
Vascular Dysfunction in Preeclampsia

 , ,
, ,
Abstract
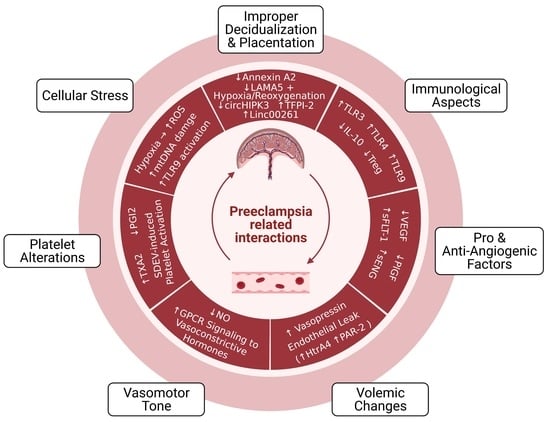
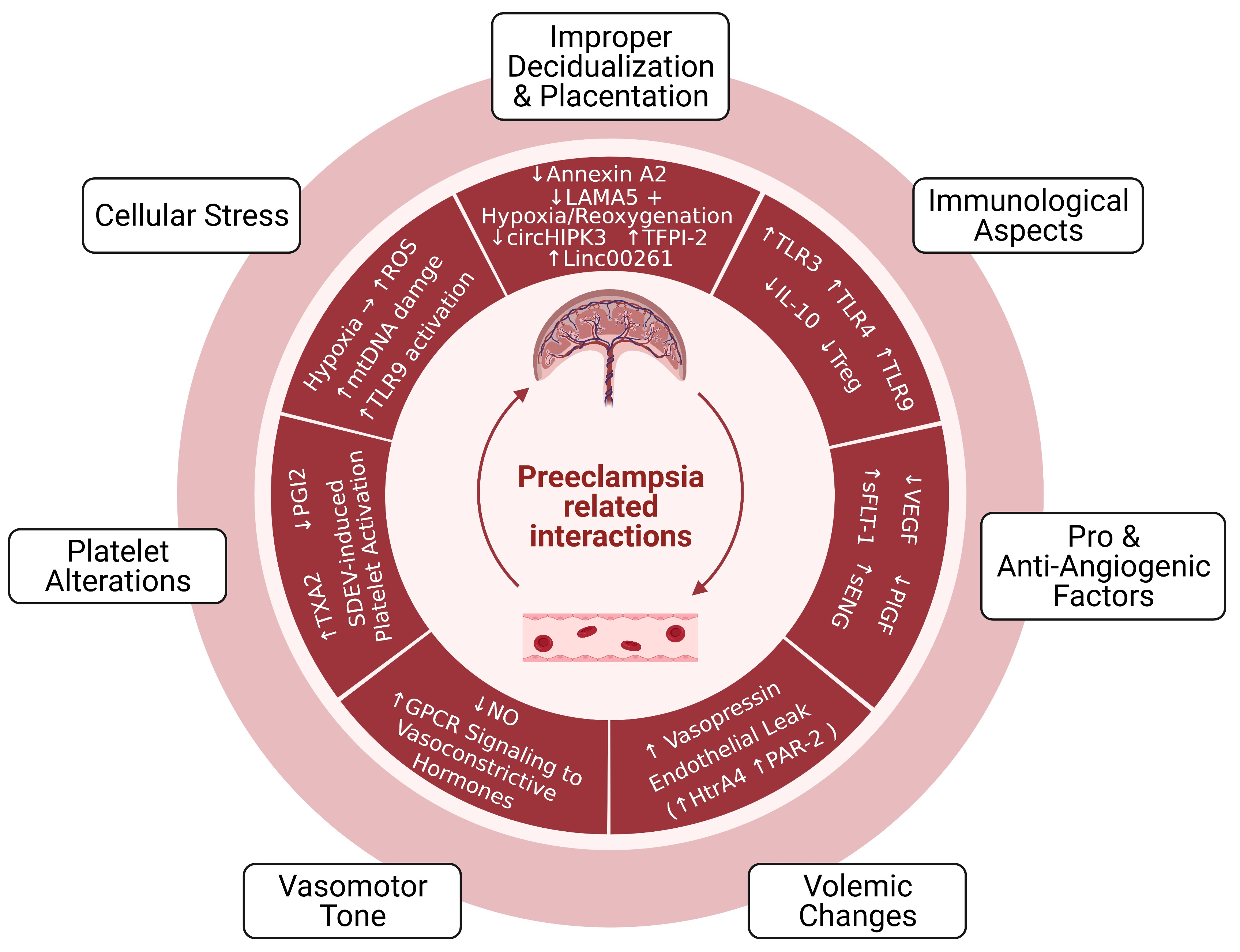
1. Introduction
2. Pathophysiology of Preeclampsia
2.1. Improper Decidualization and Placentation
2.1.1. Cellular and Molecular Aspects
2.1.2. Immunological and Vascular Aspects
3. Circulating and Placenta-Derived Vascular Substances Associated with Preeclampsia (VEGF, PlGF, sFLT-1, ENG, and sENG)
4. Endothelial Damage in Preeclampsia
4.1. Volemic Changes Associated with Endothelial Barrier Integrity
4.2. Altered Vasomotor Tone
5. Preeclampsia-Associated Platelet Alterations
6. Oxidative Stress, Mitochondrial DNA Damage, and TLR9 Activation
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obs. Gynecol 2013, 122, 1122–1131. [Google Scholar] [CrossRef]
- Rana, S.; Lemoine, E.; Granger, J.; Karumanchi, S.A. Preeclampsia: Pathophysiology, challenges, and perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.; Kitt, J.; Leeson, P.; Aye, C.Y.L.; Lewandowski, A.J. Preeclampsia: Risk Factors, Diagnosis, Management, and the Cardiovascular Impact on the Offspring. J. Clin. Med. 2019, 8, 1625. [Google Scholar] [CrossRef] [PubMed]
- Pankiewicz, K.; Szczerba, E.; Maciejewski, T.; Fijałkowska, A. Non-obstetric complications in preeclampsia. Prz. Menopauzalny 2019, 18, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Duley, L. The global impact of pre-eclampsia and eclampsia. Semin. Perinatol. 2009, 33, 130–137. [Google Scholar] [CrossRef]
- Jeyabalan, A. Epidemiology of preeclampsia: Impact of obesity. Nutr. Rev. 2013, 71. [Google Scholar] [CrossRef] [PubMed]
- Bellamy, L.; Casas, J.-P.; Hingorani, A.D.; Williams, D.J. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: Systematic review and meta-analysis. BMJ Clin. Res. Ed. 2007, 335, 974. [Google Scholar] [CrossRef]
- Aykas, F.; Solak, Y.; Erden, A.; Bulut, K.; Dogan, S.; Sarli, B.; Acmaz, G.; Afsar, B.; Siriopol, D.; Covic, A.; et al. Persistence of cardiovascular risk factors in women with previous preeclampsia: A long-term follow-up study. J. Investig. Med. 2015, 63, 641–645. [Google Scholar] [CrossRef]
- Ying, W.; Catov, J.M.; Ouyang, P. Hypertensive Disorders of Pregnancy and Future Maternal Cardiovascular Risk. J. Am. Heart Assoc. 2018, 7, e009382. [Google Scholar] [CrossRef]
- Perez Botero, J.; Reese, J.A.; George, J.N.; McIntosh, J.J. Severe thrombocytopenia and microangiopathic hemolytic anemia in pregnancy: A guide for the consulting hematologist. Am. J. Hematol. 2021. [Google Scholar] [CrossRef]
- Fingar, K.R.; Mabry-Hernandez, I.; Ngo-Metzger, Q.; Wolff, T.; Steiner, C.A.; Elixhauser, A. Delivery Hospitalizations Involving Preeclampsia and Eclampsia, 2005–2014: Statistical Brief #222. In Healthcare Cost and Utilization Project (HCUP) Statistical Briefs; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2006. [Google Scholar]
- Bell, M.J. A historical overview of preeclampsia-eclampsia. J. Obstet. Gynecol. Neonatal Nurs. JOGNN 2010, 39, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Högberg, U. The World Health Report 2005: “make every mother and child count”—including Africans. Scand. J. Public Health 2005, 33, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Ananth, C.V.; Keyes, K.M.; Wapner, R.J. Pre-eclampsia rates in the United States, 1980–2010: Age-period-cohort analysis. BMJ 2013, 347, f6564. [Google Scholar] [CrossRef] [PubMed]
- Shih, T.; Peneva, D.; Xu, X.; Sutton, A.; Triche, E.; Ehrenkranz, R.A.; Paidas, M.; Stevens, W. The Rising Burden of Preeclampsia in the United States Impacts Both Maternal and Child Health. Am. J. Perinatol. 2016, 33, 329–338. [Google Scholar] [CrossRef]
- Wallis, A.B.; Saftlas, A.F.; Hsia, J.; Atrash, H.K. Secular trends in the rates of preeclampsia, eclampsia, and gestational hypertension, United States, 1987–2004. Am. J. Hypertens 2008, 21, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Stevens, W.; Shih, T.; Incerti, D.; Ton, T.G.N.; Lee, H.C.; Peneva, D.; Macones, G.A.; Sibai, B.M.; Jena, A.B. Short-term costs of preeclampsia to the United States health care system. Am. J. Obs. Gynecol. 2017, 217, 237–248. [Google Scholar] [CrossRef]
- Duckitt, K.; Harrington, D. Risk factors for pre-eclampsia at antenatal booking: Systematic review of controlled studies. BMJ 2005, 330, 565. [Google Scholar] [CrossRef]
- Ghosh, G.; Grewal, J.; Männistö, T.; Mendola, P.; Chen, Z.; Xie, Y.; Laughon, S.K. Racial/ethnic differences in pregnancy-related hypertensive disease in nulliparous women. Ethn. Dis. 2014, 24, 283–289. [Google Scholar]
- Zhang, M.; Wan, P.; Ng, K.; Singh, K.; Cheng, T.H.; Velickovic, I.; Dalloul, M.; Wlody, D. Preeclampsia Among African American Pregnant Women: An Update on Prevalence, Complications, Etiology, and Biomarkers. Obs. Gynecol. Surv. 2020, 75, 111–120. [Google Scholar] [CrossRef]
- Shahul, S.; Tung, A.; Minhaj, M.; Nizamuddin, J.; Wenger, J.; Mahmood, E.; Mueller, A.; Shaefi, S.; Scavone, B.; Kociol, R.D.; et al. Racial Disparities in Comorbidities, Complications, and Maternal and Fetal Outcomes in Women With Preeclampsia/eclampsia. Hypertens. Pregnancy 2015, 34, 506–515. [Google Scholar] [CrossRef]
- Bezerra, P.C.; Leão, M.D.; Queiroz, J.W.; Melo, E.M.; Pereira, F.V.; Nóbrega, M.H.; Jeronimo, A.K.; Ferreira, L.C.; Jerônimo, S.M.; de Araújo, A.C. Family history of hypertension as an important risk factor for the development of severe preeclampsia. Acta Obs. Gynecol. Scand. 2010, 89, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Williams, M.A.; Leisenring, W.M.; Sorensen, T.K.; Frederick, I.O.; Dempsey, J.C.; Luthy, D.A. Family history of hypertension and type 2 diabetes in relation to preeclampsia risk. Hypertension 2003, 41, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Cho, G.J.; Jung, U.S.; Sim, J.Y.; Lee, Y.J.; Bae, N.Y.; Choi, H.J.; Park, J.H.; Kim, H.-J.; Oh, M.-J. Is preeclampsia itself a risk factor for the development of metabolic syndrome after delivery? Obs. Gynecol. Sci. 2019, 62, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, K.K.; Borowski, K.S.; Parikh, N.I.; Saftlas, A.F. Pregnancy Complications and the Risk of Metabolic Syndrome for the Offspring. Curr. Cardiovasc. Risk Rep. 2013, 7, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Muijsers, H.E.C.; Roeleveld, N.; van der Heijden, O.W.H.; Maas, A.H.E.M. Consider Preeclampsia as a First Cardiovascular Event. Curr. Cardiovasc. Risk Rep. 2019, 13, 21. [Google Scholar] [CrossRef]
- Lu, H.Q.; Hu, R. Lasting Effects of Intrauterine Exposure to Preeclampsia on Offspring and the Underlying Mechanism. AJP Rep. 2019, 9, e275–e291. [Google Scholar] [CrossRef]
- Anderson, C.M. Preeclampsia: Exposing future cardiovascular risk in mothers and their children. J. Obs. Gynecol. Neonatal. Nurs. 2007, 36, 3–8. [Google Scholar] [CrossRef]
- Redman, C.W.G.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast stress in preeclampsia: The convergence point for multiple pathways. Am. J. Obstet. Gynecol. 2021. [Google Scholar] [CrossRef]
- Roberts, J.M.; Rich-Edwards, J.W.; McElrath, T.F.; Garmire, L.; Myatt, L. Subtypes of Preeclampsia: Recognition and Determining Clinical Usefulness. Hypertension 2021, 77, 1430–1441. [Google Scholar] [CrossRef]
- Leavey, K.; Benton, S.J.; Grynspan, D.; Kingdom, J.C.; Bainbridge, S.A.; Cox, B.J. Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertension 2016, 68, 137–147. [Google Scholar] [CrossRef]
- Phipps, E.; Prasanna, D.; Brima, W.; Jim, B. Preeclampsia: Updates in Pathogenesis, Definitions, and Guidelines. Clin. J. Am. Soc. Nephrol. 2016, 11, 1102–1113. [Google Scholar] [CrossRef]
- Staff, A.C. The two-stage placental model of preeclampsia: An update. J. Reprod. Immunol. 2019, 134–135, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Escudero, C. The placenta in preeclampsia. Pregnancy Hypertens. 2012, 2, 72–83. [Google Scholar] [CrossRef]
- Fisher, S.J. Why is placentation abnormal in preeclampsia? Am. J. Obstet. Gynecol. 2015, 213, S115–S122. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gomez, T.; Quiñonero, A.; Dominguez, F.; Rubert, L.; Perales, A.; Hajjar, K.A.; Simon, C. Preeclampsia: A defect in decidualization is associated with deficiency of Annexin A2. Am. J. Obs. Gynecol. 2020, 222, 376–376.e371. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gomez, T.; Dominguez, F.; Quiñonero, A.; Diaz-Gimeno, P.; Kapidzic, M.; Gormley, M.; Ona, K.; Padilla-Iserte, P.; McMaster, M.; Genbacev, O.; et al. Defective decidualization during and after severe preeclampsia reveals a possible maternal contribution to the etiology. Proc. Natl. Acad. Sci. USA 2017, 114, E8468–E8477. [Google Scholar] [CrossRef]
- Ng, S.-W.; Norwitz, G.A.; Pavlicev, M.; Tilburgs, T.; Simón, C.; Norwitz, E.R. Endometrial Decidualization: The Primary Driver of Pregnancy Health. Int. J. Mol. Sci. 2020, 21, 4092. [Google Scholar] [CrossRef]
- Garrido-Gómez, T.; Castillo-Marco, N.; Cordero, T.; Simón, C. Decidualization resistance in the origin of preeclampsia. Am. J. Obs. Gynecol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aranguren, L.C.; Prada, C.E.; Riaño-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef]
- Okada, H.; Tsuzuki, T.; Murata, H. Decidualization of the human endometrium. Reprod. Med. Biol. 2018, 17, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Baergen, R.N. Manual of Pathology of the Human Placenta; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Wang, Y.; Zhao, S. Cell Types of the Placenta. In Vascular Biology of the Placenta; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Knight, M.; Redman, C.W.G.; Linton, E.A.; Sargent, I.L. Shedding of syncytiotrophoblast microvilli into the maternal circulation in pre-eclamptic pregnancies. BJOG Int. J. Obstet. Gynaecol. 1998, 105, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Cockell, A.P.; Learmont, J.G.; Smárason, A.K.; Redman, C.W.G.; Sargent, I.L.; Poston, L. Human placental syncytiotrophoblast microvillous membranes impair maternal vascular endothelial function. BJOG Int. J. Obstet. Gynaecol. 1997, 104, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Brkić, J.; Liu, M.; Fu, G.; Peng, C.; Wang, Y.L. Placental trophoblast cell differentiation: Physiological regulation and pathological relevance to preeclampsia. Mol. Asp. Med. 2013, 34, 981–1023. [Google Scholar] [CrossRef] [PubMed]
- Pollheimer, J.; Vondra, S.; Baltayeva, J.; Beristain, A.G.; Knöfler, M. Regulation of Placental Extravillous Trophoblasts by the Maternal Uterine Environment. Front. Immunol. 2018, 9, 2597. [Google Scholar] [CrossRef]
- Lyall, F.; Bulmer, J.N.; Duffie, E.; Cousins, F.; Theriault, A.; Robson, S.C. Human trophoblast invasion and spiral artery transformation: The role of PECAM-1 in normal pregnancy, preeclampsia, and fetal growth restriction. Am. J. Pathol. 2001, 158, 1713–1721. [Google Scholar] [CrossRef]
- Briones, M.A. Chapter 131—General Overview of the Hypercoaguable State In Transfusion Medicine and Hemostasis; Hillyer, C.D., Shaz, B.H., Zimring, J.C., Abshire, T.C., Eds.; Academic Press: San Diego, CA, USA, 2009; pp. 677–679. [Google Scholar]
- Cheng, D.; Jiang, S.; Chen, J.; Li, J.; Ao, L.; Zhang, Y. Upregulated long noncoding RNA Linc00261 in pre-eclampsia and its effect on trophoblast invasion and migration via regulating miR-558/TIMP4 signaling pathway. J. Cell. Biochem. 2019, 120, 13243–13253. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, F.; Xiao, X.; Xie, F.; Tao, D.; Huang, C.; Liu, D.; Wang, M.; Wang, L.; Zeng, F.; et al. CircHIPK3 sponges miR-558 to suppress heparanase expression in bladder cancer cells. EMBO Rep. 2017, 18, 1646–1659. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xia, L.; Dong, L.; Wang, J.; Xiao, Q.; Yu, X.; Zhu, H. CircHIPK3 Promotes Gemcitabine (GEM) Resistance in Pancreatic Cancer Cells by Sponging miR-330-5p and Targets RASSF1. Cancer Manag. Res. 2020, 12, 921–929. [Google Scholar] [CrossRef]
- Lykoudi, A.; Kolialexi, A.; Lambrou, G.I.; Braoudaki, M.; Siristatidis, C.; Papaioanou, G.K.; Tzetis, M.; Mavrou, A.; Papantoniou, N. Dysregulated placental microRNAs in Early and Late onset Preeclampsia. Placenta 2018, 61, 24–32. [Google Scholar] [CrossRef]
- Oujo, B.; Perez-Barriocanal, F.; Bernabeu, C.; Lopez-Novoa, J. Membrane and Soluble Forms of Endoglin in Preeclampsia. Curr. Mol. Med. 2013, 13, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Zheng, L.; Song, H.; Jiao, W.; Li, D.; Fang, E.; Wang, X.; Mei, H.; Pu, J.; Huang, K.; et al. microRNA-558 facilitates the expression of hypoxia-inducible factor 2 alpha through binding to 5’-untranslated region in neuroblastoma. Oncotarget 2016, 7, 40657–40673. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.M.; Li, Q.; Jiang, W.; Xiong, X.; Li, H.Y.; Zhao, J.L.; Qi, H.B. LAMA5 promotes human umbilical vein endothelial cells migration, proliferation, and angiogenesis and is decreased in preeclampsia. J. Matern. Fetal Neonatal Med. 2020, 33, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, L.; Jia, J.; Ye, L.; Wang, Y.; Zhou, B.; Zhou, R. CircHIPK3 is decreased in preeclampsia and affects migration, invasion, proliferation, and tube formation of human trophoblast cells. Placenta 2019, 85, 1–8. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, M.; Chen, C.; Gong, W.; Yin, Z.; Li, H.; Fan, J.; Zhang, X.A.; Wang, D.W.; Zuo, H. MiR-124 aggravates failing hearts by suppressing CD151-facilitated angiogenesis in heart. Oncotarget 2018, 9, 14382–14396. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Huang, J.; Su, Y.; Wang, F.; Kong, H.; Xin, H. Overexpression of tissue factor pathway inhibitor 2 attenuates trophoblast proliferation and invasion in preeclampsia. Hum. Cell 2020. [Google Scholar] [CrossRef]
- Han, C.; Seebacher, N.A.; Hornicek, F.J.; Kan, Q.; Duan, Z. Regulation of microRNAs function by circular RNAs in human cancer. Oncotarget 2017, 8, 64622–64637. [Google Scholar] [CrossRef]
- Babicheva, A.; McDermott, K.M.; Williams, S.C.; Yee, A.M.; Dash, S.; Rodriquez, M.; Ingabire, N.; Makino, A.; Yuan, J.X.-J. Pathogenic and Therapeutic Role of MicroRNA in Pulmonary Arterial Hypertension. In Diagnosis and Treatment of Pulmonary Hypertension: From Bench to Bedside; Fukumoto, Y., Ed.; Springer: Singapore, 2017; pp. 31–54. [Google Scholar]
- Albers, R.E.; Kaufman, M.R.; Natale, B.V.; Keoni, C.; Kulkarni-Datar, K.; Min, S.; Williams, C.R.; Natale, D.R.C.; Brown, T.L. Trophoblast-specific expression of hif-1α results in preeclampsia-like symptoms and fetal growth restriction. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Rathmell, W.K.; Chen, S. VHL inactivation in renal cell carcinoma: Implications for diagnosis, prognosis and treatment. Expert Rev. Anticancer Ther. 2008, 8, 63–73. [Google Scholar] [CrossRef][Green Version]
- Harati-Sadegh, M.; Kohan, L.; Teimoori, B.; Mehrabani, M.; Salimi, S. The association of the placental Hypoxia-inducible factor1-α polymorphisms and HIF1-α mRNA expression with preeclampsia. Placenta 2018, 67, 31–37. [Google Scholar] [CrossRef]
- Gupta, S.K.; Malhotra, S.S.; Malik, A.; Verma, S.; Chaudhary, P. Cell Signaling Pathways Involved During Invasion and Syncytialization of Trophoblast Cells. Am. J. Reprod. Immunol. 2016, 75, 361–371. [Google Scholar] [CrossRef]
- O’Connor, B.B.; Pope, B.D.; Peters, M.M.; Ris-Stalpers, C.; Parker, K.K. The role of extracellular matrix in normal and pathological pregnancy: Future applications of microphysiological systems in reproductive medicine. Exp. Biol. Med. 2020, 245, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular trophoblast invasion: Implications for the pathogenesis of intrauterine growth retardation and preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Matthiesen, L.; Berg, G.; Ernerudh, J.; Ekerfelt, C.; Jonsson, Y.; Sharma, S. Immunology of preeclampsia. Chem. Immunol. Allergy 2005, 89, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Harmon, A.C.; Cornelius, D.C.; Amaral, L.M.; Faulkner, J.L.; Cunningham, M.W., Jr.; Wallace, K.; LaMarca, B. The role of inflammation in the pathology of preeclampsia. Clin. Sci. 2016, 130, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Aneman, I.; Pienaar, D.; Suvakov, S.; Simic, T.P.; Garovic, V.D.; McClements, L. Mechanisms of Key Innate Immune Cells in Early- and Late-Onset Preeclampsia. Front. Immunol. 2020, 11, 1864. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, H. Role of Decidual Natural Killer Cells in Human Pregnancy and Related Pregnancy Complications. Front. Immunol. 2021, 12, 728291. [Google Scholar] [CrossRef]
- Geldenhuys, J.; Rossouw, T.M.; Lombaard, H.A.; Ehlers, M.M.; Kock, M.M. Disruption in the regulation of immune responses in the placental subtype of preeclampsia. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Whitley, G.S.; Cartwright, J.E. Trophoblast-mediated spiral artery remodelling: A role for apoptosis. J. Anat. 2009, 215, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Ishida, J.; Aoki, I.; Fukamizu, A. Pathophysiology of placentation abnormalities in pregnancy-induced hypertension. Vasc. Health Risk Manag. 2008, 4, 1301–1313. [Google Scholar] [CrossRef]
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef] [PubMed]
- Goulopoulou, S.; Davidge, S.T. Molecular mechanisms of maternal vascular dysfunction in preeclampsia. Trends Mol. Med. 2015, 21, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Prearo Moço, N.; Ribeiro de Andrade Ramos, B.; de Castro Silva, M.; Polettini, J.; Menon, R.; Guimarães da Silva, M. Spontaneous Prematurity, Innate Immune System, and Oxidative Stress at the Maternal-Fetal Interface: An Overview. In Translational Studies on Inflammation; IntechOpen: London, UK, 2020. [Google Scholar]
- Ozeki, A.; Tani, K.; Takahashi, H.; Suzuki, H.; Nagayama, S.; Hirashima, C.; Iwata, H.; Kuwayama, T.; Ohkuchi, A.; Shirasuna, K. Preeclamptic patient-derived circulating cell-free DNA activates the production of inflammatory cytokines via toll-like receptor 9 signalling in the human placenta. J. Hypertens. 2019, 37, 2452–2460. [Google Scholar] [CrossRef]
- Cotechini, T.; Komisarenko, M.; Sperou, A.; Macdonald-Goodfellow, S.; Adams, M.A.; Graham, C.H. Inflammation in rat pregnancy inhibits spiral artery remodeling leading to fetal growth restriction and features of preeclampsia. J. Exp. Med. 2014, 211, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.H.; Chiasson, V.L.; Mahajan, A.; Young, K.J.; Mitchell, B.M. Toll-like receptor 3 activation during pregnancy elicits preeclampsia-like symptoms in rats. Am. J. Hypertens 2009, 22, 1314–1319. [Google Scholar] [CrossRef]
- Xue, P.; Fan, W.; Diao, Z.; Li, Y.; Kong, C.; Dai, X.; Peng, Y.; Chen, L.; Wang, H.; Hu, Y.; et al. Up-regulation of PTEN via LPS/AP-1/NF-κB pathway inhibits trophoblast invasion contributing to preeclampsia. Mol. Immunol. 2020, 118, 182–190. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Yang, X.; Li, Y.; Huang, D.; Xu, X.; Yang, W.; Dai, Y.; Zhang, H.; Chen, Z.; Cheng, W. TLR9 (toll-like receptor 9) agonist suppresses angiogenesis by differentially regulating VEGFA (Vascular Endothelial Growth Factor A) and sFLT1 (Soluble Vascular Endothelial Growth Factor Receptor 1) in Preeclampsia. Hypertension 2018, 71, 671–680. [Google Scholar] [CrossRef]
- Kalkunte, S.; Boij, R.; Norris, W.; Friedman, J.; Lai, Z.; Kurtis, J.; Lim, K.-H.; Padbury, J.F.; Matthiesen, L.; Sharma, S. Sera from preeclampsia patients elicit symptoms of human disease in mice and provide a basis for an in vitro predictive assay. Am. J. Pathol. 2010, 177, 2387–2398. [Google Scholar] [CrossRef]
- Cheng, S.-B.; Sharma, S. Interleukin-10: A pleiotropic regulator in pregnancy. Am. J. Reprod. Immunol. 2015, 73, 487–500. [Google Scholar] [CrossRef]
- Care, A.S.; Bourque, S.L.; Morton, J.S.; Hjartarson, E.P.; Robertson, S.A.; Davidge, S.T. Reduction in regulatory t cells in early pregnancy causes uterine artery dysfunction in mice. Hypertension 2018, 72, 177–187. [Google Scholar] [CrossRef]
- Gierman, L.M.; Silva, G.B.; Pervaiz, Z.; Rakner, J.J.; Mundal, S.B.; Thaning, A.J.; Nervik, I.; Elschot, M.; Mathew, S.; Thomsen, L.C.V.; et al. TLR3 expression by maternal and fetal cells at the maternal-fetal interface in normal and preeclamptic pregnancies. J. Leukoc. Biol. 2021, 109, 173–183. [Google Scholar] [CrossRef]
- Kalkunte, S.; Nevers, T.; Norris, W.E.; Sharma, S. Vascular IL-10: A protective role in preeclampsia. J. Reprod. Immunol. 2011, 88, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Thaxton, J.E.; Sharma, S. Interleukin-10: A multi-faceted agent of pregnancy. Am. J. Reprod. Immunol. 2010, 63, 482–491. [Google Scholar] [CrossRef]
- Blois, S.M.; Freitag, N.; Tirado-González, I.; Cheng, S.B.; Heimesaat, M.M.; Bereswill, S.; Rose, M.; Conrad, M.L.; Barrientos, G.; Sharma, S. NK cell-derived IL-10 is critical for DC-NK cell dialogue at the maternal-fetal interface. Sci. Rep. 2017, 7, 2189. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, P.A.; Pilmore, H.L.; Simmons, L.A.; Painter, D.M. A Deficiency of Placental IL-10 in preeclampsia. J. Immunol. 1999, 163, 3491–3495. [Google Scholar]
- Chatterjee, P.; Chiasson, V.L.; Kopriva, S.E.; Young, K.J.; Chatterjee, V.; Jones, K.A.; Mitchell, B.M. Interleukin 10 deficiency exacerbates toll-like receptor 3-induced preeclampsia-like symptoms in mice. Hypertension 2011, 58, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Abu-Raya, B.; Michalski, C.; Sadarangani, M.; Lavoie, P.M. Maternal Immunological Adaptation During Normal Pregnancy. Front. Immunol. 2020, 11, 575197. [Google Scholar] [CrossRef]
- Corthay, A. How do regulatory t cells work? Scand. J. Immunol. 2009, 70, 326–336. [Google Scholar] [CrossRef]
- Zhao, H.; Liao, X.; Kang, Y. Tregs: Where we are and what comes next? Front. Immunol. 2017, 8, 1578. [Google Scholar] [CrossRef]
- Robertson, S.A.; Green, E.S.; Care, A.S.; Moldenhauer, L.M.; Prins, J.R.; Louise Hull, M.; Barry, S.C.; Dekker, G. Therapeutic potential of regulatory T cells in preeclampsia-opportunities and challenges. Front. Immunol. 2019, 10, 478. [Google Scholar] [CrossRef]
- Wang, A.; Rana, S.; Karumanchi, S.A. Preeclampsia: The role of angiogenic factors in its pathogenesis. Physiology 2009, 24, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Oh, M.-J.; Jung, J.W.; Lim, J.-E.; Seol, H.-J.; Lee, K.-J.; Kim, H.-J. The levels of circulating vascular endothelial growth factor and soluble Flt-1 in pregnancies complicated by preeclampsia. J. Korean Med. Sci. 2007, 22, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Karumanchi, S.A. Angiogenic factors and preeclampsia. Semin. Nephrol. 2011, 31, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Smani, T.; Gómez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.M.; Rosado, J.A. Trp channels in angiogenesis and other endothelial functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef]
- Geva, E.; Ginzinger, D.G.; Zaloudek, C.J.; Moore, D.H.; Byrne, A.; Jaffe, R.B. Human placental vascular development: Vasculogenic and angiogenic (branching and nonbranching) transformation is regulated by vascular endothelial growth factor-a, angiopoietin-1, and angiopoietin-2. J. Clin. Endocrinol. Metab. 2002, 87, 4213–4224. [Google Scholar] [CrossRef]
- Grummer, M.A.; Sullivan, J.A.; Magness, R.R.; Bird, I.M. Vascular endothelial growth factor acts through novel, pregnancy-enhanced receptor signalling pathways to stimulate endothelial nitric oxide synthase activity in uterine artery endothelial cells. Biochem. J. 2009, 417, 501–511. [Google Scholar] [CrossRef]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor–Associated Hypertension and Vascular Disease. Hypertension 2018, 71, E1–E8. [Google Scholar] [CrossRef]
- Chau, K.; Hennessy, A.; Makris, A. Placental growth factor and pre-eclampsia. J. Hum. Hypertens. 2017, 31, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Kenyon, A.; Adamson, D. Physiology. In Basic Science in Obstetrics and Gynaecology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 173–230. [Google Scholar]
- Dewerchin, M.; Carmeliet, P. PlGF: A multitasking cytokine with disease-restricted activity. Cold Spring Harb. Perspect. Med. 2012, 2, a011056. [Google Scholar] [CrossRef]
- Lobmaier, S.M.; Figueras, F.; Mercade, I.; Crovetto, F.; Peguero, A.; Parra-Saavedra, M.; Ortiz, J.U.; Crispi, F.; Gratacós, E. Levels of maternal serum angiogenic factors in third-trimester normal pregnancies: Reference ranges, influence of maternal and pregnancy factors and fetoplacental Doppler indices. Fetal. Diagn. 2014, 36, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Ng, Q.J.; Han, J.Y.; Saffari, S.E.; Yeo, G.S.-H.; Chern, B.S.M.; Tan, K.H. Longitudinal circulating placental growth factor (PlGF) and soluble FMS-like tyrosine kinase-1 (sFlt-1) concentrations during pregnancy in Asian women: A prospective cohort study. BMJ Open 2019, 9, e028321. [Google Scholar] [CrossRef]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating Angiogenic Factors and the Risk of Preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef]
- Benschop, L.; Schalekamp-Timmermans, S.; Broere-Brown, Z.A.; Roeters van Lennep, J.E.; Jaddoe, V.W.V.; Roos-Hesselink, J.W.; Ikram, M.K.; Steegers, E.A.P.; Roberts, J.M.; Gandley, R.E. Placental Growth Factor as an Indicator of Maternal Cardiovascular Risk After Pregnancy. Circulation 2019, 139, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Rajakumar, A. Preeclampsia and soluble fms-like tyrosine kinase 1. J. Clin. Endocrinol. Metab. 2009, 94, 2252–2254. [Google Scholar] [CrossRef]
- Thomas, C.P.; Andrews, J.I.; Raikwar, N.S.; Kelley, E.A.; Herse, F.; Dechend, R.; Golos, T.G.; Liu, K.Z. A recently evolved novel trophoblast-enriched secreted form of fms-like tyrosine kinase-1 variant is up-regulated in hypoxia and preeclampsia. J. Clin. Endocrinol. Metab. 2009, 94, 2524–2530. [Google Scholar] [CrossRef] [PubMed]
- Adamson, S.L. sFLT1 in preeclampsia: Trophoblast defense against a decidual VEGFA barrage? J. Clin. Investig. 2014, 124, 4690–4692. [Google Scholar] [CrossRef][Green Version]
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N. Engl. J. Med. 2016, 374, 13–22. [Google Scholar] [CrossRef]
- Sela, S.; Itin, A.; Natanson-Yaron, S.; Greenfield, C.; Goldman-Wohl, D.; Yagel, S.; Keshet, E. A novel human-specific soluble vascular endothelial growth factor receptor 1: Cell type-specific splicing and implications to vascular endothelial growth factor homeostasis and preeclampsia. Circ. Res. 2008, 102, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, T.; Fujii, T.; Kusumi, M.; Zou, L.; Yamashita, T.; Osuga, Y.; Momoeda, M.; Kozuma, S.; Taketani, Y. Cytotrophoblasts up-regulate soluble fms-like tyrosine kinase-1 expression under reduced oxygen: An implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology 2004, 145, 4838–4845. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gu, B.; Zhang, Y.; Lewis, D.F.; Wang, Y. Hypoxia-induced increase in soluble Flt-1 production correlates with enhanced oxidative stress in trophoblast cells from the human placenta. Placenta 2005, 26, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, F.; Cusimano, M.D.; Scheithauer, B.W.; Rotondo, F.; Fazio, A.; Yousef, G.M.; Syro, L.V.; Kovacs, K.; Lloyd, R.V. Endoglin (CD105): A review of its role in angiogenesis and tumor diagnosis, progression and therapy. Anticancer Res. 2011, 31, 2283–2290. [Google Scholar]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.I.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- St-Jacques, S.; Forte, M.; Lye, S.J.; Letarte, M. Localization of endoglin, a transforming growth factor-β binding protein, and of CD44 and integrins in placenta during the first trimester of pregnancy. Biol. Reprod. 1994, 51, 405–413. [Google Scholar] [CrossRef]
- Alam, S.; Ahmad, S.; Zafeer, F.; Rizvi, A.A.; Gulati, R.; Rashid, S. Role of TGF-Β1 in The Pathogenesis of Pre-Eclampsia. Ann. Int. Med. Dent. Res. 2017, 3, 1. [Google Scholar] [CrossRef]
- Caniggia, I.; Taylor, C.V.; Ritchie, J.W.K.; Lye, S.J.; Letarte, M. Endoglin regulates trophoblast differentiation along the invasive pathway in human placental villous explants. Endocrinology 1997, 138, 4977–4988. [Google Scholar] [CrossRef]
- Ayatollahi, M.; Geramizadeh, B.; Samsami, A. Transforming growth factor beta-1 influence on fetal allografts during pregnancy. Transplant. Proc. 2005, 37, 4603–4604. [Google Scholar] [CrossRef]
- Nikuei, P.; Rajaei, M.; Malekzadeh, K.; Nejatizadeh, A.; Mohseni, F.; Atashabparvar, A. Accuracy of soluble endoglin for diagnosis of preeclampsia and its severity. Iran. Biomed. J. 2017, 21, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Powe, C.E.; Levine, R.J.; Karumanchi, S.A. Preeclampsia, a disease of the maternal endothelium: The role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 2011, 123, 2856–2869. [Google Scholar] [CrossRef]
- Bell, M.J.; Roberts, J.M.; Founds, S.A.; Jeyabalan, A.; Terhorst, L.; Conley, Y.P. Variation in endoglin pathway genes is associated with preeclampsia: A case-control candidate gene association study. BMC Pregnancy Childbirth 2013, 13. [Google Scholar] [CrossRef]
- Jerkic, M.; Rivas-Elena, J.V.; Prieto, M.; Carrón, R.; Sanz-Rodríguez, F.; Pérez-Barriocanal, F.; Rodríguez-Barbero, A.; Bernabéu, C.; López-Novoa, J.M. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. 2004, 18, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Kotani, T.; Shibata, K.; Matsumura, H.; Tsuda, H.; Sumigama, S.; Yamamoto, E.; Iwase, A.; Senga, T.; Kikkawa, F. The loss of endoglin promotes the invasion of extravillous trophoblasts. Endocrinology 2011, 152, 4386–4394. [Google Scholar] [CrossRef]
- Williams, P.J.; Mistry, H.D.; Innes, B.A.; Bulmer, J.N.; Broughton Pipkin, F. Expression of AT1R, AT2R and AT4R and Their Roles in Extravillous Trophoblast Invasion in the Human. Placenta 2010, 31, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.F.; Sun, Y.L.; Hamet, P.; Inagami, T. The angiotensin II type 1 receptor and receptor-associated proteins. Cell Res. 2001, 11, 165–180. [Google Scholar] [CrossRef]
- LaMarca, B. Endothelial dysfunction. An important mediator in the pathophysiology of hypertension during pre-eclampsia. Minerva Ginecol. 2012, 64, 309–320. [Google Scholar]
- LaMarca, B.; Wallukat, G.; Llinas, M.; Herse, F.; Dechend, R.; Granger, J.P. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension 2008, 52, 1168–1172. [Google Scholar] [CrossRef]
- LaMarca, B.; Parrish, M.; Ray, L.F.; Murphy, S.R.; Roberts, L.; Glover, P.; Wallukat, G.; Wenzel, K.; Cockrell, K.; Martin, J.N., Jr.; et al. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: Role of endothelin-1. Hypertension 2009, 54, 905–909. [Google Scholar] [CrossRef]
- Parrish, M.R.; Murphy, S.R.; Rutland, S.; Wallace, K.; Wenzel, K.; Wallukat, G.; Keiser, S.; Ray, L.F.; Dechend, R.; Martin, J.N.; et al. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am. J. Hypertens. 2010, 23, 911–916. [Google Scholar] [CrossRef]
- Bezerra Maia E Holanda Moura, S.; Marques Lopes, L.; Murthi, P.; da Silva Costa, F. Prevention of preeclampsia. J. Pregnancy 2012, 2012, 435090. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.M.; Garovic, V.D. Drug treatment of hypertension in pregnancy. Drugs 2014, 74, 283–296. [Google Scholar] [CrossRef]
- Duhig, K.; Vandermolen, B.; Shennan, A. Recent advances in the diagnosis and management of pre-eclampsia. F1000Res 2018, 7, 242. [Google Scholar] [CrossRef]
- Spradley, F.T.; Tan, A.Y.; Joo, W.S.; Daniels, G.; Kussie, P.; Karumanchi, S.A.; Granger, J.P. Placental growth factor administration abolishes placental ischemia-induced hypertension. Hypertension 2016, 67, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Ren, Z.; Possomato-Vieira, J.S.; Khalil, R.A. Restoring placental growth factor-soluble fms-like tyrosine kinase-1 balance reverses vascular hyper-reactivity and hypertension in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R505–R521. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, R.; Hagmann, H.; Schaarschmidt, W.; Roth, B.; Cingoez, T.; Karumanchi, S.A.; Wenger, J.; Lucchesi, K.J.; Tamez, H.; Lindner, T.; et al. Removal of soluble fms-like tyrosine kinase-1 by dextran sulfate apheresis in preeclampsia. J. Am. Soc. Nephrol. 2016, 27, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Bridges, J.P.; Gilbert, J.S.; Colson, D.; Gilbert, S.A.; Dukes, M.P.; Ryan, M.J.; Granger, J.P. Oxidative stress contributes to soluble fms-like tyrosine kinase-1 induced vascular dysfunction in pregnant rats. Am. J. Hypertens. 2009, 22, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M. The Endothelium: Part 1: Multiple Functions of the Endothelial Cells- Focus on Endothelium-Derived Vasoactive Mediators. In Colloquium Series on Integrated Systems Physiology; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2011. [Google Scholar]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef]
- Marti, C.N.; Gheorghiade, M.; Kalogeropoulos, A.P.; Georgiopoulou, V.V.; Quyyumi, A.A.; Butler, J. Endothelial dysfunction, arterial stiffness, and heart failure. J. Am. Coll. Cardiol. 2012, 60, 1455–1469. [Google Scholar] [CrossRef]
- Sukriti, S.; Tauseef, M.; Yazbeck, P.; Mehta, D. Mechanisms regulating endothelial permeability. Pulm. Circ. 2014, 4, 535–551. [Google Scholar] [CrossRef]
- Tseng, E.; Yee Teoh, S.S.; Wang, Y.; Nie, G. Elevated protease HtrA4 in the maternal circulation of preeclampsia may contribute to endothelial barrier disruption by cleaving key junctional protein VE-cadherin. Placenta 2019, 76, 51–53. [Google Scholar] [CrossRef]
- Wang, Y.; Lewis, D.F.; Alexander, J.S.; Granger, D.N. Endothelial barrier function in preeclampsia. Front. Biosci. 2007, 12, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Hayman, R.; Warren, A.; Brockelsby, J.; Johnson, I.; Baker, P. Plasma from women with pre-eclampsia induces an in vitro alteration in the endothelium-dependent behaviour of myometrial resistance arteries. BJOG Int. J. Obstet. Gynaecol. 2000, 107, 108–115. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.J.; Derayunan, A.; Hader, S.; Lohr, N.; Beyer, A.; Gutterman, D. Impaired Microvascular Endothelial Function in Preeclampsia. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Chambers, J.C.; Fusi, L.; Malik, I.S.; Haskard, D.O.; De Swiet, M.; Kooner, J.S. Association of maternal endothelial dysfunction with preeclampsia. J. Am. Med. Assoc. 2001, 285, 1607–1612. [Google Scholar] [CrossRef]
- Sandgren, J.A.; Deng, G.; Linggonegoro, D.W.; Scroggins, S.M.; Perschbacher, K.J.; Nair, A.R.; Nishimura, T.E.; Zhang, S.Y.; Agbor, L.N.; Wu, J.; et al. Arginine vasopressin infusion is sufficient to model clinical features of preeclampsia in mice. JCI Insight 2018, 3, e99403. [Google Scholar] [CrossRef] [PubMed]
- Vanwijk, M.J.; Kublickiene, K.; Boer, K.; VanBavel, E. Vascular function in preeclampsia. Cardiovasc. Res. 2000, 47, 38–48. [Google Scholar] [CrossRef]
- Schrier, R.W. Pathogenesis of sodium and water retention in high-output and low-output cardiac failure, nephrotic syndrome, cirrhosis, and pregnancy (1). N. Engl. J. Med. 1988, 319, 1065–1072. [Google Scholar] [CrossRef]
- Tkachenko, O.; Shchekochikhin, D.; Schrier, R.W. Hormones and hemodynamics in pregnancy. Int. J. Endocrinol. Metab. 2014, 12, e14098. [Google Scholar] [CrossRef]
- Vricella, L.K. Emerging understanding and measurement of plasma volume expansion in pregnancy. Am. J. Clin. Nutr. 2017, 106, 1620S–1625S. [Google Scholar] [CrossRef] [PubMed]
- Silver, H.M.; Seebeck, M.; Carlson, R. Comparison of total blood volume in normal, preeclamptic, and nonproteinuric gestational hypertensive pregnancy by simultaneous measurement of red blood cell and plasma volumes. Am. J. Obs. Gynecol. 1998, 179, 87–93. [Google Scholar] [CrossRef]
- Salas, S.P.; Marshall, G.; Gutiérrez, B.L.; Rosso, P. Time Course of Maternal Plasma Volume and Hormonal Changes in Women With Preeclampsia or Fetal Growth Restriction. Hypertension 2006, 47, 203–208. [Google Scholar] [CrossRef]
- Soffronoff, E.C.; Kaufmann, B.M.; Connaughton, J.F. Intravascular volume determinations and fetal outcome in hypertensive diseases of pregnancy. Am. J. Obs. Gynecol. 1977, 127, 4–9. [Google Scholar] [CrossRef]
- Arias, F. Expansion of intravascular volume and fetal outcome in patients with chronic hypertension and pregnancy. Am. J. Obs. Gynecol. 1975, 123, 610–616. [Google Scholar] [CrossRef]
- Anthony, J.; Schoeman, L.K. Fluid management in pre-eclampsia. Obs. Med. 2013, 6, 100–104. [Google Scholar] [CrossRef]
- Uddin, M.N.; McLean, L.B.; Hunter, F.A.; Horvat, D.; Severson, J.; Tharakan, B.; Childs, E.W.; Puschett, J.B. Vascular leak in a rat model of preeclampsia. Am. J. Nephrol. 2009, 30, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Zhao, M.; Chen, Q.; Wang, Y.; Li, Y.; Kaitu’u-Lino, T.J.; Tong, S.; Nie, G. Human HtrA4 Expression Is Restricted to the Placenta, Is Significantly Up-Regulated in Early-Onset Preeclampsia, and High Levels of HtrA4 Cause Endothelial Dysfunction. J. Clin. Endocrinol. Metab. 2015, 100, E936–E945. [Google Scholar] [CrossRef]
- Wang, Y.; Gu, Y.; Lewis, D.F.; Alexander, J.S.; Granger, D.N. Elevated plasma chymotrypsin-like protease (chymase) activity in women with preeclampsia. Hypertens Pregnancy 2010, 29, 253–261. [Google Scholar] [CrossRef]
- Gu, Y.; Groome, L.J.; Alexander, J.S.; Wang, Y. PAR-2 triggers placenta-derived protease-induced altered VE-cadherin reorganization at endothelial junctions in preeclampsia. Placenta 2012, 33, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.L.; Shelton, R.L.; Athar, S. The osmoregulation of vasopressin. Kidney Int. 1976, 10, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Dunn, F.L.; Brennan, T.J.; Nelson, A.E.; Robertson, G.L. The role of blood osmolality and volume in regulating vasopressin secretion in the rat. J. Clin. Investig. 1973, 52, 3212–3219. [Google Scholar] [CrossRef]
- Santillan, M.K.; Santillan, D.A.; Scroggins, S.M.; Min, J.Y.; Sandgren, J.A.; Pearson, N.A.; Leslie, K.K.; Hunter, S.K.; Zamba, G.K.D.; Gibson-Corley, K.N.; et al. Vasopressin in preeclampsia: A novel very early human pregnancy biomarker and clinically relevant mouse model. Hypertension 2014, 64, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Lopes van Balen, V.A.; van Gansewinkel, T.A.G.; de Haas, S.; van Kuijk, S.M.J.; van Drongelen, J.; Ghossein-Doha, C.; Spaanderman, M.E.A. Physiological adaptation of endothelial function to pregnancy: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2017, 50, 697–708. [Google Scholar] [CrossRef]
- Choi, J.W.; Im, M.W.; Pai, S.H. Nitric oxide production increases during normal pregnancy and decreases in preeclampsia. Ann. Clin. Lab. Sci. 2002, 32, 257–263. [Google Scholar]
- Seligman, S.P.; Buyon, J.P.; Clancy, R.M.; Young, B.K.; Abramson, S.B. The role of nitric oxide in the pathogenesis of preeclampsia. Am. J. Obstet. Gynecol. 1994, 171, 944–948. [Google Scholar] [CrossRef]
- Lind, L. Endothelium-dependent Vasodilation in Hypertension: A Review. Blood Press. 2000, 9, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Boeldt, D.S.; Krupp, J.; Yi, F.-X.; Khurshid, N.; Shah, D.M.; Bird, I.M. Positive versus negative effects of VEGF165 on Ca2+ signaling and NO production in human endothelial cells. Am. J. Physiol. -Heart Circ. Physiol. 2016, 312, H173–H181. [Google Scholar] [CrossRef]
- Fleming, I.; Busse, R. Signal transduction of eNOS activation. Cardiovasc. Res. 1999, 43, 532–541. [Google Scholar] [CrossRef]
- Cale, J.M.; Bird, I.M. Dissociation of endothelial nitric oxide synthase phosphorylation and activity in uterine artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2006, 290. [Google Scholar] [CrossRef] [PubMed]
- Boeldt, D.S.; Yi, F.X.; Bird, I.M. eNOS activation and NO function: Pregnancy adaptive programming of capacitative entry responses alters nitric oxide (NO) output in vascular endothelium-new insights into eNOS regulation through adaptive cell signaling. J. Endocrinol. 2011, 210, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Boeldt, D.S.; Grummer, M.A.; Yi, F.X.; Magness, R.R.; Bird, I.M. Phosphorylation of Ser-279/282 and Tyr-265 positions on Cx43 as possible mediators of VEGF-165 inhibition of pregnancy-adapted Ca2+ burst function in ovine uterine artery endothelial cells. Mol. Cell. Endocrinol. 2015, 412, 73–84. [Google Scholar] [CrossRef][Green Version]
- Feliers, D.; Chen, X.; Akis, N.; Choudhury, G.G.; Madaio, M.; Kasinath, B.S. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef]
- Craici, I.M.; Wagner, S.J.; Weissgerber, T.L.; Grande, J.P.; Garovic, V.D. Advances in the pathophysiology of pre-eclampsia and related podocyte injury. Kidney Int. 2014, 86, 275–285. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Shen, X.; Kevil, C.G. A tale of two gases: NO and H2S, foes or friends for life? Redox Biol. 2013, 1, 313–318. [Google Scholar] [CrossRef]
- Naik, J.S.; Osmond, J.M.; Walker, B.R.; Kanagy, N.L. Hydrogen sulfide-induced vasodilation mediated by endothelial TRPV4 channels. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1437–H1444. [Google Scholar] [CrossRef]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Módis, K.; Panopoulos, P.; Asimakopoulou, A.; Gerö, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Xue, W.-L.; Wang, M.-J.; Zhou, Y.; Zhang, C.-C.; Sun, C.; Zhu, L.; Liang, K.; Chen, Y.; Tao, B.-B.; et al. H2S regulates endothelial nitric oxide synthase protein stability by promoting microRNA-455-3p expression. Sci. Rep. 2017, 7, 44807. [Google Scholar] [CrossRef]
- Bir, S.C.; Kolluru, G.K.; McCarthy, P.; Shen, X.; Pardue, S.; Pattillo, C.B.; Kevil, C.G. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1α and vascular endothelial growth factor-dependent angiogenesis. J. Am. Heart Assoc. 2012, 1, e004093. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A. Molecular mechanisms and therapeutic implications of the carbon monoxide/hmox1 and the hydrogen sulfide/CSE pathways in the prevention of pre-eclampsia and fetal growth restriction. Pregnancy Hypertens 2014, 4, 243–244. [Google Scholar] [CrossRef]
- Wang, K.; Ahmad, S.; Cai, M.; Rennie, J.; Fujisawa, T.; Crispi, F.; Baily, J.; Miller, M.R.; Cudmore, M.; Hadoke, P.W.; et al. Dysregulation of hydrogen sulfide producing enzyme cystathionine γ-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013, 127, 2514–2522. [Google Scholar] [CrossRef]
- Possomato-Vieira, J.S.; Palei, A.C.; Pinto-Souza, C.C.; Cavalli, R.; Dias-Junior, C.A.; Sandrim, V. Circulating levels of hydrogen sulphide negatively correlate to nitrite levels in gestational hypertensive and preeclamptic pregnant women. Clin. Exp. Pharm. Physiol. 2021, 48, 1224–1230. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, S.; Shen, X.; Glawe, J.; Kolluru, G.K.; Kevil, C.G. Nitric Oxide and Hydrogen Sulfide Regulation of Ischemic Vascular Growth and Remodeling. Compr. Physiol. 2019, 9, 1213–1247. [Google Scholar] [CrossRef]
- Holwerda, K.M.; Bos, E.M.; Rajakumar, A.; Ris-Stalpers, C.; van Pampus, M.G.; Timmer, A.; Erwich, J.J.; Faas, M.M.; van Goor, H.; Lely, A.T. Hydrogen sulfide producing enzymes in pregnancy and preeclampsia. Placenta 2012, 33, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Szabo, C.; Ichinose, F.; Ahmed, A.; Whiteman, M.; Papapetropoulos, A. The role of H2S bioavailability in endothelial dysfunction. Trends Pharm. Sci 2015, 36, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Sankaralingam, S.; Xu, H.; Davidge, S.T. Arginase contributes to endothelial cell oxidative stress in response to plasma from women with preeclampsia. Cardiovasc. Res. 2010, 85, 194–203. [Google Scholar] [CrossRef]
- Speer, P.D.; Powers, R.W.; Frank, M.P.; Harger, G.; Markovic, N.; Roberts, J.M. Elevated asymmetric dimethylarginine concentrations precede clinical preeclampsia, but not pregnancies with small-for-gestational-age infants. Am. J. Obstet. Gynecol. 2008, 198, 112.e111–112.e127. [Google Scholar] [CrossRef]
- Braekke, K.; Ueland, P.M.; Harsem, N.K.; Staff, A.C. Asymmetric Dimethylarginine in the Maternal and Fetal Circulation in Preeclampsia. Pediatric Res. 2009, 66, 411–415. [Google Scholar] [CrossRef]
- Davidge, S.T.; Baker, P.N.; Roberts, J.M. NOS expression is increased in endothelial cells exposed to plasma from women with preeclampsia. Am. J. Physiol. Heart Circ. Physiol. 1995, 269. [Google Scholar] [CrossRef]
- Aggarwal, P.K.; Chandel, N.; Jain, V.; Jha, V. The relationship between circulating endothelin-1, soluble fms-like tyrosine kinase-1 and soluble endoglin in preeclampsia. J. Hum. Hypertens. 2012, 26, 236–241. [Google Scholar] [CrossRef]
- Gant, N.F.; Daley, G.L.; Chand, S.; Whalley, P.J.; MacDonald, P.C. A study of angiotensin II pressor response throughout primigravid pregnancy. J. Clin. Investig. 1973, 52, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, T.F. Angiotensin, ACE-inhibitors and endothelial control of vasomotor tone. Basic Res. Cardiol. 1993, 88 (Suppl 1), 15–24. [Google Scholar] [CrossRef]
- Leaños-Miranda, A.; Inova, C.-G.; Méndez-Aguilar, F.; Molina-Pérez, C.J.; Ramírez-Valenzuela, K.L.; Sillas-Pardo, L.J.; Uraga-Camacho, N.C.; Isordia-Salas, I.; Berumen-Lechuga, M.G. Lower circulating angiotensin II levels are related to the severity of preeclampsia and its risk as disclosed by a specific bioassay. Medicine 2018, 97, e12498. [Google Scholar] [CrossRef] [PubMed]
- Stanhewicz, A.E.; Jandu, S.; Santhanam, L.; Alexander, L.M. Increased angiotensin II sensitivity contributes to microvascular dysfunction in women who have had preeclampsia. Hypertension 2017, 70, 382–389. [Google Scholar] [CrossRef]
- Jain, A. Endothelin-1: A key pathological factor in pre-eclampsia? Reprod. Biomed. Online 2012, 25, 443–449. [Google Scholar] [CrossRef]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Sudano, I.; Magagna, A.; Salvetti, A. Role of Endothelin in the Control of Peripheral Vascular Tone in Human Hypertension. Heart Fail. Rev. 2001, 6, 277–285. [Google Scholar] [CrossRef]
- Dechanet, C.; Fort, A.; Barbero-Camps, E.; Dechaud, H.; Richard, S.; Virsolvy, A. Endothelin-Dependent Vasoconstriction in Human Uterine Artery: Application to Preeclampsia. PLoS ONE 2011, 6, e16540. [Google Scholar] [CrossRef]
- Saleh, L.; Verdonk, K.; Visser, W.; van den Meiracker, A.H.; Danser, A.H.J. The emerging role of endothelin-1 in the pathogenesis of pre-eclampsia. Adv. Cardiovasc. Dis. 2016, 10, 282–293. [Google Scholar] [CrossRef]
- Alexander, B.T.; Rinewalt, A.N.; Cockrell, K.L.; Massey, M.B.; Bennett, W.A.; Granger, J.P. Endothelin type a receptor blockade attenuates the hypertension in response to chronic reductions in uterine perfusion pressure. Hypertension 2001, 37, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Thorin, E.; Webb, D.J. Endothelium-derived endothelin-1. Pflug. Arch. 2010, 459, 951–958. [Google Scholar] [CrossRef]
- Wynne, B.M.; Chiao, C.-W.; Webb, R.C. Vascular smooth muscle cell signaling mechanisms for contraction to angiotensin II and endothelin-1. J. Am. Soc. Hypertens 2009, 3, 84–95. [Google Scholar] [CrossRef]
- Hoffert, J.D.; Pisitkun, T.; Saeed, F.; Song, J.H.; Chou, C.-L.; Knepper, M.A. Dynamics of the G protein-coupled vasopressin V2 receptor signaling network revealed by quantitative phosphoproteomics. Mol. Cell Proteom. 2012, 11, M111.014613. [Google Scholar] [CrossRef]
- Kimple, A.J.; Bosch, D.E.; Giguère, P.M.; Siderovski, D.P. Regulators of G-protein signaling and their Gα substrates: Promises and challenges in their use as drug discovery targets. Pharm. Rev. 2011, 63, 728–749. [Google Scholar] [CrossRef]
- Osei-Owusu, P.; Blumer, K.J. Regulator of G protein signaling 2: A versatile regulator of vascular function. Prog. Mol. Biol. Transl. Sci. 2015, 133, 77–92. [Google Scholar]
- Perschbacher, K.J.; Deng, G.; Sandgren, J.A.; Walsh, J.W.; Witcher, P.C.; Sapouckey, S.A.; Owens, C.E.; Zhang, S.Y.; Scroggins, S.M.; Pearson, N.A. Reduced mRNA Expression of RGS2 (Regulator of G Protein Signaling-2) in the Placenta Is Associated With Human Preeclampsia and Sufficient to Cause Features of the Disorder in Mice. Hypertension 2020, 75, 569–579. [Google Scholar] [CrossRef]
- Perschbacher, K.J.; Deng, G.; Fisher, R.A.; Gibson-Corley, K.N.; Santillan, M.K.; Grobe, J.L. Regulators of G protein signaling in cardiovascular function during pregnancy. Physiol. Genom. 2018, 50, 590–604. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.N.; Dahlen, S.A.; Owens, E.A.; Osei-Owusu, P.; Osei-Owusu, P.; N, K.J.; A, D.S.; A, O.E.; Patrick, O.O. Regulator of G protein signaling 2 facilitates uterine artery adaptation during pregnancy in mice. J. Am. Heart Assoc. 2019, 8, e010917. [Google Scholar] [CrossRef]
- Osei-Owusu, P.; Sabharwal, R.; Kaltenbronn, K.M.; Rhee, M.H.; Chapleau, M.W.; Dietrich, H.H.; Blumer, K.J. Regulator of G protein signaling 2 deficiency causes endothelial dysfunction and impaired endothelium-derived hyperpolarizing factor-mediated relaxation by dysregulating G i/o signaling. J. Biol. Chem. 2012, 287, 12541–12549. [Google Scholar] [CrossRef] [PubMed]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-P.; Hasan, A.A.; Zeng, S.; Hocher, B. Plasma ET-1 concentrations are elevated in pregnant women with hypertension -meta-analysis of clinical studies. Kidney Blood Press. Res. 2017, 42, 654–663. [Google Scholar] [CrossRef]
- Shah, D.M. The role of RAS in the pathogenesis of preeclampsia. Curr. Hypertens. Rep. 2006, 8, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Eastabrook, G.; Brown, M.; Sargent, I. The origins and end-organ consequence of pre-eclampsia. Best Pr. Res. Clin. Obs. Gynaecol. 2011, 25, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Lambert, G.; Brichant, J.F.; Hartstein, G.; Bonhomme, V.; Dewandre, P.Y. Preeclampsia: An update. Acta Anaesthesiol. Belg. 2014, 65, 137–149. [Google Scholar]
- Can, M.; Guven, B.; Bektas, S.; Arikan, I. Oxidative stress and apoptosis in preeclampsia. Tissue Cell 2014, 46, 477–481. [Google Scholar] [CrossRef]
- Cornelius, D.C. Preeclampsia: From inflammation to immunoregulation. Clin. Med. Insights Blood Disord. 2018, 11. [Google Scholar] [CrossRef]
- Toniolo, A.; Buccellati, C.; Pinna, C.; Gaion, R.M.; Sala, A.; Bolego, C. Cyclooxygenase-1 and prostacyclin production by endothelial cells in the presence of mild oxidative stress. PLoS ONE 2013, 8, e56683. [Google Scholar] [CrossRef]
- Brock, T.G.; McNish, R.W.; Peters-Golden, M. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J. Biol. Chem. 1999, 274, 11660–11666. [Google Scholar] [CrossRef]
- Walsh, S.W. Preeclampsia: An imbalance in placental prostacyclin and thromboxane Production. Am. J. Obstet. Gynecol. 1985, 152, 335–340. [Google Scholar] [CrossRef]
- Atallah, A.; Lecarpentier, E.; Goffinet, F.; Doret-Dion, M.; Gaucherand, P.; Tsatsaris, V. Aspirin for Prevention of Preeclampsia. Drugs 2017, 77, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.F.; Canzoneri, B.J.; Gu, Y.; Zhao, S.; Wang, Y. Maternal Levels of Prostacyclin, Thromboxane, ICAM, and VCAM in Normal and Preeclamptic Pregnancies. Am. J. Reprod. Immunol. 2010, 64, 376–383. [Google Scholar] [CrossRef]
- Walsh, S.W. Low-Dose Aspirin: Treatment for the Imbalance of Increased Thromboxane and Decreased Prostacyclin in Preeclampsia. Am. J. Perinatol. 1989, 6, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Tannetta, D.S.; Hunt, K.; Jones, C.I.; Davidson, N.; Coxon, C.H.; Ferguson, D.; Redman, C.W.; Gibbins, J.M.; Sargent, I.L.; Tucker, K.L. Syncytiotrophoblast Extracellular Vesicles from Pre-Eclampsia Placentas Differentially Affect Platelet Function. PLoS ONE 2015, 10, e0142538. [Google Scholar] [CrossRef]
- Duley, L.; Meher, S.; Hunter, K.E.; Seidler, A.L.; Askie, L.M. Antiplatelet agents for preventing pre-eclampsia and its complications. Cochrane Database Syst. Rev. 2007, CD004659. [Google Scholar] [CrossRef] [PubMed]
- ACOG Committee Opinion No. 743: Low-Dose Aspirin Use During Pregnancy. Obs. Gynecol 2018, 132, e44–e52. [CrossRef]
- Periayah, M.H.; Halim, A.S.; Saad, A.Z.M. Mechanism action of platelets and crucial blood coagulation pathways in Hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar]
- Jakobsen, C.; Larsen, J.B.; Fuglsang, J.; Hvas, A.M. Platelet function in preeclampsia–a systematic review and meta-analysis. Platelets 2019, 30, 549–562. [Google Scholar] [CrossRef]
- Goksu Erol, A.Y.; Nazli, M.; Yildiz, S.E. Significance of platelet endothelial cell adhesion molecule-1 (PECAM-1) and intercellular adhesion molecule-1 (ICAM-1) expressions in preeclamptic placentae. Endocrine 2012, 42, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.A.; Peck, J.D.; Deschamps, D.R.; McIntosh, J.J.; Knudtson, E.J.; Terrell, D.R.; Vesely, S.K.; George, J.N. Platelet counts during pregnancy. N. Engl. J. Med. 2018, 379, 32–43. [Google Scholar] [CrossRef]
- Reese, J.A.; Peck, J.D.; Yu, Z.; Scordino, T.A.; Deschamps, D.R.; McIntosh, J.J.; Terrell, D.R.; Vesely, S.K.; George, J.N. Platelet sequestration and consumption in the placental intervillous space contribute to lower platelet counts during pregnancy. Am. J. Hematol. 2019, 94, E8–E11. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Levine, L.D. Thrombocytopenia in pregnancy. Blood 2017, 130, 2271–2277. [Google Scholar] [CrossRef]
- Ciobanu, A.M.; Colibaba, S.; Cimpoca, B.; Peltecu, G.; Panaitescu, A.M. Thrombocytopenia in Pregnancy. Maedica 2016, 11, 55–60. [Google Scholar]
- Heilmann, L.; Rath, W.; Pollow, K. Hemostatic abnormalities in patients with severe preeclampsia. Clin. Appl. Thromb. Hemost. 2007, 13, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Liu, X.; Li, H.; Zou, J.; Yang, Z.; Han, J.; Huang, W.; Yu, L.; Zheng, Y.; Li, L. Blood coagulation parameters and platelet indices: Changes in normal and preeclamptic pregnancies and predictive values for preeclampsia. PLoS ONE 2014, 9, e114488. [Google Scholar] [CrossRef]
- Youssef, L.; Miranda, J.; Blasco, M.; Paules, C.; Crovetto, F.; Palomo, M.; Torramade-Moix, S.; García-Calderó, H.; Tura-Ceide, O.; Dantas, A.P.; et al. Complement and coagulation cascades activation is the main pathophysiological pathway in early-onset severe preeclampsia revealed by maternal proteomics. Sci. Rep. 2021, 11, 3048. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.R.; Bonnar, J.; Norris, L.A.; Darling, M.R.; Walshe, J.J. The effect of pre-eclampsia on coagulation and fibrinolytic activation in the neonate. Thromb. Res. 2000, 99, 567–570. [Google Scholar] [CrossRef]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- Tenório, M.B.; Ferreira, R.C.; Moura, F.A.; Bueno, N.B.; de Oliveira, A.C.M.; Goulart, M.O.F. Cross-Talk between Oxidative Stress and Inflammation in Preeclampsia. Oxidative Med. Cell. Longev. 2019, 2019, 8238727. [Google Scholar] [CrossRef]
- Walsh, S.W. Plasma from preeclamptic women stimulates transendothelial migration of neutrophils. Reprod. Sci. 2009, 16, 320–325. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Williamson, R.D.; McCarthy, F.P.; Kenny, L.C.; McCarthy, C.M. Activation of a TLR9 mediated innate immune response in preeclampsia. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Nieminen, A.L.; Qian, T.; Trost, L.C.; Elmore, S.P.; Nishimura, Y.; Crowe, R.A.; Cascio, W.E.; Bradham, C.A.; Brenner, D.A.; et al. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim. Et Biophys. Acta Bioenerg. 1998, 1366, 177–196. [Google Scholar] [CrossRef]
- Van Houten, B.; Hunter, S.E.; Meyer, J.N. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front. Biosci. 2016, 21, 42–54. [Google Scholar] [CrossRef]
- Thurairajah, K.; Briggs, G.D.; Balogh, Z.J. The source of cell-free mitochondrial DNA in trauma and potential therapeutic strategies. Eur. J. Trauma Emerg. Surg. 2018, 44, 325–334. [Google Scholar] [CrossRef]
- Chockalingam, A.; Brooks, J.C.; Cameron, J.L.; Blum, L.K.; Leifer, C.A. TLR9 traffics through the Golgi complex to localize to endolysosomes and respond to CpG DNA. Immunol. Cell Biol. 2009, 87, 209–217. [Google Scholar] [CrossRef]
- Goulopoulou, S.; Matsumoto, T.; Bomfim, G.F.; Webb, R.C. Toll-like receptor 9 activation: A novel mechanism linking placenta-derived mitochondrial DNA and vascular dysfunction in pre-eclampsia. Clin. Sci. 2012, 123, 429–435. [Google Scholar] [CrossRef]
- Bao, W.; Xia, H.; Liang, Y.; Ye, Y.; Lu, Y.; Xu, X.; Duan, A.; He, J.; Chen, Z.; Wu, Y.; et al. Toll-like Receptor 9 Can be Activated by Endogenous Mitochondrial DNA to Induce Podocyte Apoptosis. Sci. Rep. 2016, 6, 22579. [Google Scholar] [CrossRef] [PubMed]
- Marschalek, J.; Wohlrab, P.; Ott, J.; Wojta, J.; Speidl, W.; Klein, K.U.; Kiss, H.; Pateisky, P.; Zeisler, H.; Kuessel, L. Maternal serum mitochondrial DNA (mtDNA) levels are elevated in preeclampsia–a matched case-control study. Pregnancy Hypertens. 2018, 14, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Panda, B.; Panda, A.; Ueda, I.; Abrahams, V.M.; Norwitz, E.R.; Stanic, A.K.; Young, B.C.; Ecker, J.L.; Altfeld, M.; Shaw, A.C.; et al. Dendritic cells in the circulation of women with preeclampsia demonstrate a pro-inflammatory bias secondary to dysregulation of TLR receptors. J. Reprod. Immunol. 2012, 94, 210–215. [Google Scholar] [CrossRef]
- Redman, C. Pre-eclampsia: A complex and variable disease. Pregnancy Hypertens 2014, 4, 241–242. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, G.; Pussetto, M.; Rose, M.; Staff, A.C.; Blois, S.M.; Toblli, J.E. Defective trophoblast invasion underlies fetal growth restriction and preeclampsia-like symptoms in the stroke-prone spontaneously hypertensive rat. Mol. Hum. Reprod. 2017, 23, 509–519. [Google Scholar] [CrossRef]
- Ma, Y.; Ye, Y.; Zhang, J.; Ruan, C.C.; Gao, P.J. Immune imbalance is associated with the development of preeclampsia. Medicine 2019, 98, e15080. [Google Scholar] [CrossRef]
- Raijmakers, M.T.; Dechend, R.; Poston, L. Oxidative stress and preeclampsia: Rationale for antioxidant clinical trials. Hypertension 2004, 44, 374–380. [Google Scholar] [CrossRef]
- Enkhmaa, D.; Wall, D.; Mehta, P.K.; Stuart, J.J.; Rich-Edwards, J.W.; Merz, C.N.B.; Shufelt, C. Preeclampsia and Vascular Function: A Window to Future Cardiovascular Disease Risk. J. Womens Health 2016, 25, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Romundstad, P.R.; Magnussen, E.B.; Smith, G.D.; Vatten, L.J. Hypertension in pregnancy and later cardiovascular risk: Common antecedents? Circulation 2010, 122, 579–584. [Google Scholar] [CrossRef]
- Redman, C.W. Stress responses and pre-eclampsia. Pregnancy Hypertens 2013, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Roman, D.L.; Traynor, J.R. Regulators of G protein signaling (RGS) proteins as drug targets: Modulating G-protein-coupled receptor (GPCR) signal transduction. J. Med. Chem. 2011, 54, 7433–7440. [Google Scholar] [CrossRef]
- Vangrieken, P.; Al-Nasiry, S.; Bast, A.; Leermakers, P.A.; Tulen, C.B.M.; Schiffers, P.M.H.; van Schooten, F.J.; Remels, A.H.V. Placental Mitochondrial Abnormalities in Preeclampsia. Reprod. Sci. 2021, 28, 2186–2199. [Google Scholar] [CrossRef] [PubMed]
- Cushen, S.C.; Osikoya, O.; Blessing, A.; Phillips, N.R.; Goulopoulou, S. Placental Exposure to Hypoxia and Oxidative Stress Causes Mitochondrial DNA Release into the Extracellular Space. FASEB J. 2019, 33, 703. [Google Scholar] [CrossRef]
- Marín, R.; Chiarello, D.I.; Abad, C.; Rojas, D.; Toledo, F.; Sobrevia, L. Oxidative stress and mitochondrial dysfunction in early-onset and late-onset preeclampsia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165961. [Google Scholar] [CrossRef] [PubMed]
- Turco, M.Y.; Gardner, L.; Kay, R.G.; Hamilton, R.S.; Prater, M.; Hollinshead, M.S.; McWhinnie, A.; Esposito, L.; Fernando, R.; Skelton, H.; et al. Trophoblast organoids as a model for maternal-fetal interactions during human placentation. Nature 2018, 564, 263–267. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preeclampsia | Physiological Effects | Reference |
|---|---|---|
| ↑ TLR3 | Elevated systolic blood pressure | [81] |
| Reduced aortic vasodilation | [81] | |
| Increased urinary protein concentrations | [81] | |
| ↑ TLR4 | Impaired endovascular spiral artery structure | [80] |
| Suppressed trophoblast migration | [82] | |
| ↑ TLR9 | Suppressed trophoblast migration | [83] |
| Decreased VEGFA, increased sFLT-1 | [83] | |
| ↓ IL-10 | Decreased buffering of antiangiogenic factors | [84] |
| Increased endoplasmic reticulum stress | [85] | |
| ↓ Treg cells | Increased uterine artery vasoconstriction | [86] |
| Elevated endothelin-1 production | [86] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Opichka, M.A.; Rappelt, M.W.; Gutterman, D.D.; Grobe, J.L.; McIntosh, J.J. Vascular Dysfunction in Preeclampsia. Cells 2021, 10, 3055. https://doi.org/10.3390/cells10113055
Opichka MA, Rappelt MW, Gutterman DD, Grobe JL, McIntosh JJ. Vascular Dysfunction in Preeclampsia. Cells. 2021; 10(11):3055. https://doi.org/10.3390/cells10113055
Chicago/Turabian StyleOpichka, Megan A., Matthew W. Rappelt, David D. Gutterman, Justin L. Grobe, and Jennifer J. McIntosh. 2021. "Vascular Dysfunction in Preeclampsia" Cells 10, no. 11: 3055. https://doi.org/10.3390/cells10113055
APA StyleOpichka, M. A., Rappelt, M. W., Gutterman, D. D., Grobe, J. L., & McIntosh, J. J. (2021). Vascular Dysfunction in Preeclampsia. Cells, 10(11), 3055. https://doi.org/10.3390/cells10113055