
The Challenges and Opportunities in the Development of MicroRNA Therapeutics: A Multidisciplinary Viewpoint


 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
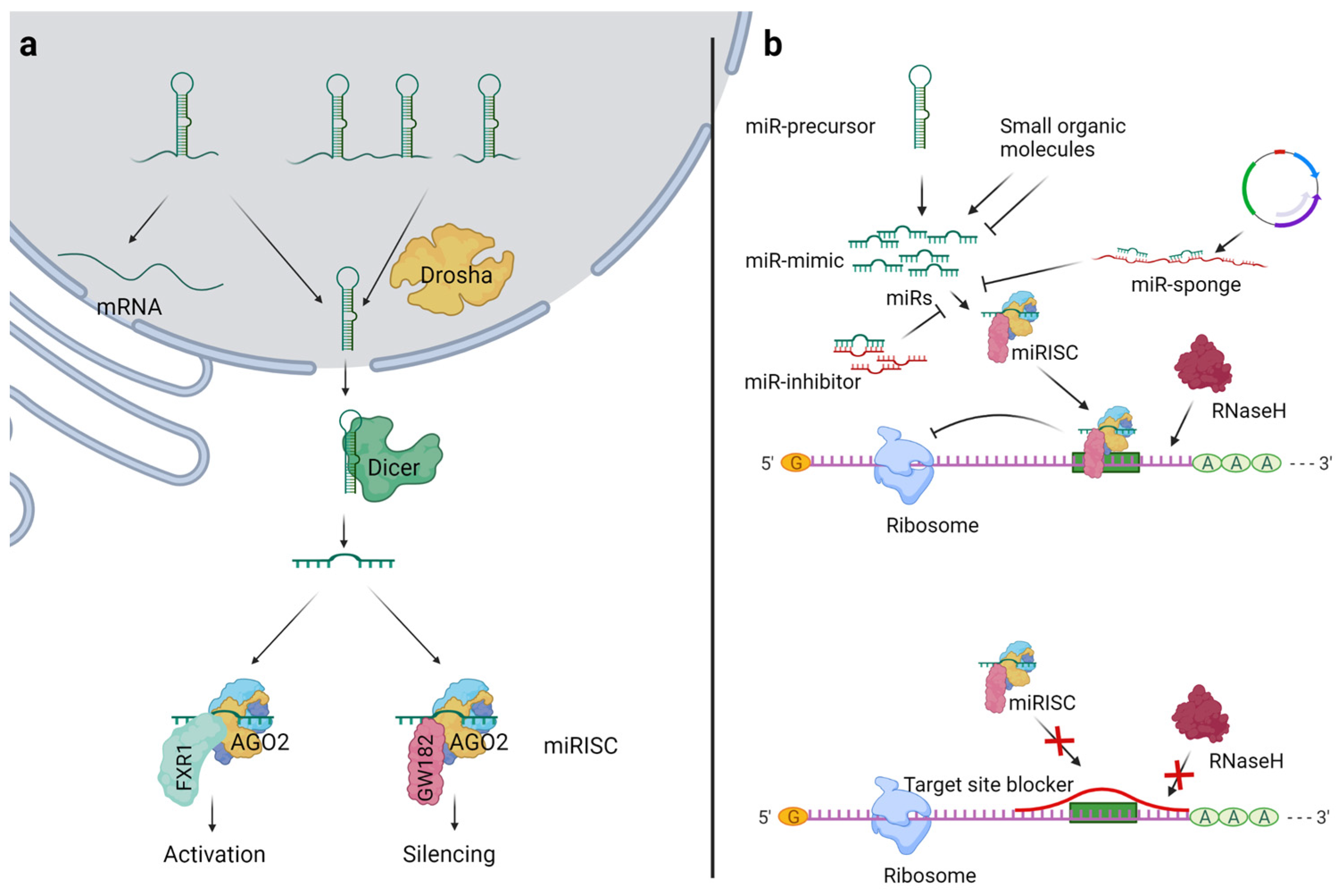
2. The MiR Biogenesis and Mechanism of Action
3. MiRs: Therapeutic Target
3.1. MiR Modulation
3.1.1. Deletion of Processing Enzymes
3.1.2. MiR Mimics and MiR Precursors
3.1.3. MiR Inhibitors and MiR Sponges
3.1.4. Target Site Blockers (TSBs)
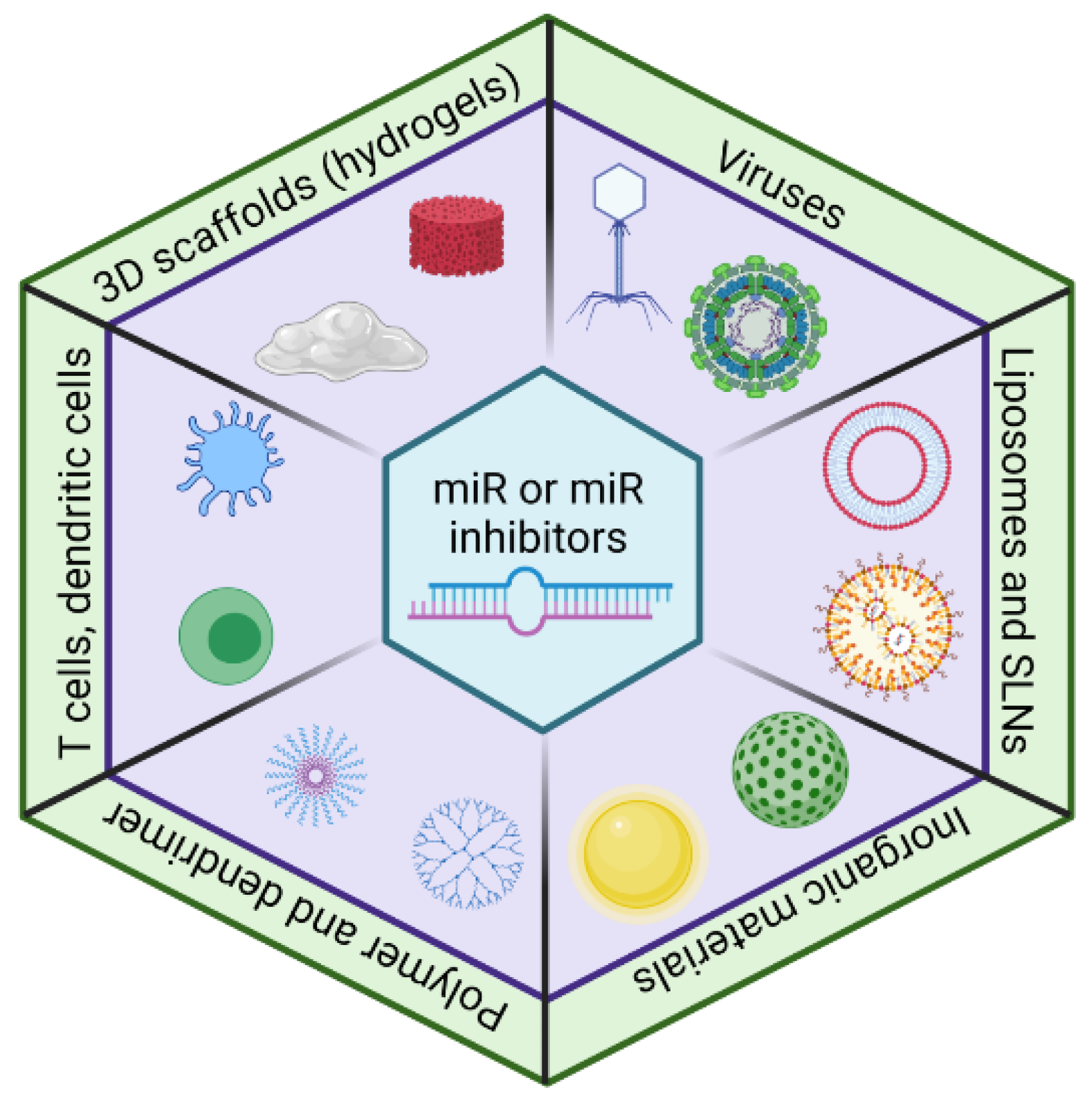
3.2. Delivery of MiR Modulators
3.2.1. Viral Vector-Based Delivery System
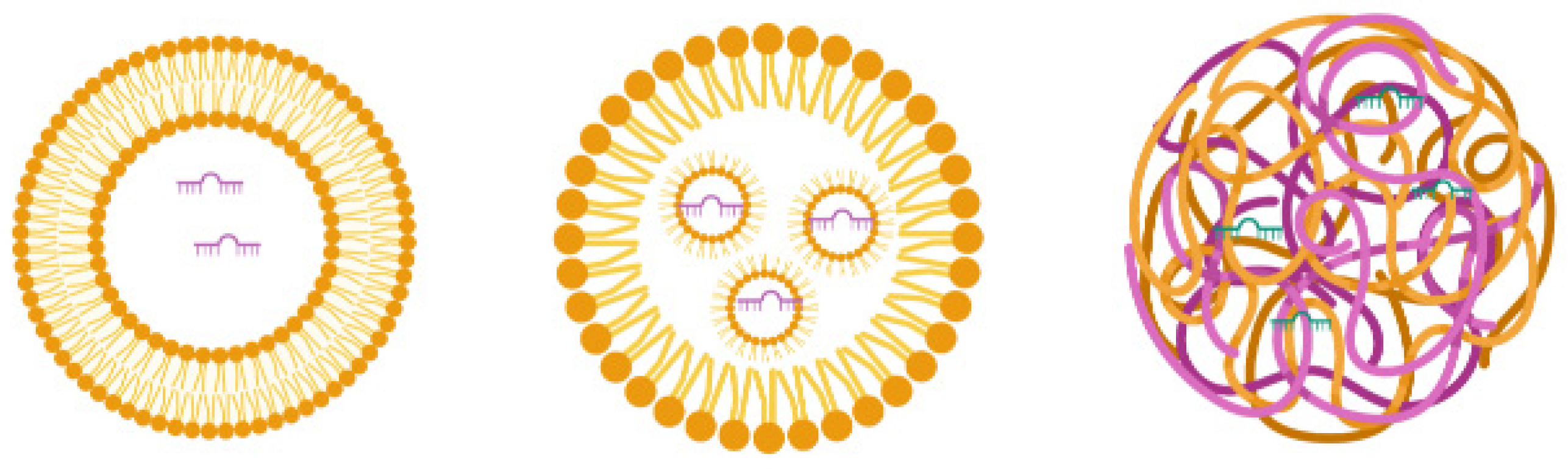
3.2.2. Inorganic Material-Based Delivery System
3.2.3. D Scaffold-Based Delivery System
3.2.4. Polymeric- and Dendrimer-Based Delivery Systems
3.2.5. Lipid-Based Delivery Systems
3.2.6. Cell-Based Delivery Systems
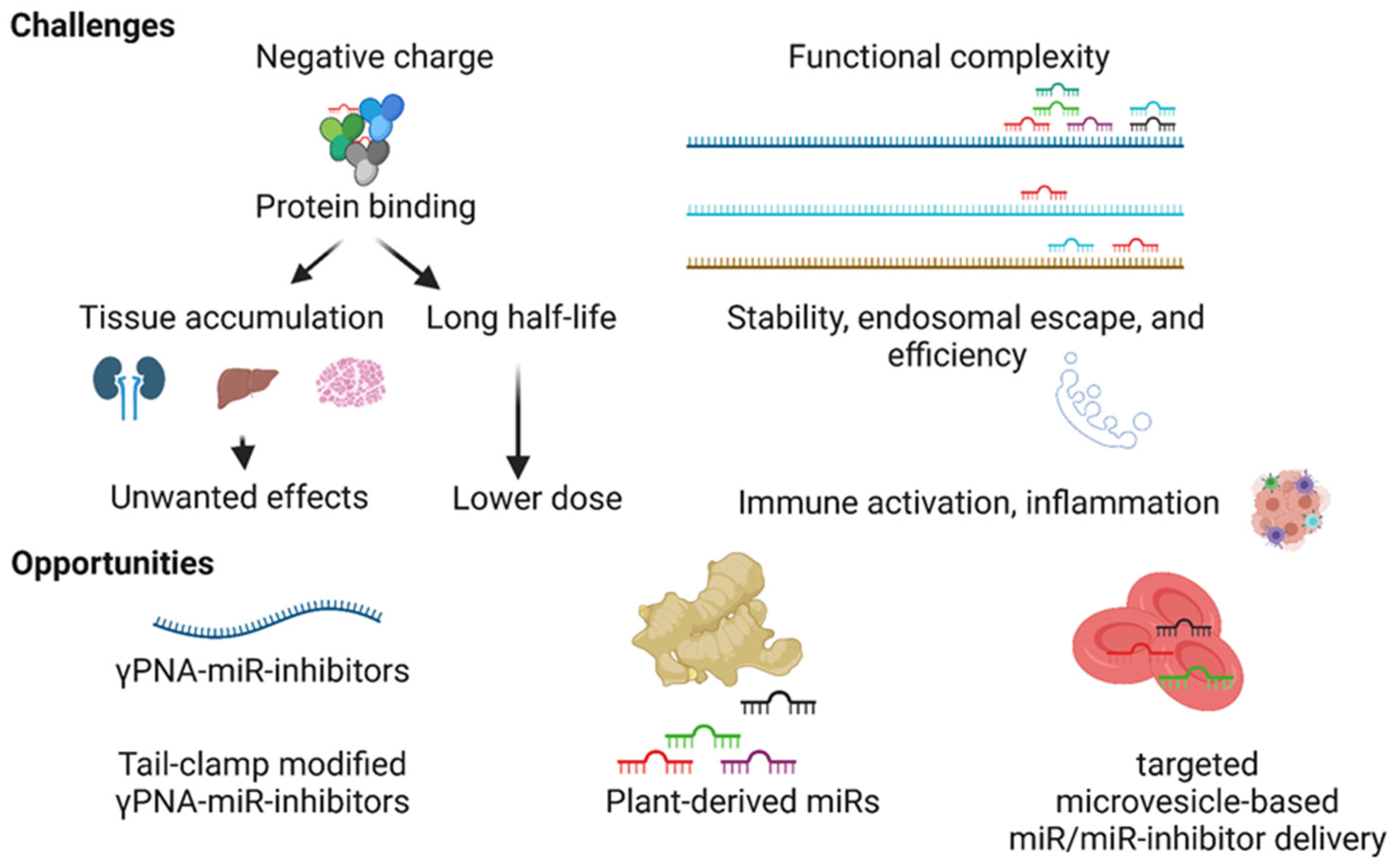
4. Challenges in the Development of MiR Therapeutics
4.1. The Negative Charge of the MiR Inhibitors
4.2. Functional Complexity and Off-Target Effects
4.3. Stability of MiR Modulators in the Blood and Endosomal Escape
4.4. Chemical Modifications and Cellular Entry of MiR Inhibitors
5. Opportunities for the Development of MiR-Therapeutics
5.1. Gamma-Modified PNA-Based MiR Inhibitors
5.2. Plant-Derived MiRs
5.3. Cell-Targeted Delivery Using Microvesicles and Exosomes
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef]
- Horvitz, H.R.; Sulston, J.E. Isolation and genetic characterization of cell-lineage mutants of the nematode Caenorhabditis elegans. Genetics 1980, 96, 435–454. [Google Scholar] [CrossRef]
- Moran, Y.; Agron, M.; Praher, D.; Technau, U. The evolutionary origin of plant and animal microRNAs. Nat. Ecol. Evol. 2017, 1, 27. [Google Scholar] [CrossRef]
- Moroz, L.L.; Kocot, K.M.; Citarella, M.R.; Dosung, S.; Norekian, T.P.; Povolotskaya, I.S.; Grigorenko, A.P.; Dailey, C.; Berezikov, E.; Buckley, K.M.; et al. The ctenophore genome and the evolutionary origins of neural systems. Nature 2014, 510, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grasser, F.A.; Lenhof, H.P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.S. Therapeutic advances of miRNAs: A preclinical and clinical update. J. Adv. Res. 2021, 28, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Shaham, O.; Gueta, K.; Mor, E.; Oren-Giladi, P.; Grinberg, D.; Xie, Q.; Cvekl, A.; Shomron, N.; Davis, N.; Keydar-Prizant, M.; et al. Pax6 regulates gene expression in the vertebrate lens through miR-204. PLoS Genet. 2013, 9, e1003357. [Google Scholar] [CrossRef] [Green Version]
- Krol, J.; Busskamp, V.; Markiewicz, I.; Stadler, M.B.; Ribi, S.; Richter, J.; Duebel, J.; Bicker, S.; Fehling, H.J.; Schubeler, D.; et al. Characterizing light-regulated retinal microRNAs reveals rapid turnover as a common property of neuronal microRNAs. Cell 2010, 141, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karali, M.; Peluso, I.; Marigo, V.; Banfi, S. Identification and characterization of microRNAs expressed in the mouse eye. Investig. Ophthalmol. Vis. Sci. 2007, 48, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deo, M.; Yu, J.Y.; Chung, K.H.; Tippens, M.; Turner, D.L. Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev. Dyn. 2006, 235, 2538–2548. [Google Scholar] [CrossRef] [Green Version]
- Vikram, A.; Kim, Y.R.; Kumar, S.; Li, Q.; Kassan, M.; Jacobs, J.S.; Irani, K. Vascular microRNA-204 is remotely governed by the microbiome and impairs endothelium-dependent vasorelaxation by downregulating Sirtuin1. Nat. Commun. 2016, 7, 12565. [Google Scholar] [CrossRef]
- Mikhaylova, O.; Stratton, Y.; Hall, D.; Kellner, E.; Ehmer, B.; Drew, A.F.; Gallo, C.A.; Plas, D.R.; Biesiada, J.; Meller, J.; et al. VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 2012, 21, 532–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, L.; Bourachot, B.; Mateescu, B.; Reyal, F.; Mechta-Grigoriou, F. Regulation of miR-200c/141 expression by intergenic DNA-looping and transcriptional read-through. Nat. Commun. 2016, 7, 8959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, E.; Guttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Provost, P.; Dishart, D.; Doucet, J.; Frendewey, D.; Samuelsson, B.; Radmark, O. Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J. 2002, 21, 5864–5874. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Pi, J.; Zou, D.; Wang, X.; Xu, J.; Yu, S.; Zhang, T.; Li, F.; Zhang, X.; Zhao, H.; et al. microRNA arm-imbalance in part from complementary targets mediated decay promotes gastric cancer progression. Nat. Commun. 2019, 10, 4397. [Google Scholar] [CrossRef] [Green Version]
- Li, S.C.; Liao, Y.L.; Ho, M.R.; Tsai, K.W.; Lai, C.H.; Lin, W.C. miRNA arm selection and isomiR distribution in gastric cancer. BMC Genom. 2012, 13 (Suppl. S1), S13. [Google Scholar] [CrossRef] [Green Version]
- Griffiths-Jones, S.; Hui, J.H.; Marco, A.; Ronshaugen, M. MicroRNA evolution by arm switching. EMBO Rep. 2011, 12, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, M.H.; Shin, S.; Jung, S.R.; Kim, E.; Song, J.J.; Hohng, S. Human Argonaute 2 Has Diverse Reaction Pathways on Target RNAs. Mol. Cell 2015, 59, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Janas, M.M.; Wang, B.; Harris, A.S.; Aguiar, M.; Shaffer, J.M.; Subrahmanyam, Y.V.; Behlke, M.A.; Wucherpfennig, K.W.; Gygi, S.P.; Gagnon, E.; et al. Alternative RISC assembly: Binding and repression of microRNA-mRNA duplexes by human Ago proteins. RNA 2012, 18, 2041–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederichs, S.; Haber, D.A. Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell 2007, 131, 1097–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.S.; Maurin, T.; Robine, N.; Rasmussen, K.D.; Jeffrey, K.L.; Chandwani, R.; Papapetrou, E.P.; Sadelain, M.; O’Carroll, D.; Lai, E.C. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15163–15168. [Google Scholar] [CrossRef] [Green Version]
- Berezikov, E.; Chung, W.J.; Willis, J.; Cuppen, E.; Lai, E.C. Mammalian mirtron genes. Mol. Cell 2007, 28, 328–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiring, A.M.; Harb, J.G.; Neviani, P.; Garton, C.; Oaks, J.J.; Spizzo, R.; Liu, S.; Schwind, S.; Santhanam, R.; Hickey, C.J.; et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell 2010, 140, 652–665. [Google Scholar] [CrossRef] [Green Version]
- Bukhari, S.I.A.; Truesdell, S.S.; Lee, S.; Kollu, S.; Classon, A.; Boukhali, M.; Jain, E.; Mortensen, R.D.; Yanagiya, A.; Sadreyev, R.I.; et al. A Specialized Mechanism of Translation Mediated by FXR1a-Associated MicroRNP in Cellular Quiescence. Mol. Cell 2016, 61, 760–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truesdell, S.S.; Mortensen, R.D.; Seo, M.; Schroeder, J.C.; Lee, J.H.; LeTonqueze, O.; Vasudevan, S. MicroRNA-mediated mRNA translation activation in quiescent cells and oocytes involves recruitment of a nuclear microRNP. Sci. Rep. 2012, 2, 842. [Google Scholar] [CrossRef] [Green Version]
- Cheung, T.H.; Quach, N.L.; Charville, G.W.; Liu, L.; Park, L.; Edalati, A.; Yoo, B.; Hoang, P.; Rando, T.A. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 2012, 482, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Katz, S.; Cussigh, D.; Urban, N.; Blomfield, I.; Guillemot, F.; Bally-Cuif, L.; Coolen, M. A Nuclear Role for miR-9 and Argonaute Proteins in Balancing Quiescent and Activated Neural Stem Cell States. Cell Rep. 2016, 17, 1383–1398. [Google Scholar] [CrossRef] [Green Version]
- Coll, M.; El Taghdouini, A.; Perea, L.; Mannaerts, I.; Vila-Casadesus, M.; Blaya, D.; Rodrigo-Torres, D.; Affo, S.; Morales-Ibanez, O.; Graupera, I.; et al. Integrative miRNA and Gene Expression Profiling Analysis of Human Quiescent Hepatic Stellate Cells. Sci. Rep. 2015, 5, 11549. [Google Scholar] [CrossRef] [Green Version]
- Orom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef]
- Hammond, S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicerna Pharmaceuticals Announces First Patient Dosed in Phase 1b/2 Clinical Trial of DCR-MYC, an Investigational RNAi Therapeutic Targeting the MYC Oncogene, in Patients with Advanced Hepatocellular Carcinoma. Available online: https://investors.dicerna.com/news-releases/news-release-details/dicerna-pharmaceuticals-announces-first-patient-dosed-phase-1b2 (accessed on 11 March 2021).
- Da Costa Martins, P.A.; Bourajjaj, M.; Gladka, M.; Kortland, M.; van Oort, R.J.; Pinto, Y.M.; Molkentin, J.D.; De Windt, L.J. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation 2008, 118, 1567–1576. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.F.; Murchison, E.P.; Tang, R.; Callis, T.E.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.M.; Schneider, M.D.; Selzman, C.H.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2008, 105, 2111–2116. [Google Scholar] [CrossRef] [Green Version]
- De Pietri Tonelli, D.; Pulvers, J.N.; Haffner, C.; Murchison, E.P.; Hannon, G.J.; Huttner, W.B. miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development 2008, 135, 3911–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Teta, M.; Choi, Y.S.; Okegbe, T.; Wong, G.; Tam, O.H.; Chong, M.M.; Seykora, J.T.; Nagy, A.; Littman, D.R.; Andl, T.; et al. Inducible deletion of epidermal Dicer and Drosha reveals multiple functions for miRNAs in postnatal skin. Development 2012, 139, 1405–1416. [Google Scholar] [CrossRef] [Green Version]
- Gaddam, R.R.; Jecobsen, V.P.; Kim, Y.R.; Kumar, S.; Gabani, M.; Jacobs, J.S.; Dhuri, K.; Kassan, M.; Li, Q.; Bahal, R.; et al. Microbiota-governed microRNA-204 impairs endothelial function and blood pressure decline during inactivity in db/db mice. Sci. Rep. 2020, 10, 10065. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.H.; Hart, C.C.; Al-Bassam, S.; Avery, A.; Taylor, J.; Patel, P.D.; Vojtek, A.B.; Turner, D.L. Polycistronic RNA polymerase II expression vectors for RNA interference based on BIC/miR-155. Nucleic Acids Res. 2006, 34, e53. [Google Scholar] [CrossRef] [Green Version]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallant-Behm, C.L.; Piper, J.; Lynch, J.M.; Seto, A.G.; Hong, S.J.; Mustoe, T.A.; Maari, C.; Pestano, L.A.; Dalby, C.M.; Jackson, A.L.; et al. A MicroRNA-29 Mimic (Remlarsen) Represses Extracellular Matrix Expression and Fibroplasia in the Skin. J. Investig. Dermatol. 2019, 139, 1073–1081. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Trujillo, R.D.; Yue, S.B.; Tang, Y.; O’Gorman, W.E.; Chen, C.Z. The potential functions of primary microRNAs in target recognition and repression. EMBO J. 2010, 29, 3272–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Min, H.; Yue, S.; Chen, C.Z. Pre-miRNA loop nucleotides control the distinct activities of mir-181a-1 and mir-181c in early T cell development. PLoS ONE 2008, 3, e3592. [Google Scholar] [CrossRef] [Green Version]
- Jafari, N.; Abediankenari, S.; Hossein-Nataj, H. miR-34a mimic or pre-mir-34a, which is the better option for cancer therapy? KatoIII as a model to study miRNA action in human gastric cancer cells. Cancer Cell Int. 2021, 21, 178. [Google Scholar] [CrossRef]
- Shan, G.; Li, Y.; Zhang, J.; Li, W.; Szulwach, K.E.; Duan, R.; Faghihi, M.A.; Khalil, A.M.; Lu, L.; Paroo, Z.; et al. A small molecule enhances RNA interference and promotes microRNA processing. Nat. Biotechnol. 2008, 26, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Young, D.D.; Connelly, C.M.; Grohmann, C.; Deiters, A. Small molecule modifiers of microRNA miR-122 function for the treatment of hepatitis C virus infection and hepatocellular carcinoma. J. Am. Chem. Soc. 2010, 132, 7976–7981. [Google Scholar] [CrossRef] [PubMed]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef] [PubMed]
- Dragulescu-Andrasi, A.; Rapireddy, S.; Frezza, B.M.; Gayathri, C.; Gil, R.R.; Ly, D.H. A simple gamma-backbone modification preorganizes peptide nucleic acid into a helical structure. J. Am. Chem. Soc. 2006, 128, 10258–10267. [Google Scholar] [CrossRef]
- Lundin, P.; Johansson, H.; Guterstam, P.; Holm, T.; Hansen, M.; Langel, Ü.; El Andaloussi, S. Distinct uptake routes of cell-penetrating peptide conjugates. Bioconjug. Chem. 2008, 19, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Swenson, C.S.; Heemstra, J.M. Peptide nucleic acids harness dual information codes in a single molecule. Chem. Commun. Camb. 2020, 56, 1926–1935. [Google Scholar] [CrossRef]
- Zhou, P.; Wang, M.; Du, L.; Fisher, G.W.; Waggoner, A.; Ly, D.H. Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (GPNA). J. Am. Chem. Soc. 2003, 125, 6878–6879. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [Green Version]
- De Santi, C.; Fernandez Fernandez, E.; Gaul, R.; Vencken, S.; Glasgow, A.; Oglesby, I.K.; Hurley, K.; Hawkins, F.; Mitash, N.; Mu, F.; et al. Precise Targeting of miRNA Sites Restores CFTR Activity in CF Bronchial Epithelial Cells. Mol. Ther. 2020, 28, 1190–1199. [Google Scholar] [CrossRef]
- Hawez, A.; Al-Haidari, A.; Madhi, R.; Rahman, M.; Thorlacius, H. MiR-155 Regulates PAD4-Dependent Formation of Neutrophil Extracellular Traps. Front. Immunol. 2019, 10, 2462. [Google Scholar] [CrossRef] [Green Version]
- Wisse, E.; Jacobs, F.; Topal, B.; Frederik, P.; De Geest, B. The size of endothelial fenestrae in human liver sinusoids: Implications for hepatocyte-directed gene transfer. Gene Ther. 2008, 15, 1193–1199. [Google Scholar] [CrossRef] [Green Version]
- Seth, P.P.; Tanowitz, M.; Bennett, C.F. Selective tissue targeting of synthetic nucleic acid drugs. J. Clin. Investig. 2019, 129, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Bass, D.M. Viral infections: New and emerging. Curr. Opin. Gastroenterol. 2010, 26, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. The ancient Virus World and evolution of cells. Biol. Direct 2006, 1, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, I.; Chatterjee, A. Recent Advances in miRNA Delivery Systems. Methods Protoc. 2021, 4, 10. [Google Scholar] [CrossRef]
- Xie, J.; Ameres, S.L.; Friedline, R.; Hung, J.H.; Zhang, Y.; Xie, Q.; Zhong, L.; Su, Q.; He, R.; Li, M.; et al. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat. Methods 2012, 9, 403–409. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Jiang, Y.M.; Huang, Z.; Doi, H.; Matsumoto, S.; Kondo, N.; Iida, M.; et al. Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat. Med. 2012, 18, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Yla-Herttuala, S. Endgame: Glybera finally recommended for approval as the first gene therapy drug in the European union. Mol. Ther. 2012, 20, 1831–1832. [Google Scholar] [CrossRef] [Green Version]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2011, 12, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Chen, H.; Wei, M.; Chen, X.; Zhang, Y.; Cao, L.; Yuan, P.; Wang, F.; Yang, G.; Ma, J. Gold nanoparticle-based miR155 antagonist macrophage delivery restores the cardiac function in ovariectomized diabetic mouse model. Int. J. Nanomed. 2017, 12, 4963–4979. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhu, L.; Zhu, L.; Wang, P.; Xu, W.; Huang, J. Recent Developments in Delivery of MicroRNAs Utilizing Nanosystems for Metabolic Syndrome Therapy. Int. J. Mol. Sci. 2021, 22, 7855. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Duo, Y.; Bi, J.; Zeng, X.; Mei, L.; Bao, S.; He, L.; Shan, A.; Zhang, Y.; Yu, X. Targeted delivery of anti-miR-155 by functionalized mesoporous silica nanoparticles for colorectal cancer therapy. Int. J. Nanomed. 2018, 13, 1241–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, D.W.; Kim, H.Y.; Li, F.; Park, J.Y.; Kim, D.; Park, J.H.; Han, H.S.; Byun, J.W.; Lee, Y.S.; Jeong, J.M.; et al. In vivo visualization of endogenous miR-21 using hyaluronic acid-coated graphene oxide for targeted cancer therapy. Biomaterials 2017, 121, 144–154. [Google Scholar] [CrossRef]
- Sun, S.; Wang, Y.; Zhou, R.; Deng, Z.; Han, Y.; Han, X.; Tao, W.; Yang, Z.; Shi, C.; Hong, D.; et al. Targeting and Regulating of an Oncogene via Nanovector Delivery of MicroRNA using Patient-Derived Xenografts. Theranostics 2017, 7, 677–693. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.K.; Jeon, O.; Dang, P.N.; Huynh, C.T.; Varghai, D.; Riazi, H.; McMillan, A.; Herberg, S.; Alsberg, E. RNA interfering molecule delivery from in situ forming biodegradable hydrogels for enhancement of bone formation in rat calvarial bone defects. Acta Biomater. 2018, 75, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Oliva, N.; Atilano, M.; Song, H.S.; Artzi, N. Self-assembled RNA-triple-helix hydrogel scaffold for microRNA modulation in the tumour microenvironment. Nat. Mater. 2016, 15, 353–363. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Chen, Y.E.; Chen, J.; Ma, P.X. Cell-free 3D scaffold with two-stage delivery of miRNA-26a to regenerate critical-sized bone defects. Nat. Commun. 2016, 7, 10376. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Tian, H.; Guo, Y.; Li, Y.; Guo, Z.; Zhu, X.; Chen, X. miRNA oligonucleotide and sponge for miRNA-21 inhibition mediated by PEI-PLL in breast cancer therapy. Acta Biomater. 2015, 25, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Xie, Y.; Wu, L.; Chen, X.; Liu, H.; Zhou, Y.; Zou, H.; Liu, D.; Zhao, Y.; Kong, X.; et al. PLGA-based dual targeted nanoparticles enhance miRNA transfection efficiency in hepatic carcinoma. Sci. Rep. 2017, 7, 46250. [Google Scholar] [CrossRef]
- Lee, D.W.; Lim, C.; Israelachvili, J.N.; Hwang, D.S. Strong adhesion and cohesion of chitosan in aqueous solutions. Langmuir 2013, 29, 14222–14229. [Google Scholar] [CrossRef] [Green Version]
- Higashi, N.; Morikawa, A.; Fujioka, K.; Fujita, Y.; Sano, Y.; Miyata-Takeuchi, M.; Suzuki, N.; Irimura, T. Human macrophage lectin specific for galactose/N-acetylgalactosamine is a marker for cells at an intermediate stage in their differentiation from monocytes into macrophages. Int. Immunol. 2002, 14, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Ma, J.; Chen, M.; Jiang, H.; Fu, Y.; Gan, J.; Dong, L.; Zhang, J.; Chen, J. Dual TNF-alpha/IL-12p40 Interference as a Strategy to Protect against Colitis Based on miR-16 Precursors with Macrophage Targeting Vectors. Mol. Ther. 2015, 23, 1611–1621. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhang, B.; Zhou, L.; Shi, Y.; Li, Z.; Xia, Y.; Tian, J. Imaging Dendrimer-Grafted Graphene Oxide Mediated Anti-miR-21 Delivery with an Activatable Luciferase Reporter. ACS Appl. Mater. Interfaces 2016, 8, 9014–9021. [Google Scholar] [CrossRef]
- Khan, D.R.; Rezler, E.M.; Lauer-Fields, J.; Fields, G.B. Effects of drug hydrophobicity on liposomal stability. Chem. Biol. Drug Des. 2008, 71, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.P.; Huang, L. Time-dependent maturation of cationic liposome-DNA complex for serum resistance. Gene Ther. 1998, 5, 380–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Khan, W.; Hosseinkhani, H.; Ickowicz, D.; Hong, P.D.; Yu, D.S.; Domb, A.J. Polysaccharide gene transfection agents. Acta Biomater. 2012, 8, 4224–4232. [Google Scholar] [CrossRef]
- Cui, S.; Wang, Y.; Gong, Y.; Lin, X.; Zhao, Y.; Zhi, D.; Zhou, Q.; Zhang, S. Correlation of the cytotoxic effects of cationic lipids with their headgroups. Toxicol. Res. Camb. 2018, 7, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, D.M.; Tajmir-Riahi, H.A. Study on the interaction of cationic lipids with bovine serum albumin. J. Phys. Chem. B 2010, 114, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.L.; Ewert, K.K.; Majzoub, R.N.; Hwu, Y.K.; Liang, K.S.; Leal, C.; Safinya, C.R. Optimizing cationic and neutral lipids for efficient gene delivery at high serum content. J. Gene Med. 2014, 16, 84–96. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Li, X.Q.; Duan, J.L.; Bao, C.J.; Cui, Y.N.; Su, Z.B.; Xu, J.R.; Luo, Q.; Chen, M.; Xie, Y.; et al. Nanosized functional miRNA liposomes and application in the treatment of TNBC by silencing Slug gene. Int. J. Nanomed. 2019, 14, 3645–3667. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Song, K.; Lu, X.; Feng, W.; Di, W. Liposomal Delivery of MicroRNA-7 Targeting EGFR to Inhibit the Growth, Invasion, and Migration of Ovarian Cancer. ACS Omega 2021, 6, 11669–11678. [Google Scholar] [CrossRef]
- Zhang, W.; Peng, F.; Zhou, T.; Huang, Y.; Zhang, L.; Ye, P.; Lu, M.; Yang, G.; Gai, Y.; Yang, T.; et al. Targeted delivery of chemically modified anti-miR-221 to hepatocellular carcinoma with negatively charged liposomes. Int. J. Nanomed. 2015, 10, 4825–4836. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Ryals, R.C.; Weller, K.K.; Pennesi, M.E.; Sahay, G. Lipid nanoparticles for delivery of messenger RNA to the back of the eye. J. Control. Release 2019, 303, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Vhora, I.; Lalani, R.; Bhatt, P.; Patil, S.; Misra, A. Lipid-nucleic acid nanoparticles of novel ionizable lipids for systemic BMP-9 gene delivery to bone-marrow mesenchymal stem cells for osteoinduction. Int. J. Pharm. 2019, 563, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Fenton, O.S.; Olafson, K.N.; Pillai, P.S.; Mitchell, M.J.; Langer, R. Advances in Biomaterials for Drug Delivery. Adv. Mater. 2018, 30, e1705328. [Google Scholar] [CrossRef]
- Manjunath, K.; Reddy, J.S.; Venkateswarlu, V. Solid lipid nanoparticles as drug delivery systems. Methods Find. Exp. Clin. Pharmacol. 2005, 27, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Liu, C.; Ye, J.; Zou, W.; Zhang, N.; Xu, W. Novel cationic SLN containing a synthesized single-tailed lipid as a modifier for gene delivery. Nanotechnology 2009, 20, 215102. [Google Scholar] [CrossRef]
- Muller, R.H.; Mader, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery—A review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Bunjes, H. Lipid nanoparticles for the delivery of poorly water-soluble drugs. J. Pharm. Pharmacol. 2010, 62, 1637–1645. [Google Scholar] [CrossRef]
- Freitas, C.; Muller, R.H. Correlation between long-term stability of solid lipid nanoparticles (SLN) and crystallinity of the lipid phase. Eur. J. Pharm. Biopharm. 1999, 47, 125–132. [Google Scholar] [CrossRef]
- Tabatt, K.; Sameti, M.; Olbrich, C.; Muller, R.H.; Lehr, C.M. Effect of cationic lipid and matrix lipid composition on solid lipid nanoparticle-mediated gene transfer. Eur. J. Pharm. Biopharm. 2004, 57, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Meng, T.; Yuan, M.; Wen, L.; Cheng, B.; Liu, N.; Huang, X.; Hong, Y.; Yuan, H.; Hu, F. MicroRNA-200c delivered by solid lipid nanoparticles enhances the effect of paclitaxel on breast cancer stem cell. Int. J. Nanomed. 2016, 11, 6713–6725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.J.; Zhong, Z.R.; Liu, J.; Zhang, Z.R.; Sun, X.; Gong, T. Solid lipid nanoparticles loaded with anti-microRNA oligonucleotides (AMOs) for suppression of microRNA-21 functions in human lung cancer cells. Pharm. Res. 2012, 29, 97–109. [Google Scholar] [CrossRef]
- Surman, M.; Drozdz, A.; Stepien, E.; Przybylo, M. Extracellular Vesicles as Drug Delivery Systems—Methods of Production and Potential Therapeutic Applications. Curr. Pharm. Des. 2019, 25, 132–154. [Google Scholar] [CrossRef]
- Badeau, B.A.; Comerford, M.P.; Arakawa, C.K.; Shadish, J.A.; DeForest, C.A. Engineered modular biomaterial logic gates for environmentally triggered therapeutic delivery. Nat. Chem. 2018, 10, 251–258. [Google Scholar] [CrossRef]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Fan, F.; Li, R.; Niu, B.; Zhu, L.; Yu, S.; Wang, S.; Li, C.; Wang, D. Different Erythrocyte MicroRNA Profiles in Low- and High-Altitude Individuals. Front. Physiol. 2018, 9, 1099. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ukidve, A.; Gao, Y.; Kim, J.; Mitragotri, S. Erythrocyte leveraged chemotherapy (ELeCt): Nanoparticle assembly on erythrocyte surface to combat lung metastasis. Sci. Adv. 2019, 5, eaax9250. [Google Scholar] [CrossRef] [Green Version]
- Muzykantov, V.R. Drug delivery by red blood cells: Vascular carriers designed by mother nature. Expert Opin. Drug Deliv. 2010, 7, 403–427. [Google Scholar] [CrossRef] [Green Version]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Azad, A.; et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parayath, N.N.; Stephan, S.B.; Koehne, A.L.; Nelson, P.S.; Stephan, M.T. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun. 2020, 11, 6080. [Google Scholar] [CrossRef]
- Soundara Rajan, T.; Gugliandolo, A.; Bramanti, P.; Mazzon, E. In Vitro-Transcribed mRNA Chimeric Antigen Receptor T Cell (IVT mRNA CAR T) Therapy in Hematologic and Solid Tumor Management: A Preclinical Update. Int. J. Mol. Sci. 2020, 21, 6514. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2018, 9, 3176. [Google Scholar] [CrossRef]
- Firdessa-Fite, R.; Creusot, R.J. Nanoparticles versus Dendritic Cells as Vehicles to Deliver mRNA Encoding Multiple Epitopes for Immunotherapy. Mol. Ther. Methods Clin. Dev. 2020, 16, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, X.; Tian, B.; Liu, J.; Yang, L.; Zeng, L.; Chen, T.; Hong, A.; Wang, X. Nucleolin-targeted Extracellular Vesicles as a Versatile Platform for Biologics Delivery to Breast Cancer. Theranostics 2017, 7, 1360–1372. [Google Scholar] [CrossRef]
- Wayne, E.C.; Long, C.; Haney, M.J.; Batrakova, E.V.; Leisner, T.M.; Parise, L.V.; Kabanov, A.V. Targeted Delivery of siRNA Lipoplexes to Cancer Cells Using Macrophage Transient Horizontal Gene Transfer. Adv. Sci. Weinh. 2019, 6, 1900582. [Google Scholar] [CrossRef] [Green Version]
- Akao, Y.; Iio, A.; Itoh, T.; Noguchi, S.; Itoh, Y.; Ohtsuki, Y.; Naoe, T. Microvesicle-mediated RNA molecule delivery system using monocytes/macrophages. Mol. Ther. 2011, 19, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Wise, P.M.; Murtadha, M.; Liu, C.W.; Wu, C.H.; Jong, A.Y.; Seeger, R.C.; Fabbri, M. Natural Killer-Derived Exosomal miR-186 Inhibits Neuroblastoma Growth and Immune Escape Mechanisms. Cancer Res. 2019, 79, 1151–1164. [Google Scholar] [CrossRef]
- Sun, H.; Shi, K.; Qi, K.; Kong, H.; Zhang, J.; Dai, S.; Ye, W.; Deng, T.; He, Q.; Zhou, M. Natural Killer Cell-Derived Exosomal miR-3607-3p Inhibits Pancreatic Cancer Progression by Targeting IL-26. Front. Immunol. 2019, 10, 2819. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Cheng, Z.; Wang, Y.; Han, T. The Risks of miRNA Therapeutics: In a Drug Target Perspective. Drug Des. Devel. Ther. 2021, 15, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, B.M.; Mays, D.; Lipsky, J.; Stewart, J.A.; Fauq, A.; Richelson, E. Pharmacokinetics and tissue distribution of a peptide nucleic acid after intravenous administration. Antisense Nucleic Acid Drug Dev. 2002, 12, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.; Lum, B.L.; Fisher, G.A.; Halsey, J.; Geary, R.S.; Holmlund, J.T.; Kwoh, T.J.; Dorr, F.A.; Sikic, B.I. A phase I trial of aprinocarsen (ISIS 3521/LY900003), an antisense inhibitor of protein kinase C-alpha administered as a 24-hour weekly infusion schedule in patients with advanced cancer. Investig. New Drugs 2005, 23, 467–477. [Google Scholar] [CrossRef]
- Desai, A.A.; Schilsky, R.L.; Young, A.; Janisch, L.; Stadler, W.M.; Vogelzang, N.J.; Cadden, S.; Wright, J.A.; Ratain, M.J. A phase I study of antisense oligonucleotide GTI-2040 given by continuous intravenous infusion in patients with advanced solid tumors. Ann. Oncol. 2005, 16, 958–965. [Google Scholar] [CrossRef]
- Kahan, B.D.; Stepkowski, S.; Kilic, M.; Katz, S.M.; Van Buren, C.T.; Welsh, M.S.; Tami, J.A.; Shanahan, W.R., Jr. Phase I and phase II safety and efficacy trial of intercellular adhesion molecule-1 antisense oligodeoxynucleotide (ISIS 2302) for the prevention of acute allograft rejection. Transplantation 2004, 78, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Iannitti, T.; Morales-Medina, J.C.; Palmieri, B. Phosphorothioate oligonucleotides: Effectiveness and toxicity. Curr. Drug Targets 2014, 15, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, Y.W.; Brewer, G.; Jing, Q. MicroRNA degradation and turnover: Regulating the regulators. Wiley Interdiscip. Rev. RNA 2012, 3, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Segal, M.; Biscans, A.; Gilles, M.E.; Anastasiadou, E.; De Luca, R.; Lim, J.; Khvorova, A.; Slack, F.J. Hydrophobically Modified let-7b miRNA Enhances Biodistribution to NSCLC and Downregulates HMGA2 In Vivo. Mol. Ther. Nucleic Acids 2020, 19, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar] [CrossRef] [Green Version]
- Segal, M.; Slack, F.J. Challenges identifying efficacious miRNA therapeutics for cancer. Expert Opin. Drug Discov. 2020, 15, 987–992. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, Y.; Guo, Y.; Zou, L.; Jin, H.; Zhong, L.; Wu, Y.; Zhang, L.; Yang, Z. Exploring directional invasion of serum nuclease into siRNA duplexes by asymmetrical terminal modifications. ChemMedChem 2014, 9, 2111–2119. [Google Scholar] [CrossRef]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef]
- Linsley, C.S.; Wu, B.M. Recent advances in light-responsive on-demand drug-delivery systems. Ther. Deliv. 2017, 8, 89–107. [Google Scholar] [CrossRef] [Green Version]
- Christie, R.J.; Nishiyama, N.; Kataoka, K. Delivering the code: Polyplex carriers for deoxyribonucleic acid and ribonucleic acid interference therapies. Endocrinology 2010, 151, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Mollaei, H.; Safaralizadeh, R.; Rostami, Z. MicroRNA replacement therapy in cancer. J. Cell Physiol. 2019, 234, 12369–12384. [Google Scholar] [CrossRef]
- Zein, R.; Sharrouf, W.; Selting, K. Physical Properties of Nanoparticles That Result in Improved Cancer Targeting. J. Oncol. 2020, 2020, 5194780. [Google Scholar] [CrossRef] [PubMed]
- Masarudin, M.J.; Cutts, S.M.; Evison, B.J.; Phillips, D.R.; Pigram, P.J. Factors determining the stability, size distribution, and cellular accumulation of small, monodisperse chitosan nanoparticles as candidate vectors for anticancer drug delivery: Application to the passive encapsulation of [(14)C]-doxorubicin. Nanotechnol. Sci. Appl. 2015, 8, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nat. Lond. 1993, 365, 566–568. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Demidov, V.V.; Potaman, V.N.; Frank-Kamenetskii, M.D.; Egholm, M.; Buchard, O.; Sonnichsen, S.H.; Nielsen, P.E. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem. Pharmacol. 1994, 48, 1310–1313. [Google Scholar] [CrossRef]
- He, G.; Rapireddy, S.; Bahal, R.; Sahu, B.; Ly, D.H. Strand Invasion of Extended, Mixed-Sequence B-DNA by γPNAs. J. Am. Chem. Soc. 2009, 131, 12088–12090. [Google Scholar] [CrossRef] [Green Version]
- Sahu, B.; Sacui, I.; Rapireddy, S.; Zanotti, K.J.; Bahal, R.; Armitage, B.A.; Ly, D.H. Synthesis and Characterization of Conformationally Preorganized, (R)-Diethylene Glycol-Containing γ-Peptide Nucleic Acids with Superior Hybridization Properties and Water Solubility. J. Org. Chem. 2011, 76, 5614–5627. [Google Scholar] [CrossRef] [Green Version]
- Sahu, B.; Chenna, V.; Lathrop, K.L.; Thomas, S.M.; Zon, G.; Livak, K.J.; Ly, D.H. Synthesis of conformationally preorganized and cell-permeable guanidine-based gamma-peptide nucleic acids (gammaGPNAs). J. Org. Chem. 2009, 74, 1509–1516. [Google Scholar] [CrossRef] [Green Version]
- Dhuri, K.; Gaddam, R.R.; Vikram, A.; Slack, F.J.; Bahal, R. Therapeutic potential of chemically-modified, synthetic, triplex peptide nucleic acid-based oncomiR inhibitors for cancer therapy. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Kaplan, A.R.; Pham, H.; Liu, Y.; Oyaghire, S.; Bahal, R.; Engelman, D.M.; Glazer, P.M. Ku80-Targeted pH-Sensitive Peptide-PNA Conjugates Are Tumor Selective and Sensitize Cancer Cells to Ionizing Radiation. Mol. Cancer Res. 2020, 18, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Dhuri, K.; Vyas, R.N.; Blumenfeld, L.; Verma, R.; Bahal, R. Nanoparticle Delivered Anti-miR-141-3p for Stroke Therapy. Cells 2021, 10, 1011. [Google Scholar] [CrossRef] [PubMed]
- Wahane, A.; Malik, S.; Shih, K.C.; Gaddam, R.R.; Chen, C.; Liu, Y.; Nieh, M.P.; Vikram, A.; Bahal, R. Dual-Modality Poly-l-histidine Nanoparticles to Deliver Peptide Nucleic Acids and Paclitaxel for In Vivo Cancer Therapy. ACS Appl. Mater. Interfaces 2021, 13, 45244–45258. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Quijano, E.; Liu, Y.; Bahal, R.; Scanlon, S.E.; Song, E.; Hsieh, W.C.; Braddock, D.E.; Ly, D.H.; Saltzman, W.M.; et al. Anti-tumor Activity of miniPEG-gamma-Modified PNAs to Inhibit MicroRNA-210 for Cancer Therapy. Mol. Ther. Nucleic Acids 2017, 9, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, A.; Rapireddy, S.; Ly, D.H.; Meller, A. Electronic Barcoding of a Viral Gene at the Single-Molecule Level. Nano Lett. 2012, 12, 1722–1728. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Pearse, A.; Liu, Y.; Taylor, R.E. Modular self-assembly of gamma-modified peptide nucleic acids in organic solvent mixtures. Nat. Commun. 2020, 11, 2960. [Google Scholar] [CrossRef]
- Bahal, R.; Ali McNeer, N.; Quijano, E.; Liu, Y.; Sulkowski, P.; Turchick, A.; Lu, Y.C.; Bhunia, D.C.; Manna, A.; Greiner, D.L.; et al. In vivo correction of anaemia in beta-thalassemic mice by gammaPNA-mediated gene editing with nanoparticle delivery. Nat. Commun. 2016, 7, 13304. [Google Scholar] [CrossRef] [PubMed]
- Bahal, R.; Quijano, E.; McNeer, N.A.; Liu, Y.; Bhunia, D.C.; Lopez-Giraldez, F.; Fields, R.J.; Saltzman, W.M.; Ly, D.H.; Glazer, P.M. Single-stranded gammaPNAs for in vivo site-specific genome editing via Watson-Crick recognition. Curr. Gene Ther. 2014, 14, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, A.S.; Bahal, R.; Farrelly, J.S.; Quijano, E.; Bianchi, A.H.; Luks, V.L.; Putman, R.; Lopez-Giraldez, F.; Coskun, S.; Song, E.; et al. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun. 2018, 9, 2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeer, N.A.; Anandalingam, K.; Fields, R.J.; Caputo, C.; Kopic, S.; Gupta, A.; Quijano, E.; Polikoff, L.; Kong, Y.; Bahal, R.; et al. Correction of F508del CFTR in airway epithelium using nanoparticles delivering triplex-forming PNAs. Nat. Commun. 2015, 6, 6952. [Google Scholar] [CrossRef]
- Thomas, S.M.; Sahu, B.; Rapireddy, S.; Bahal, R.; Wheeler, S.E.; Procopio, E.M.; Kim, J.; Joyce, S.C.; Contrucci, S.; Wang, Y.; et al. Antitumor effects of EGFR antisense guanidine-based peptide nucleic acids in cancer models. ACS Chem. Biol. 2013, 8, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Beavers, K.R.; Werfel, T.A.; Shen, T.; Kavanaugh, T.E.; Kilchrist, K.V.; Mares, J.W.; Fain, J.S.; Wiese, C.B.; Vickers, K.C.; Weiss, S.M.; et al. Porous Silicon and Polymer Nanocomposites for Delivery of Peptide Nucleic Acids as Anti-MicroRNA Therapies. Adv. Mater. 2016, 28, 7984–7992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budak, H.; Bulut, R.; Kantar, M.; Alptekin, B. MicroRNA nomenclature and the need for a revised naming prescription. Brief. Funct. Genom. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Chin, A.R.; Fong, M.Y.; Somlo, G.; Wu, J.; Swiderski, P.; Wu, X.; Wang, S.E. Cross-kingdom inhibition of breast cancer growth by plant miR159. Cell Res. 2016, 26, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, H.; Yuan, Y.; Etheridge, A.; Zhou, Y.; Huang, D.; Wilmes, P.; Galas, D. The complex exogenous RNA spectra in human plasma: An interface with human gut biota? PLoS ONE 2012, 7, e51009. [Google Scholar] [CrossRef]
- Wuriyanghan, H.; Rosa, C.; Falk, B.W. Oral delivery of double-stranded RNAs and siRNAs induces RNAi effects in the potato/tomato psyllid, Bactericerca cockerelli. PLoS ONE 2011, 6, e27736. [Google Scholar] [CrossRef] [Green Version]
- Pirro, S.; Zanella, L.; Kenzo, M.; Montesano, C.; Minutolo, A.; Potesta, M.; Sobze, M.S.; Canini, A.; Cirilli, M.; Muleo, R.; et al. MicroRNA from Moringa oleifera: Identification by High Throughput Sequencing and Their Potential Contribution to Plant Medicinal Value. PLoS ONE 2016, 11, e0149495. [Google Scholar] [CrossRef]
- Yang, J.; Hotz, T.; Broadnax, L.; Yarmarkovich, M.; Elbaz-Younes, I.; Hirschi, K.D. Anomalous uptake and circulatory characteristics of the plant-based small RNA MIR2911. Sci. Rep. 2016, 6, 26834. [Google Scholar] [CrossRef]
- Mlotshwa, S.; Pruss, G.J.; MacArthur, J.L.; Endres, M.W.; Davis, C.; Hofseth, L.J.; Pena, M.M.; Vance, V. A novel chemopreventive strategy based on therapeutic microRNAs produced in plants. Cell Res. 2015, 25, 521–524. [Google Scholar] [CrossRef]
- Teng, Y.; Xu, F.; Zhang, X.; Mu, J.; Sayed, M.; Hu, X.; Lei, C.; Sriwastva, M.; Kumar, A.; Sundaram, K.; et al. Plant-derived exosomal microRNAs inhibit lung inflammation induced by exosomes SARS-CoV-2 Nsp12. Mol. Ther. 2021, 29, 2424–2440. [Google Scholar] [CrossRef]
- Potesta, M.; Roglia, V.; Fanelli, M.; Pietrobono, E.; Gismondi, A.; Vumbaca, S.; Nguedia Tsangueu, R.G.; Canini, A.; Colizzi, V.; Grelli, S.; et al. Effect of microvesicles from Moringa oleifera containing miRNA on proliferation and apoptosis in tumor cell lines. Cell Death Discov. 2020, 6, 43. [Google Scholar] [CrossRef]
- Rakhmetullina, A.; Pyrkova, A.; Aisina, D.; Ivashchenko, A. In silico prediction of human genes as potential targets for rice miRNAs. Comput. Biol. Chem. 2020, 87, 107305. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.; Mu, J.; Dokland, T.; Zhuang, X.; Wang, Q.; Jiang, H.; Xiang, X.; Deng, Z.B.; Wang, B.; Zhang, L.; et al. Grape exosome-like nanoparticles induce intestinal stem cells and protect mice from DSS-induced colitis. Mol. Ther. 2013, 21, 1345–1357. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Zhang, F.; Dong, L.; Wu, H.; Xu, J.; Li, H.; Wang, J.; Zhou, Z.; Liu, C.; Wang, Y.; et al. SIDT1-dependent absorption in the stomach mediates host uptake of dietary and orally administered microRNAs. Cell Res. 2021, 31, 247–258. [Google Scholar] [CrossRef]
- Diez-Sainz, E.; Lorente-Cebrian, S.; Aranaz, P.; Riezu-Boj, J.I.; Martinez, J.A.; Milagro, F.I. Potential Mechanisms Linking Food-Derived MicroRNAs, Gut Microbiota and Intestinal Barrier Functions in the Context of Nutrition and Human Health. Front. Nutr. 2021, 8, 586564. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.; Zhang, Y.; Petrick, J.S.; Heck, G.; Ivashuta, S.; Marshall, W.S. Lack of detectable oral bioavailability of plant microRNAs after feeding in mice. Nat. Biotechnol. 2013, 31, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Davis, C.D.; Wang, T.T.Y. Extensive Degradation and Low Bioavailability of Orally Consumed Corn miRNAs in Mice. Nutrients 2018, 10, 215. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Elbaz-Younes, I.; Primo, C.; Murungi, D.; Hirschi, K.D. Intestinal permeability, digestive stability and oral bioavailability of dietary small RNAs. Sci. Rep. 2018, 8, 10253. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Bang-Berthelsen, C.H.; Holm, A.; Houben, A.J.; Muller, A.H.; Thymann, T.; Pociot, F.; Estivill, X.; Friedlander, M.R. Survey of 800+ data sets from human tissue and body fluid reveals xenomiRs are likely artifacts. RNA 2017, 23, 433–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zen, K.; Zhang, C.Y. Reply to Lack of detectable oral bioavailability of plant microRNAs after feeding in mice. Nat. Biotechnol. 2013, 31, 967–969. [Google Scholar] [CrossRef]
- Sundaram, G.M. Dietary non-coding RNAs from plants: Fairy tale or treasure? Noncoding RNA Res. 2019, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Sanchita; Trivedi, R.; Asif, M.H.; Trivedi, P.K. Dietary plant miRNAs as an augmented therapy: Cross-kingdom gene regulation. RNA Biol. 2018, 15, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Z.; Tan, J.; Miao, Y.; Zhang, Q. The role of microvesicles containing microRNAs in vascular endothelial dysfunction. J. Cell Mol. Med. 2019, 23, 7933–7945. [Google Scholar] [CrossRef] [PubMed]
- Vikram, A.; Tripathi, D.N.; Kumar, A.; Singh, S. Oxidative stress and inflammation in diabetic complications. Int. J. Endocrinol. 2014, 2014, 679754. [Google Scholar] [CrossRef]
- Yuan, H.; Ma, Q.; Ye, L.; Piao, G. The Traditional Medicine and Modern Medicine from Natural Products. Molecules 2016, 21, 559. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Momin, M.Y.; Gaddam, R.R.; Kravitz, M.; Gupta, A.; Vikram, A. The Challenges and Opportunities in the Development of MicroRNA Therapeutics: A Multidisciplinary Viewpoint. Cells 2021, 10, 3097. https://doi.org/10.3390/cells10113097
Momin MY, Gaddam RR, Kravitz M, Gupta A, Vikram A. The Challenges and Opportunities in the Development of MicroRNA Therapeutics: A Multidisciplinary Viewpoint. Cells. 2021; 10(11):3097. https://doi.org/10.3390/cells10113097
Chicago/Turabian StyleMomin, Mohammad Yahya, Ravinder Reddy Gaddam, Madeline Kravitz, Anisha Gupta, and Ajit Vikram. 2021. "The Challenges and Opportunities in the Development of MicroRNA Therapeutics: A Multidisciplinary Viewpoint" Cells 10, no. 11: 3097. https://doi.org/10.3390/cells10113097
APA StyleMomin, M. Y., Gaddam, R. R., Kravitz, M., Gupta, A., & Vikram, A. (2021). The Challenges and Opportunities in the Development of MicroRNA Therapeutics: A Multidisciplinary Viewpoint. Cells, 10(11), 3097. https://doi.org/10.3390/cells10113097