neuroendocrinol. 2006, 18, 298–303. [Google Scholar] [CrossRef]
- McFadden, S.A.; Menchella, J.A.; Chalmers, J.A.; Centeno, M.L.; Belsham, D.D. Glucose responsiveness in a novel adult-derived GnRH cell line, mHypoA-GnRH/GFP: Involvement of AMP-activated protein kinase. Mol. Cell. Endocrinol. 2013, 377, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Graham, I.; Hastings, R.; Gunewardena, S.; Brinkmeier, M.L.; Conn, P.M.; Camper, S.A.; Kumar, T.R. Gonadotrope-specific deletion of Dicer results in severely suppressed gonadotropins and fertility defects. J. Biol. Chem. 2015, 290, 2699–2714. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Elías, M.D.; Rainero Cáceres, T.S.; Giaccagli, M.M.; Guazzone, V.A.; Dalton, G.N.; De Siervi, A.; Cuasnicú, P.S.; Cohen, D.J.; Da Ros, V.G. Association between high-fat diet feeding and male fertility in high reproductive performance mice. Sci. Rep. 2019, 9, 18546. [Google Scholar] [CrossRef]
- Soulis, G.; Kitraki, E.; Gerozissis, K. Early neuroendocrine alterations in female rats following a diet moderately enriched in fat. Cell. Mol. Neurobiol. 2005, 25, 869–880. [Google Scholar] [CrossRef]
- DiVall, S.A.; Herrera, D.; Sklar, B.; Wu, S.; Wondisford, F.; Radovick, S.; Wolfe, A. Insulin receptor signaling in the GnRH neuron plays a role in the abnormal GnRH pulsatility of obese female mice. PLoS ONE 2015, 10, e0119995. [Google Scholar] [CrossRef]
- Balasubramanian, P.; Jagannathan, L.; Mahaley, R.E.; Subramanian, M.; Gilbreath, E.T.; Mohankumar, P.S.; Mohankumar, S.M. High fat diet affects reproductive functions in female diet-induced obese and dietary resistant rats. J. Neuroendocrinol. 2012, 24, 748–755. [Google Scholar] [CrossRef]
- Hussain, M.A.; Abogresha, N.M.; Hassan, R.; Tamany, D.A.; Lotfy, M. Effect of feeding a high-fat diet independently of caloric intake on reproductive function in diet-induced obese female rats. Arch. Med. Sci. 2016, 12, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Levi, N.J.; Wilson, C.W.; Redweik, G.A.J.; Gray, N.W.; Grzybowski, C.W.; Lenkey, J.A.; Moseman, A.W.; Bertsch, A.D.; Dao, N.; Walsh, H.E. Obesity-related cellular stressors regulate gonadotropin releasing hormone gene expression via c-Fos/AP-1. Mol. Cell. Endocrinol. 2018, 478, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Barabas, K.; Szabo-Meleg, E.; Abraham, I.M. Effect of inflammation on female gonadotropin-releasing hormone (GnRH) neurons: Mechanisms and consequences. Int. J. Mol. Sci. 2020, 21, 529. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.; Sarchielli, E.; Comeglio, P.; Filippi, S.; Vignozzi, L.; Marini, M.; Rastrelli, G.; Maneschi, E.; Cellai, I.; Persani, L.; et al. Metabolic syndrome induces inflammation and impairs gonadotropin-releasing hormone neurons in the preoptic area of the hypothalamus in rabbits. Mol. Cell. Endocrinol. 2014, 382, 107–119. [Google Scholar] [CrossRef]
- Lainez, N.M.; Jonak, C.R.; Nair, M.G.; Ethell, I.M.; Wilson, E.H.; Carson, M.J.; Coss, D. Diet-induced obesity elicits macrophage infiltration and reduction in spine density in the hypothalami of male but not female mice. Front. Immunol. 2018, 9, 1992. [Google Scholar] [CrossRef] [PubMed]
- McIlwraith, E.K.; Loganathan, N.; Belsham, D.D. Phoenixin expression is Regulated by the fatty acids palmitate, docosahexaenoic acid and oleate, and the endocrine disrupting chemical bisphenol a in immortalized hypothalamic neurons. Front. Neurosci. 2018, 12, 838. [Google Scholar] [CrossRef]
- McIlwraith, E.K.; Loganathan, N.; Belsham, D.D. Regulation of Gpr173 expression, a putative phoenixin receptor, by saturated fatty acid palmitate and endocrine-disrupting chemical bisphenol A through a p38-mediated mechanism in immortalized hypothalamic neurons. Mol. Cell. Endocrinol. 2019, 485, 54–60. [Google Scholar] [CrossRef]
- Zhai, L.; Zhao, J.; Zhu, Y.; Liu, Q.; Niu, W.; Liu, C.; Wang, Y. Downregulation of leptin receptor and kisspeptin/GPR54 in the murine hypothalamus contributes to male hypogonadism caused by high-fat diet-induced obesity. Endocrine 2018, 62, 195–206. [Google Scholar] [CrossRef]
- Barchitta, M.; Maugeri, A.; Quattrocchi, A.; Agrifoglio, O.; Agodi, A. The role of miRNAs as biomarkers for pregnancy outcomes: A comprehensive review. Int. J. Genomics 2017, 2017, 8067972. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.; Gahlay, G.K. Using miRNAs as diagnostic biomarkers for male infertility: Opportunities and challenges. Mol. Hum. Reprod. 2020, 26, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Messina, A.; Langlet, F.; Chachlaki, K.; Roa, J.; Rasika, S.; Jouy, N.; Gallet, S.; Gaytan, F.; Parkash, J.; Tena-Sempere, M.; et al. A microRNA switch regulates the rise in hypothalamic GnRH production before puberty. Nat. Neurosci. 2016, 19, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Odle, A.K.; MacNicol, M.C.; Childs, G.V.; MacNicol, A.M. Post-transcriptional regulation of Gnrhr: A checkpoint for metabolic control of female reproduction. Int. J. Mol. Sci. 2021, 22, 3312. [Google Scholar] [CrossRef] [PubMed]
- Heras, V.; Sangiao-Alvarellos, S.; Manfredi-Lozano, M.; Sanchez-Tapia, M.J.; Ruiz-Pino, F.; Roa, J.; Lara-Chica, M.; Morrugares-Carmona, R.; Jouy, N.; Abreu, A.P.; et al. Hypothalamic miR-30 regulates puberty onset via repression of the puberty-suppressing factor, Mkrn3. PLoS Biol. 2019, 17, e3000532. [Google Scholar] [CrossRef] [PubMed]
- Perdices López, C.M.; Avendaño Herrador, M.S.; Vázquez Villar, M.S.; Romero, A.B.; Ruiz-Pino, F.; Morrugares-Carmona, R.; Sáez, A.C.; Calzado, M.A.; Rodríguez, J.M.C.; Alonso, A.M.B.; et al. Identification of a novel hypothalamic miRNA/Kisspeptin pathway as pathophysiological mechanism and putative target for management of obesity-induced hypogonadism. In Proceedings of the 22nd European Congress of Endocrinology, Online, 5–9 September 2020. [Google Scholar]
- Saper, C.B.; Lu, J.; Chou, T.C.; Gooley, J. The hypothalamic integrator for circadian rhythms. Trends Neurosci. 2005, 28, 152–157. [Google Scholar] [CrossRef]
- Valenzuela, F.J.; Vera, J.; Venegas, C.; Munoz, S.; Oyarce, S.; Munoz, K.; Lagunas, C. Evidences of polymorphism associated with circadian system and risk of pathologies: A review of the literature. Int. J. Endocrinol. 2016, 2016, 2746909. [Google Scholar] [CrossRef]
- Di Lorenzo, L.; De Pergola, G.; Zocchetti, C.; L’Abbate, N.; Basso, A.; Pannacciulli, N.; Cignarelli, M.; Giorgino, R.; Soleo, L. Effect of shift work on body mass index: Results of a study performed in 319 glucose-tolerant men working in a Southern Italian industry. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 1353–1358. [Google Scholar] [CrossRef]
- Karlsson, B.; Knutsson, A.; Lindahl, B. Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occup. Environ. Med. 2001, 58, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Oosterman, J.E.; Kalsbeek, A.; la Fleur, S.E.; Belsham, D.D. Impact of nutrients on circadian rhythmicity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R337–R350. [Google Scholar] [CrossRef]
- Buijs, F.N.; Guzman-Ruiz, M.; Leon-Mercado, L.; Basualdo, M.C.; Escobar, C.; Kalsbeek, A.; Buijs, R.M. Suprachiasmatic nucleus interaction with the arcuate nucleus; essential for organizing physiological rhythms. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Buhr, E.D.; Takahashi, J.S. Molecular components of the Mammalian circadian clock. Handb. Exp. Pharmacol. 2013, 3–27. [Google Scholar] [CrossRef]
- Kinoshita, C.; Aoyama, K.; Nakaki, T. Neuroprotection afforded by circadian regulation of intracellular glutathione levels: A key role for miRNAs. Free Radic. Biol. Med. 2018, 119, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Mo, G.; Tan, Y. Micro RNAs and the biological clock: A target for diseases associated with a loss of circadian regulation. Afr. Health Sci. 2020, 20, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Taouis, M. MicroRNAs in the hypothalamus. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Arble, D.M.; Bass, J.; Laposky, A.D.; Vitaterna, M.H.; Turek, F.W. Circadian timing of food intake contributes to weight gain. Obesity 2009, 17, 2100–2102. [Google Scholar] [CrossRef] [PubMed]
- Brown-Grant, K.; Raisman, G. Abnormalities in reproductive function associated with the destruction of the suprachiasmatic nuclei in female rats. Proc. R. Soc. Lond. B. Biol. Sci. 1977, 198, 279–296. [Google Scholar] [CrossRef]
- Simonneaux, V. A Kiss to drive rhythms in reproduction. Eur. J. Neurosci. 2018, 51, 509–530. [Google Scholar] [CrossRef]
- Kim, E.R.; Xu, Y.; Cassidy, R.M.; Lu, Y.; Yang, Y.; Tian, J.; Li, D.P.; Van Drunen, R.; Ribas-Latre, A.; Cai, Z.L.; et al. Paraventricular hypothalamus mediates diurnal rhythm of metabolism. Nat. Commun. 2020, 11, 3794. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, J.A.; Lin, S.Y.; Martino, T.A.; Arab, S.; Liu, P.; Husain, M.; Sole, M.J.; Belsham, D.D. Diurnal profiling of neuroendocrine genes in murine heart, and shift in proopiomelanocortin gene expression with pressure-overload cardiac hypertrophy. J. Mol. Endocrinol. 2008, 41, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Stutz, A.M.; Staszkiewicz, J.; Ptitsyn, A.; Argyropoulos, G. Circadian expression of genes regulating food intake. Obesity 2007, 15, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Fick, L.J.; Fick, G.H.; Belsham, D.D. Rhythmic clock and neuropeptide gene expression in hypothalamic mHypoE-44 neurons. Mol.Cell. Endocrinol. 2010, 323, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Cedernaes, J.; Huang, W.; Ramsey, K.M.; Waldeck, N.; Cheng, L.; Marcheva, B.; Omura, C.; Kobayashi, Y.; Peek, C.B.; Levine, D.C.; et al. Transcriptional basis for rhythmic control of hunger and metabolism within the AgRP neuron. Cell. Metab. 2019, 29, 1078–1091.e5. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, A.; Laposky, A.D.; Ramsey, K.M.; Estrada, C.; Joshu, C.; Kobayashi, Y.; Turek, F.W.; Bass, J. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell. Metab. 2007, 6, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Greco, J.A.; Oosterman, J.E.; Belsham, D.D. Differential effects of omega-3 fatty acid docosahexaenoic acid and palmitate on the circadian transcriptional profile of clock genes in immortalized hypothalamic neurons. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R1049–R1060. [Google Scholar] [CrossRef]
- Oosterman, J.E.; Belsham, D.D. Glucose Alters Per2 Rhythmicity Independent of AMPK, Whereas AMPK Inhibitor Compound C Causes Profound Repression of Clock Genes and AgRP in mHypoE-37 Hypothalamic Neurons. PLoS ONE 2016, 11, e0146969. [Google Scholar] [CrossRef]
- Chen, R.; Zuo, Z.; Li, Q.; Wang, H.; Li, N.; Zhang, H.; Yu, X.; Liu, Z. DHA substitution overcomes high-fat diet-induced disturbance in the circadian rhythm of lipid metabolism. Food Funct. 2020, 11, 3621–3631. [Google Scholar] [CrossRef]
- Tal, Y.; Chapnik, N.; Froy, O. Non-obesogenic doses of palmitate disrupt circadian metabolism in adipocytes. Adipocyte 2019, 8, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Zocchi, L.; Kinoshita, C.; Borrelli, E.; Sassone-Corsi, P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS ONE 2010, 5, e8561. [Google Scholar] [CrossRef] [PubMed]
- Dang, F.; Sun, X.; Ma, X.; Wu, R.; Zhang, D.; Chen, Y.; Xu, Q.; Wu, Y.; Liu, Y. Insulin post-transcriptionally modulates Bmal1 protein to affect the hepatic circadian clock. Nat. Commun. 2016, 7, 12696. [Google Scholar] [CrossRef]
- Wada, T.; Ichihashi, Y.; Suzuki, E.; Kosuge, Y.; Ishige, K.; Uchiyama, T.; Makishima, M.; Nakao, R.; Oishi, K.; Shimba, S. Deletion of Bmal1 prevents diet-induced ectopic fat accumulation by controlling oxidative capacity in the skeletal muscle. Int. J. Mol. Sci. 2018, 19, 2813. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Kooijman, S.; Gao, Y.; Tzeplaeff, L.; Cosquer, B.; Milanova, I.; Wolff, S.E.C.; Korpel, N.; Champy, M.F.; Petit-Demoulière, B.; et al. Microglia-specific knock-down of Bmal1 improves memory and protects mice from high fat diet-induced obesity. Mol. Psychiatry 2021. [Google Scholar] [CrossRef] [PubMed]
- Moraes, J.C.; Coope, A.; Morari, J.; Cintra, D.E.; Roman, E.A.; Pauli, J.R.; Romanatto, T.; Carvalheira, J.B.; Oliveira, A.L.; Saad, M.J.; et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS ONE 2009, 4, e5045. [Google Scholar] [CrossRef]
- Tran, A.; He, W.; Jiang, N.; Chen, J.T.C.; Belsham, D.D. NAMPT and BMAL1 are independently involved in the palmitate-mediated induction of neuroinflammation in hypothalamic neurons. Front. Endocrinol. 2020, 11, 351. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Morris, A.C.; Kahn, D.E.; McLean, F.H.; Hay, E.A.; Kubitz, P.; MacKenzie, A.; Martinoli, M.G.; Drew, J.E.; Williams, L.M. Palmitic acid triggers inflammatory responses in N42 cultured hypothalamic cells partially via ceramide synthesis but not via TLR4. Nutr. Neurosci. 2020, 23, 321–334. [Google Scholar] [CrossRef]
- Adlanmerini, M.; Nguyen, H.C.; Krusen, B.M.; Teng, C.W.; Geisler, C.E.; Peed, L.C.; Carpenter, B.J.; Hayes, M.R.; Lazar, M.A. Hypothalamic REV-ERB nuclear receptors control diurnal food intake and leptin sensitivity in diet-induced obese mice. J. Clin. Investig. 2021, 131, e140424. [Google Scholar] [CrossRef]
- Chen, R.; D’Alessandro, M.; Lee, C. miRNAs are required for generating a time delay critical for the circadian oscillator. Curr. Biol. 2013, 23, 1959–1968. [Google Scholar] [CrossRef]
- Shende, V.R.; Goldrick, M.M.; Ramani, S.; Earnest, D.J. Expression and rhythmic modulation of circulating microRNAs targeting the clock gene Bmal1 in mice. PLoS ONE 2011, 6, e22586. [Google Scholar] [CrossRef]
- Shende, V.R.; Neuendorff, N.; Earnest, D.J. Role of miR-142-3p in the post-transcriptional regulation of the clock gene Bmal1 in the mouse SCN. PLoS ONE 2013, 8, e65300. [Google Scholar] [CrossRef]
- Chemello, F.; Grespi, F.; Zulian, A.; Cancellara, P.; Hebert-Chatelain, E.; Martini, P.; Bean, C.; Alessio, E.; Buson, L.; Bazzega, M.; et al. Transcriptomic analysis of single isolated myofibers identifies miR-27a-3p and miR-142-3p as regulators of metabolism in skeletal muscle. Cell. Rep. 2019, 26, 3784–3797.e8. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Fonken, L.K.; Gushchina, L.V.; Aubrecht, T.G.; Maurya, S.K.; Periasamy, M.; Nelson, R.J.; Popovich, P.G. miR-155 Deletion in female mice prevents diet-induced obesity. Sci. Rep. 2016, 6, 22862. [Google Scholar] [CrossRef]
- Zhou, L.; Miller, C.; Miraglia, L.J.; Romero, A.; Mure, L.S.; Panda, S.; Kay, S.A. A genome-wide microRNA screen identifies the microRNA-183/96/182 cluster as a modulator of circadian rhythms. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Yang, W.M.; Min, K.H.; Lee, W. Induction of miR-96 by dietary saturated fatty acids exacerbates hepatic insulin resistance through the suppression of INSR and IRS-1. PLoS ONE 2016, 11, e0169039. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.Y.; Papp, J.W.; Varlamova, O.; Dziema, H.; Russell, B.; Curfman, J.P.; Nakazawa, T.; Shimizu, K.; Okamura, H.; Impey, S.; et al. microRNA modulation of circadian-clock period and entrainment. Neuron 2007, 54, 813–829. [Google Scholar] [CrossRef]
- Alvarez-Saavedra, M.; Antoun, G.; Yanagiya, A.; Oliva-Hernandez, R.; Cornejo-Palma, D.; Perez-Iratxeta, C.; Sonenberg, N.; Cheng, H.Y. miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum. Mol. Genet. 2011, 20, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Casals, N.; Keung, W.; Moran, T.H.; Lopaschuk, G.D. Differential effects of central ghrelin on fatty acid metabolism in hypothalamic ventral medial and arcuate nuclei. Physiol. Behav. 2013, 118, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Salazar, T.E.; Dominguez, J.M.; Nguyen, D.V.; Li Calzi, S.; Bhatwadekar, A.D.; Qi, X.; Busik, J.V.; Boulton, M.E.; Grant, M.B. Dicer expression exhibits a tissue-specific diurnal pattern that is lost during aging and in diabetes. PLoS ONE 2013, 8, e80029. [Google Scholar] [CrossRef]
- Dasgupta, I.; Chatterjee, A. Recent advances in miRNA delivery systems. Methods Protoc. 2021, 4, 10. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lieu, C.V.; Loganathan, N.; Belsham, D.D. Mechanisms Driving Palmitate-Mediated Neuronal Dysregulation in the Hypothalamus. Cells 2021, 10, 3120. https://doi.org/10.3390/cells10113120
Lieu CV, Loganathan N, Belsham DD. Mechanisms Driving Palmitate-Mediated Neuronal Dysregulation in the Hypothalamus. Cells. 2021; 10(11):3120. https://doi.org/10.3390/cells10113120
Chicago/Turabian StyleLieu, Calvin V., Neruja Loganathan, and Denise D. Belsham. 2021. "Mechanisms Driving Palmitate-Mediated Neuronal Dysregulation in the Hypothalamus" Cells 10, no. 11: 3120. https://doi.org/10.3390/cells10113120
APA StyleLieu, C. V., Loganathan, N., & Belsham, D. D. (2021). Mechanisms Driving Palmitate-Mediated Neuronal Dysregulation in the Hypothalamus. Cells, 10(11), 3120. https://doi.org/10.3390/cells10113120