
In Silico Designing of a Multitope Vaccine against Rhizopus microsporus with Potential Activity against Other Mucormycosis Causing Fungi

 , , ,
, , ,  , ,
, ,  ,
,  and
and
Abstract
1. Introduction
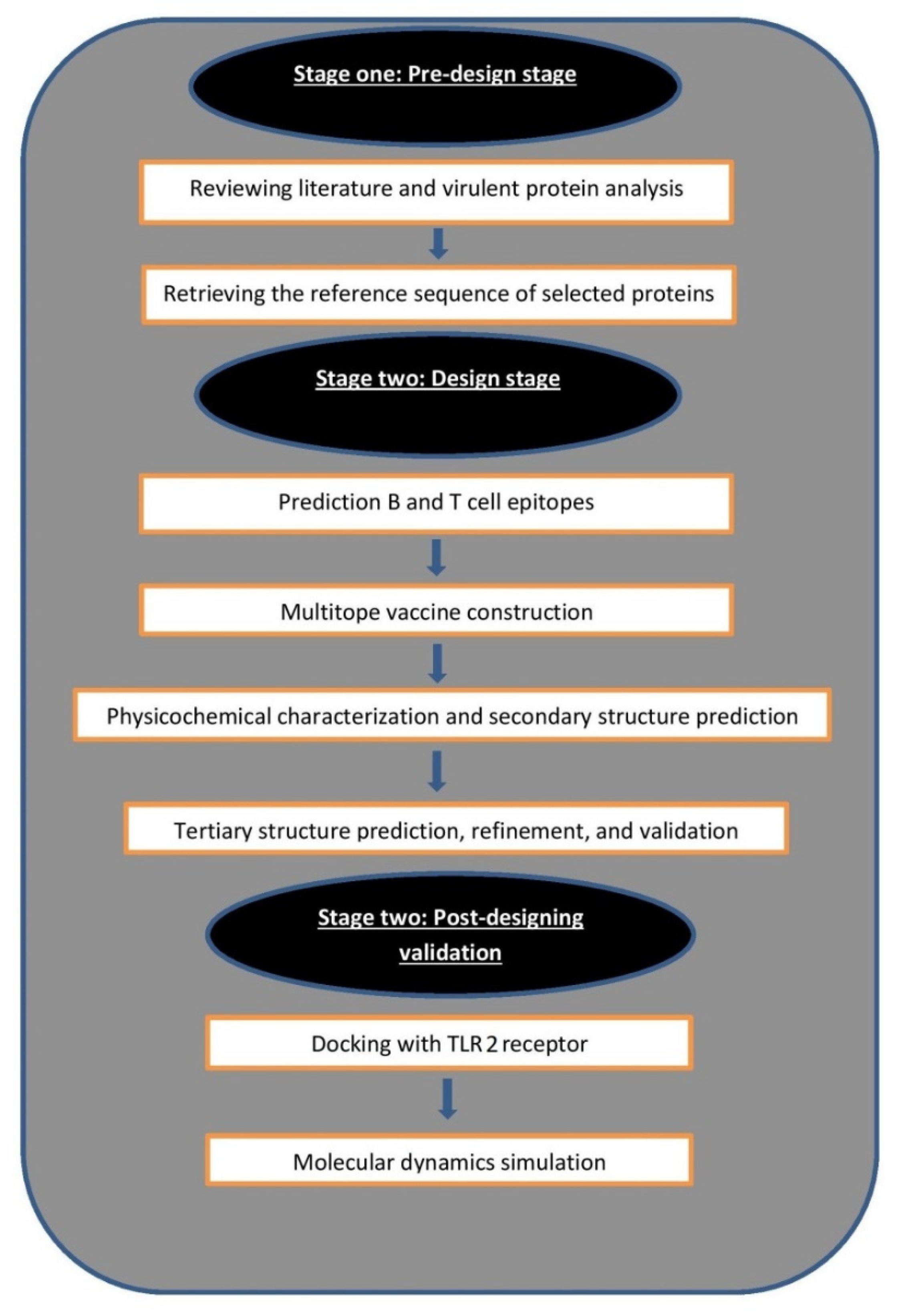
2. Materials and Methods
2.1. Selection of Proteins
2.2. Prediction of B and T Cell Epitopes
2.3. Multitope Vaccine Construction
2.4. Assessment of Vaccine’s Solubility, Physicochemical Features, and 2ry Structure
2.5. Vaccine 3D Structure Prediction and Validation
2.6. Vaccine Disulfide Engineering
2.7. Docking Analysis between Predicted Vaccine 3D Structure and TLR-2
2.8. Normal Mode Analysis
2.9. Molecular Dynamics Simulation
2.10. Reverse Translation and Codon Adaptation
2.11. Immune Simulation of the Chimeric Peptide Vaccine
3. Results
3.1. Nomination of Two Proteins as Vaccine Candidates
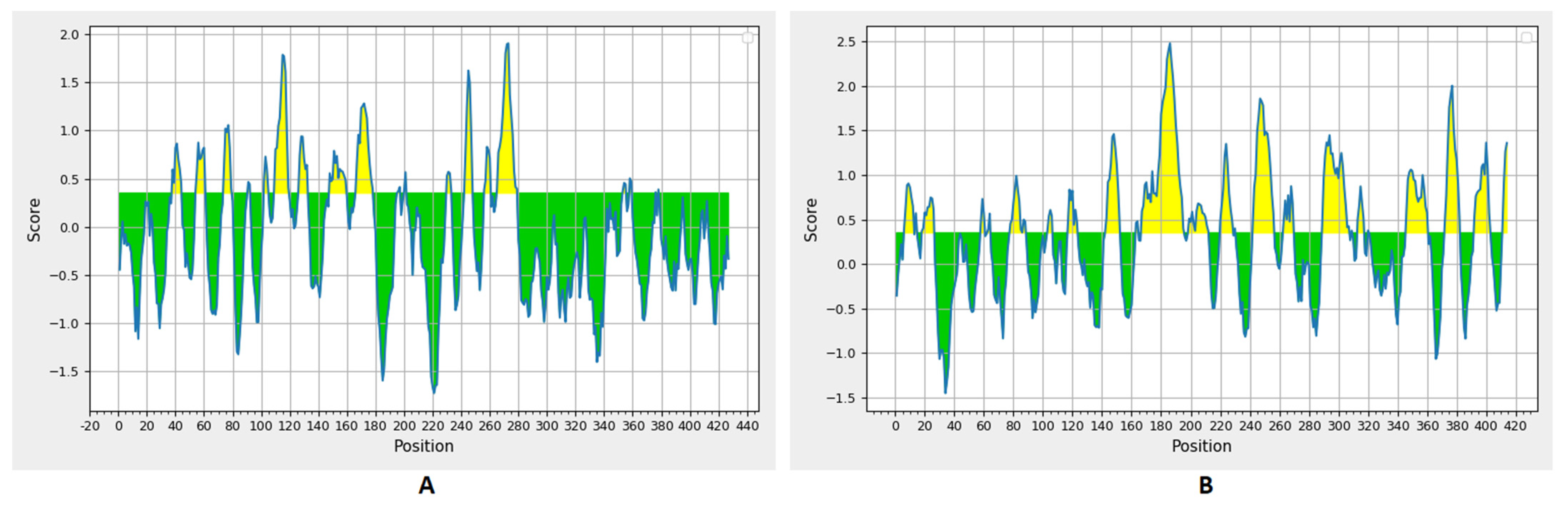
3.2. Prediction of B Cell Epitopes
3.3. Prediction of MHC-I and MHC-II Epitopes
3.4. Molecular Docking of T Cell Epitopes and Assessment of Selected Epitopes Conservation
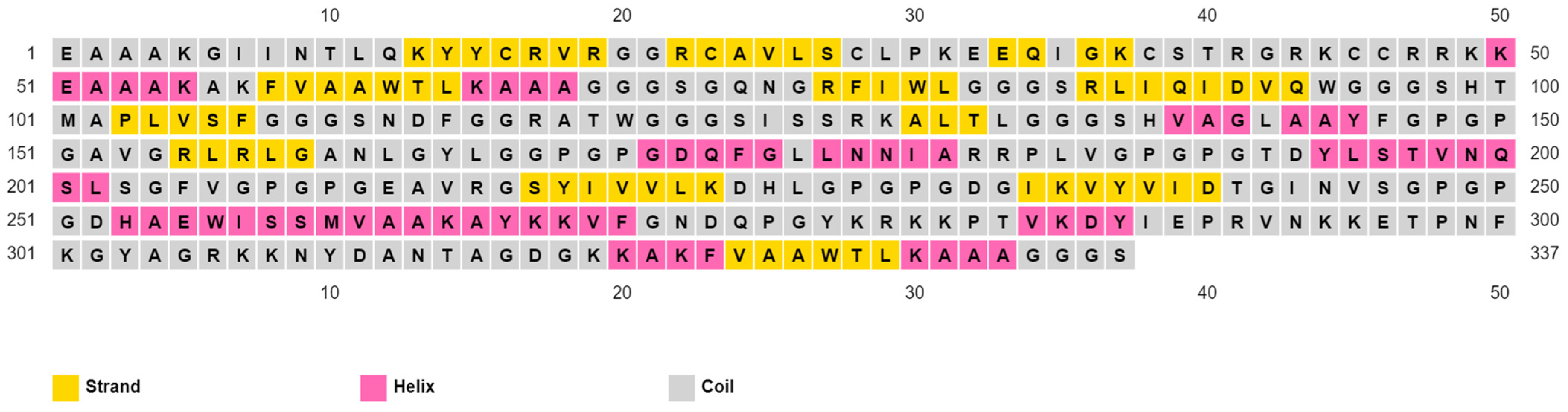
3.5. Assembly of the Multitope Vaccine and Assessment of Its Physicochemical Properties
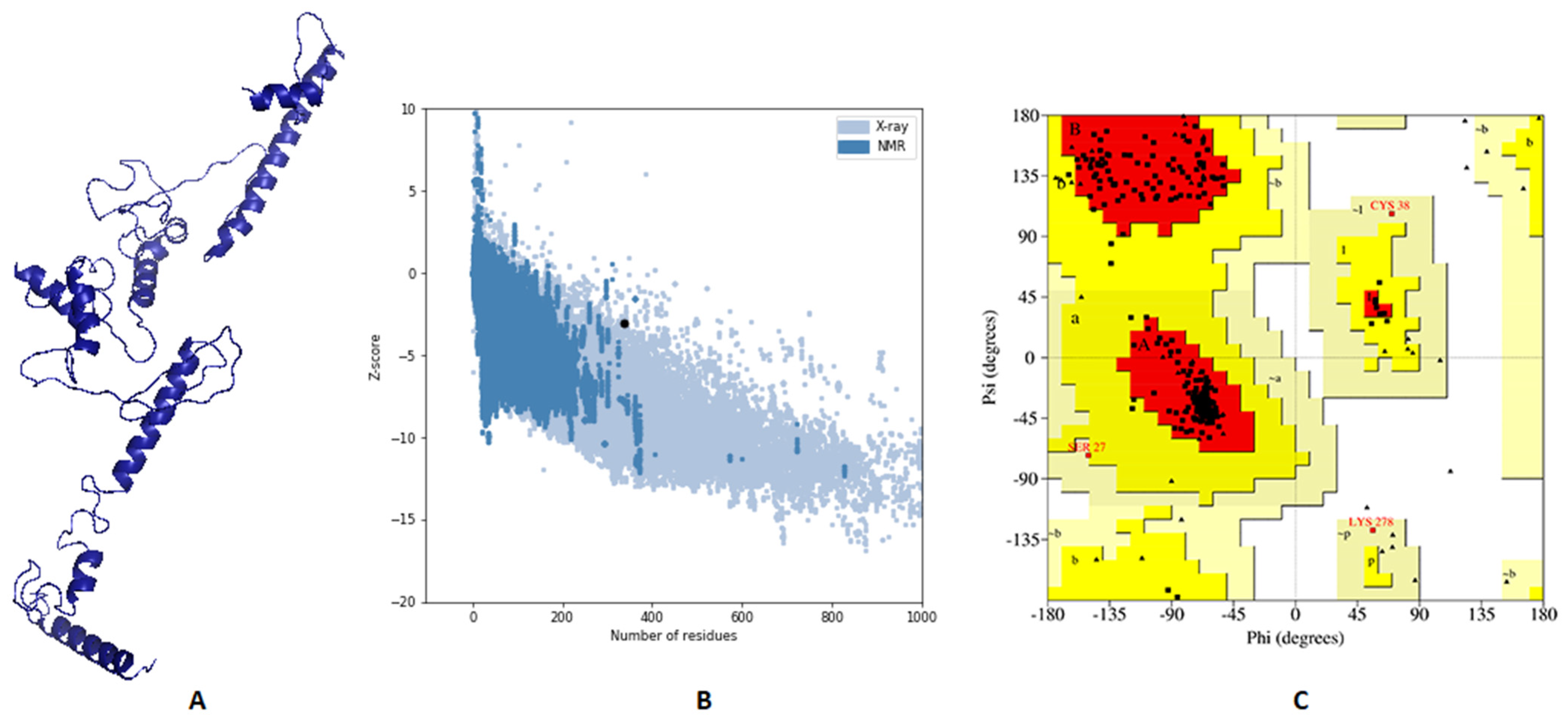
3.6. Vaccine’s Predicted 3D Structure and Its Validation
3.7. Vaccine Disulfide Engineering
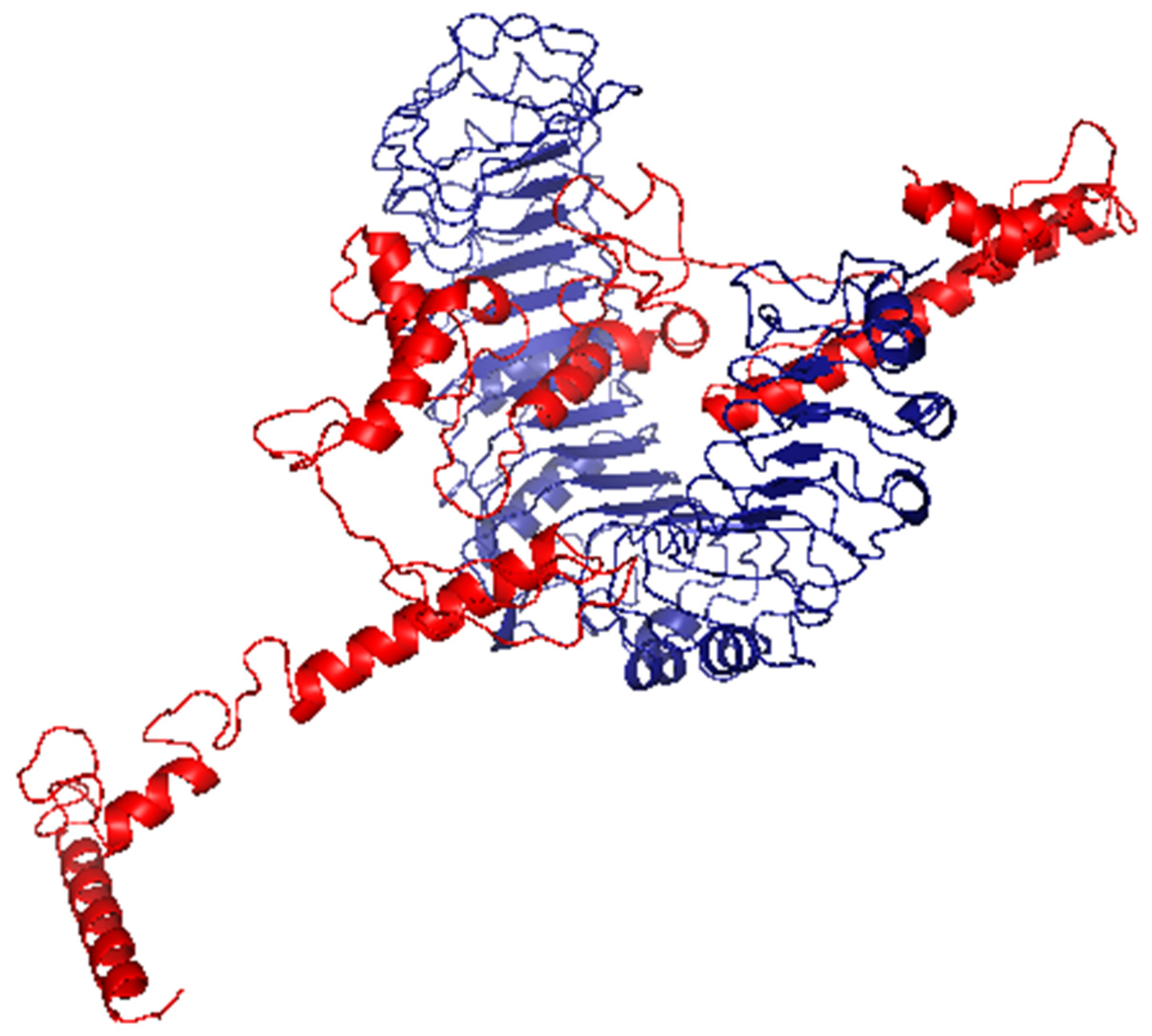
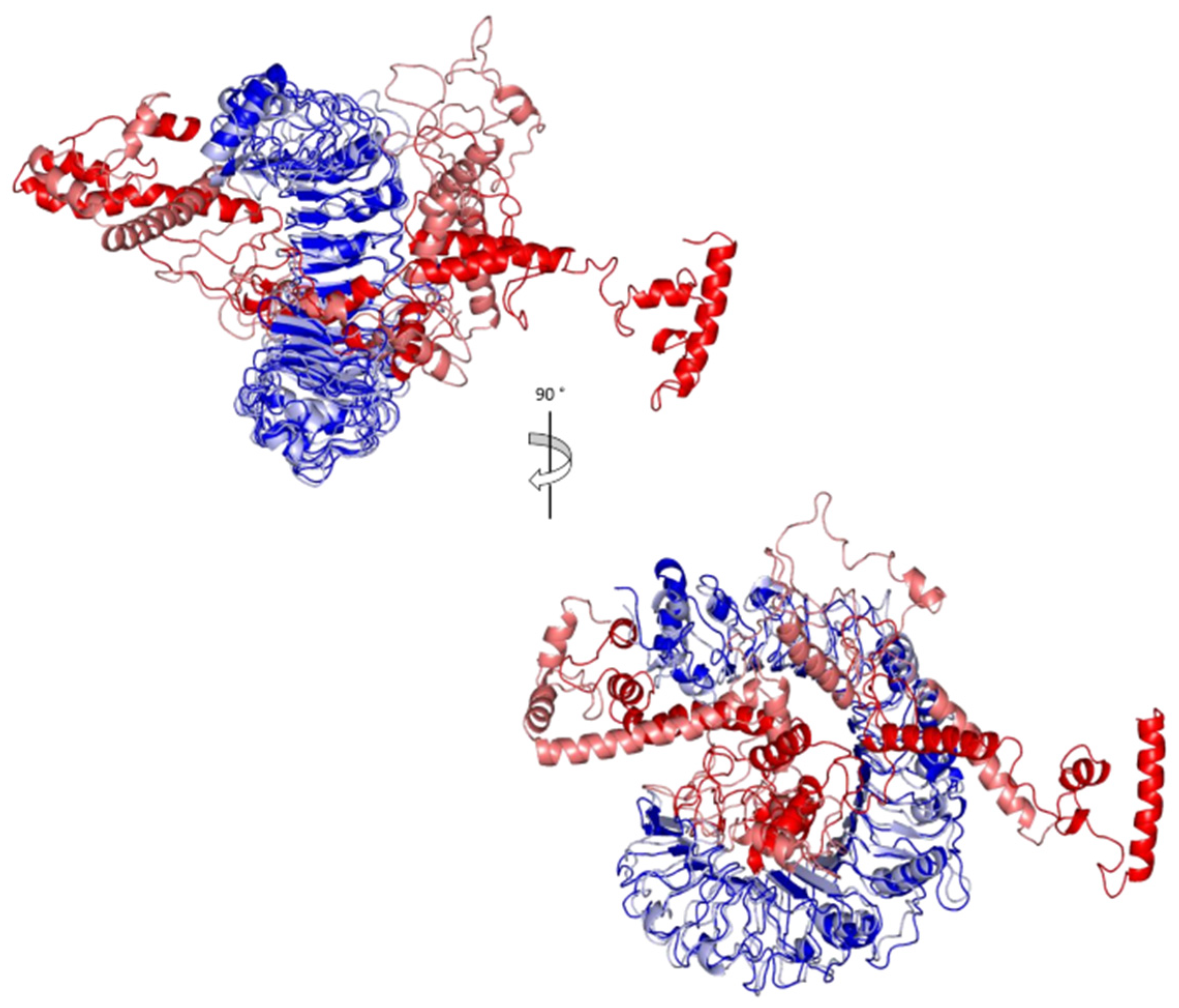
3.8. Molecular Docking of the Vaccine with TLR2
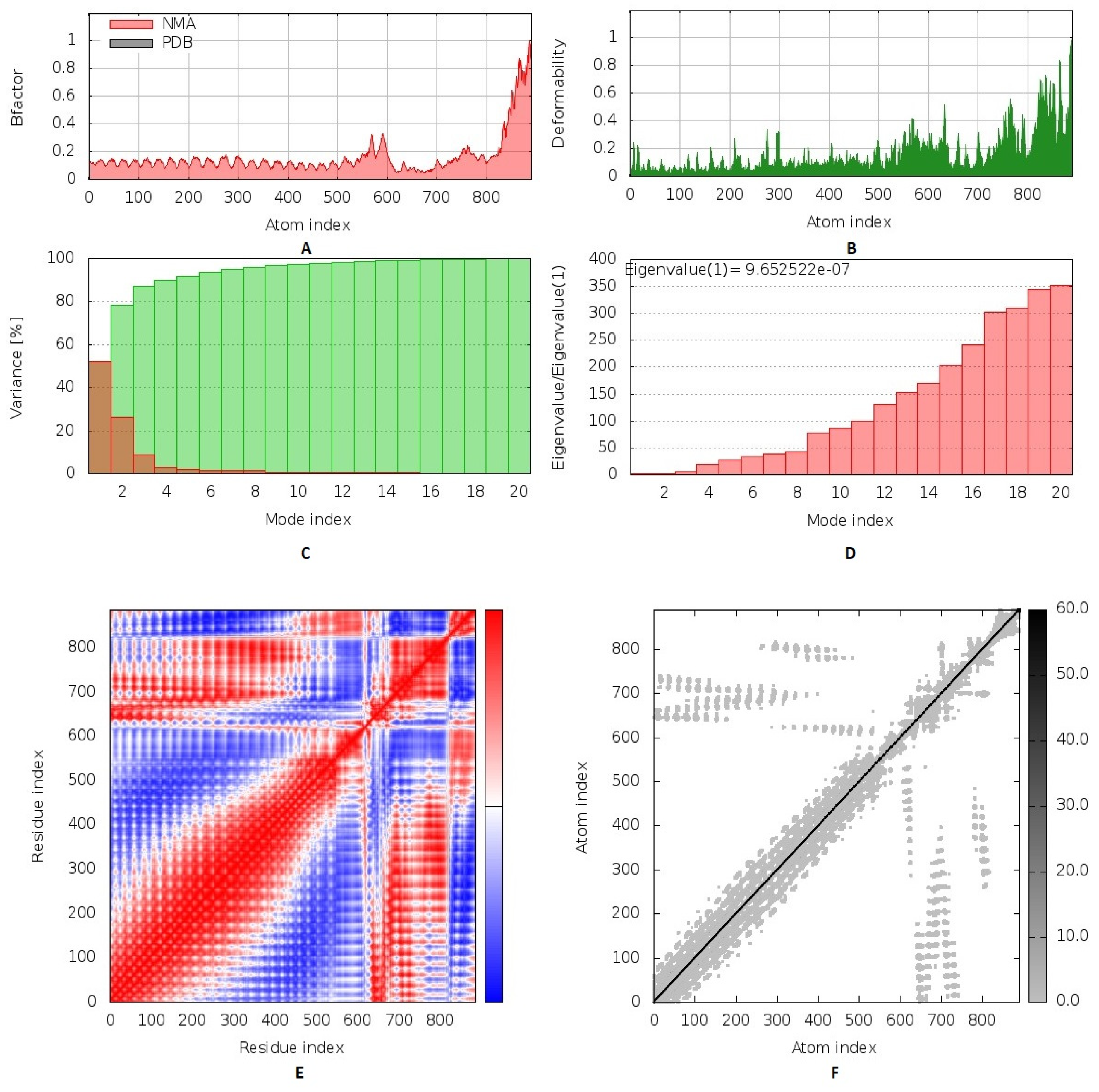
3.9. Inherited Flexibility Analysis Using Normal Mode Analysis within Dihedral Coordinates
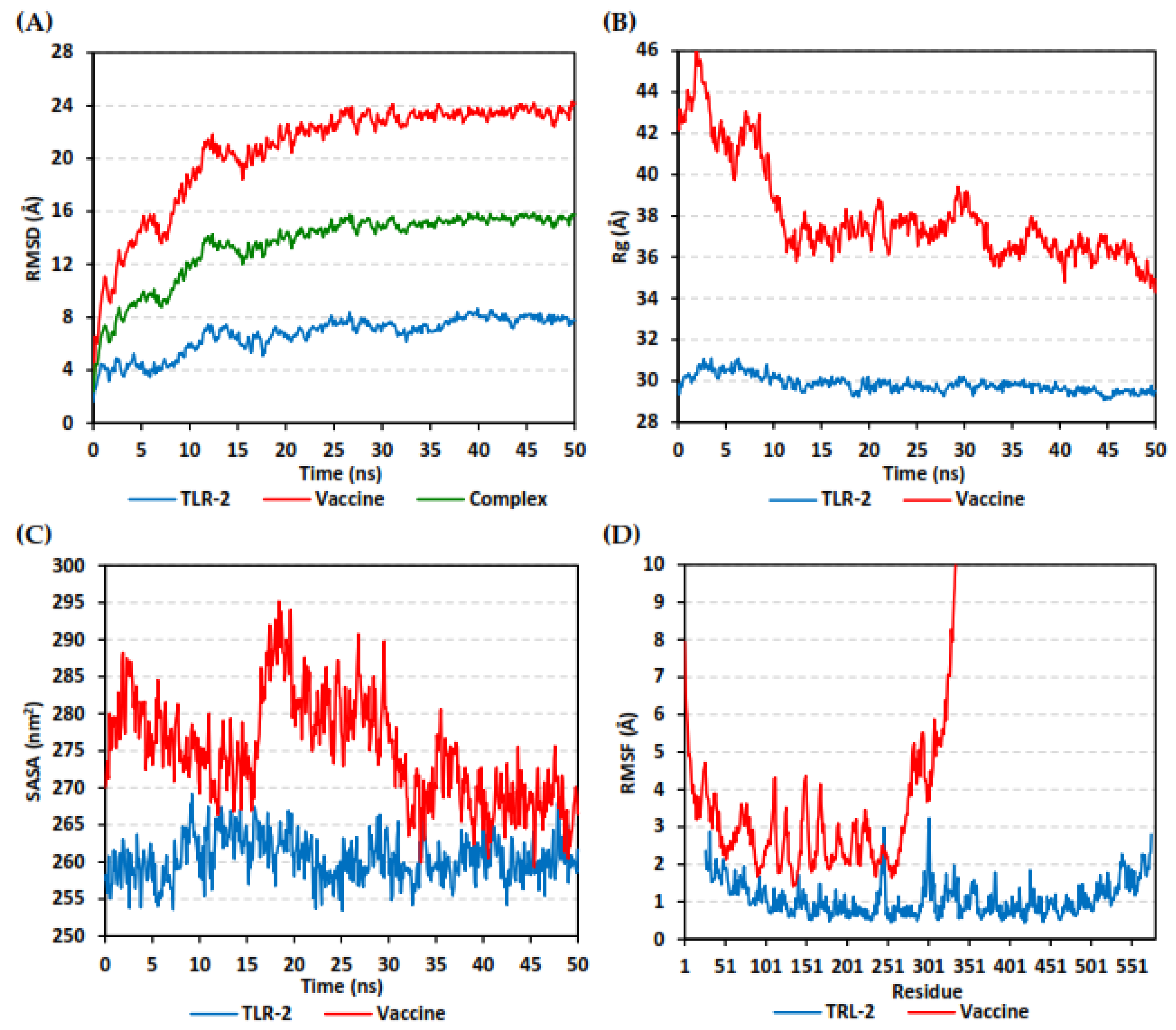
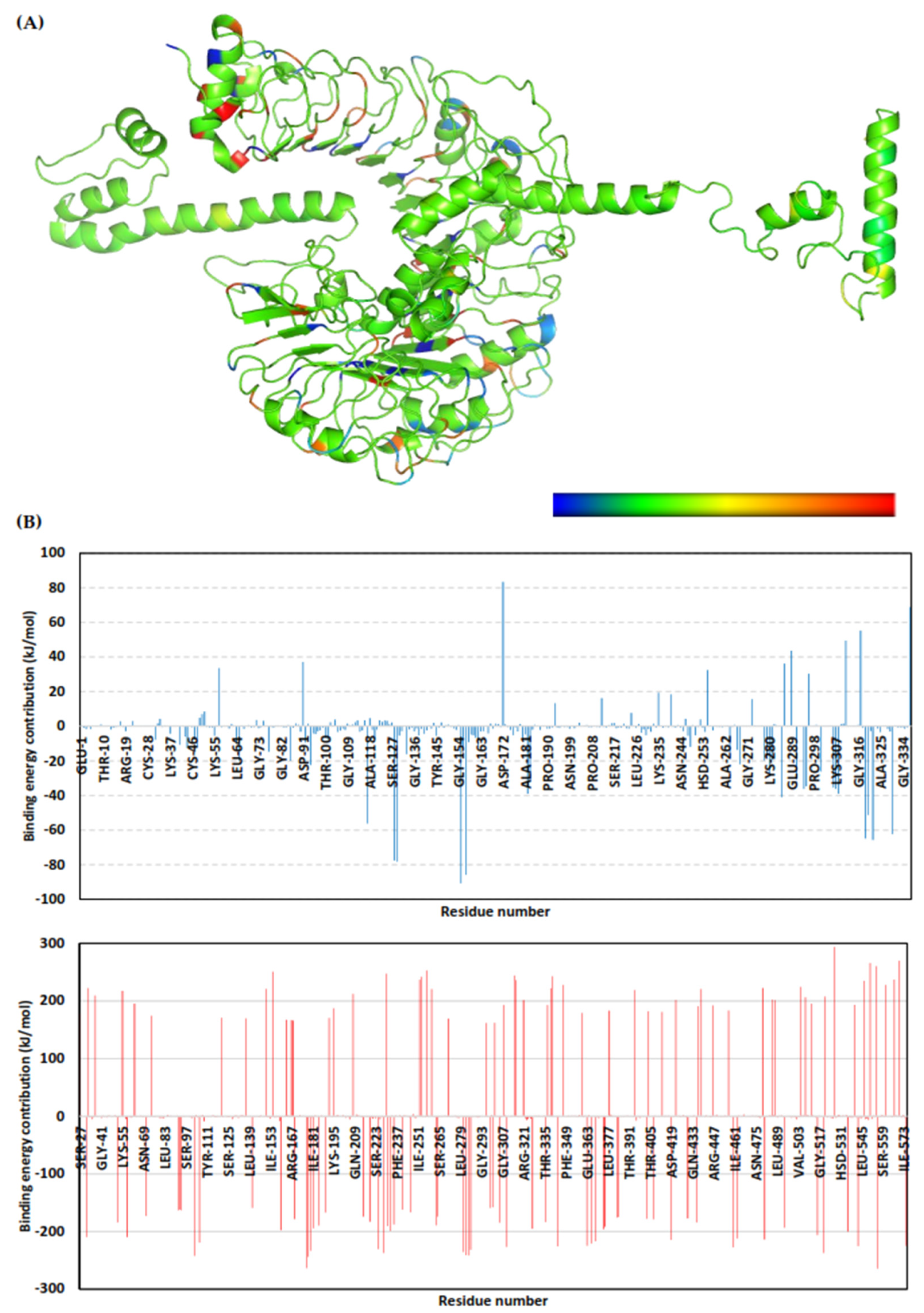
3.10. Molecular Dynamics Simulations
3.11. Vaccine Reverse Translation and Codon Optimization
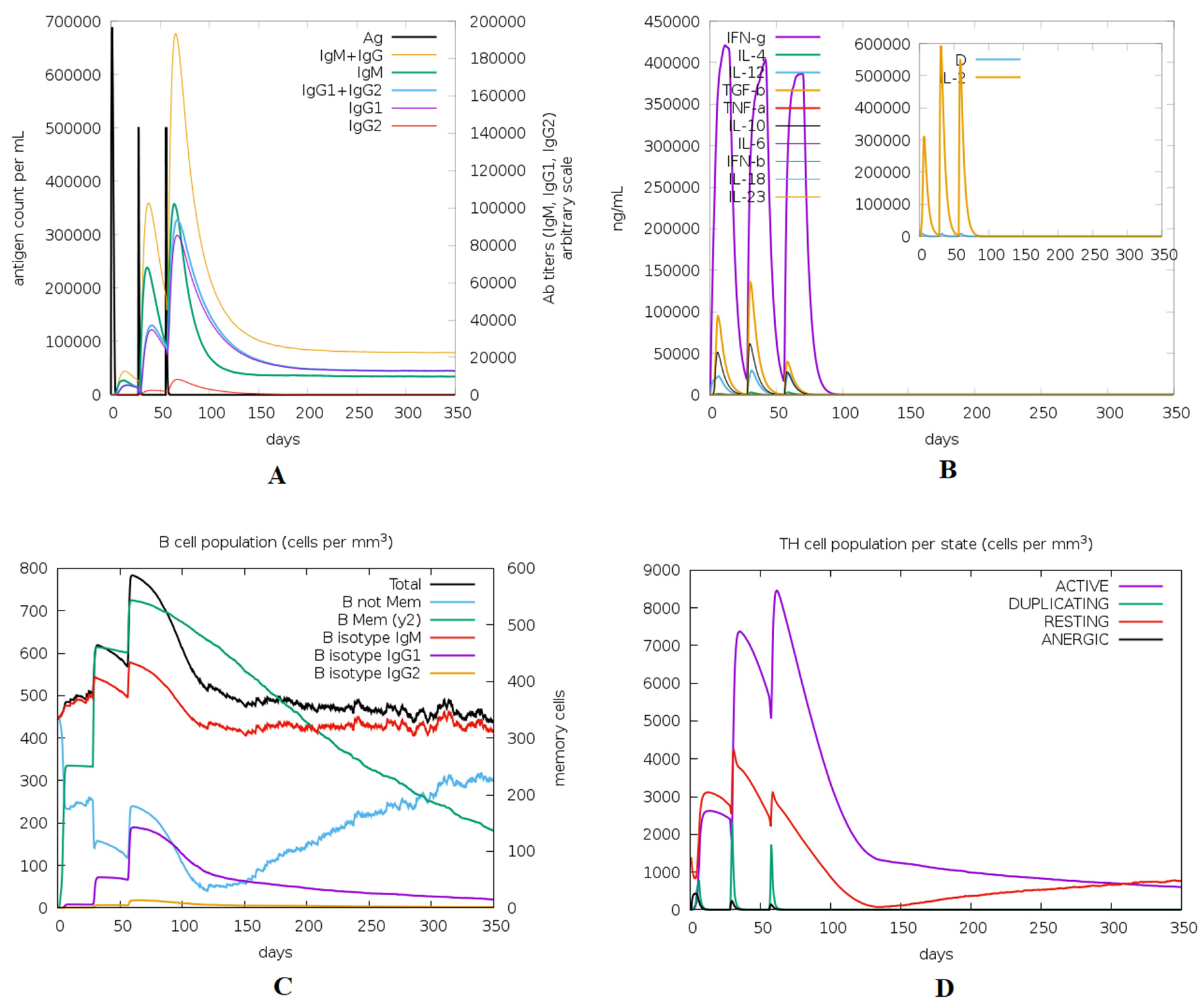
3.12. Immune Simulation of the Designed Vaccine
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Jacobsen, I.D. Animal Models to Study Mucormycosis. J. Fungi 2019, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M. The ecology of the Zygomycetes and its impact on environmental exposure. Clin. Microbiol. Infect. 2009, 15, 2–9. [Google Scholar] [CrossRef]
- Jeong, W.; Keighley, C.; Wolfe, R.; Lee, W.L.; Slavin, M.A.; Kong, D.C.M.; Chen, S.C.-A. The epidemiology and clinical manifestations of mucormycosis: A systematic review and meta-analysis of case reports. Clin. Microbiol. Infect. 2019, 25, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.E.; Kontoyiannis, D.P. Epidemiology and treatment of mucormycosis. Future Microbiol. 2013, 8, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Risum, M.; Helweg-Larsen, J.; Petersen, S.; Kampmann, P.; Overgaard, U.M.; El Fassi, D.; Nielsen, O.J.; Brabrand, M.; Rubek, N.; Munksgaard, L.; et al. Introduction of a comprehensive diagnostic and interdisciplinary management approach in haematological patients with mucormycosis: A pre and post-intervention analysis. J. Fungi 2020, 6, 268. [Google Scholar] [CrossRef] [PubMed]
- Prakash, H.; Chakrabarti, A. Epidemiology of Mucormycosis in India. Microorganisms 2021, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Sungurtekin, H.; Sargin, F.; Akbulut, M.; Karaduman, S. Severe Rhinocerebral Mucormycosis Case Developed after COVID-19. J. Bacteriol. Parasitol. 2021, 12, 1000386. [Google Scholar]
- Kanwar, A.; Jordan, A.; Olewiler, S.; Wehberg, K.; Cortes, M.; Jackson, B. A Fatal Case of Rhizopus azygosporus Pneumonia Following COVID-19. J. Fungi 2021, 7, 174. [Google Scholar] [CrossRef] [PubMed]
- Sipsas, N.V.; Gamaletsou, M.N.; Anastasopoulou, A.; Kontoyiannis, D.P. Therapy of Mucormycosis. J. Fungi 2018, 4, 90. [Google Scholar] [CrossRef]
- Dannaoui, E. Antifungal resistance in mucorales. Int. J. Antimicrob. Agents 2017, 50, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; Edwards, J.E.; Bryant, R.; Spellberg, B. Economic burden of mucormycosis in the United States: Can a vaccine be cost-effective? Med. Mycol. 2009, 47, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Gebremariam, T.C.; Clemons, K.V.; Stevens, D.A.; Ibrahim, A.S. Heat-killed yeast protects diabetic ketoacidotic-steroid treated mice from pulmonary mucormycosis. Vaccine 2014, 32, 3573–3576. [Google Scholar] [CrossRef] [PubMed]
- Backert, L.; Kohlbacher, O. Immunoinformatics and epitope prediction in the age of genomic medicine. Genome Med. 2015, 7, 119. [Google Scholar] [CrossRef] [PubMed]
- Oli, A.N.; Obialor, W.O.; Ifeanyichukwu, M.O.; Odimegwu, D.C.; Okoyeh, J.N.; Emechebe, G.O.; Adejumo, S.A.; Ibeanu, G.C. Immunoinformatics and Vaccine Development: An Overview. ImmunoTargets Ther. 2020, 9, 13–30. [Google Scholar] [CrossRef]
- Soltan, M.A.; Magdy, D.; Solyman, S.M.; Hanora, A. Design of Staphylococcus aureus New Vaccine Candidates with B and T Cell Epitope Mapping, Reverse Vaccinology, and Immunoinformatics. OMICS A J. Integr. Biol. 2020, 24, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Soltan, M.A.; Elbassiouny, N.; Gamal, H.; Elkaeed, E.B.; Eid, R.A.; Eldeen, M.A.; Al-Karmalawy, A.A. In Silico Prediction of a Multitope Vaccine against Moraxella catarrhalis: Reverse Vaccinology and Immunoinformatics. Vaccines 2021, 9, 669. [Google Scholar] [CrossRef]
- Prasasty, V.D.; Grazzolie, K.; Rosmalena, R.; Yazid, F.; Ivan, F.X.; Sinaga, E. Peptide-based subunit vaccine design of T-and b-cells multi-epitopes against zika virus using immunoinformatics approaches. Microorganisms 2019, 7, 226. [Google Scholar] [CrossRef] [PubMed]
- Tarang, S.; Kesherwani, V.; Latendresse, B.; Lindgren, L.; Rocha-Sanchez, S.M.; Weston, M.D. In silico Design of a Multivalent Vaccine Against Candida albicans. Sci. Rep. 2020, 10, 1066. [Google Scholar] [CrossRef]
- Aminnezhad, S.; Abdi-Ali, A.; Ghazanfari, T.; Bandehpour, M.; Zarrabi, M. Immunoinformatics design of multivalent chimeric vaccine for modulation of the immune system in Pseudomonas aeruginosa infection. Infect. Genet. Evol. 2020, 85, 104462. [Google Scholar] [CrossRef]
- Gu, Y.; Sun, X.; Li, B.; Huang, J.; Zhan, B.; Zhu, X. Vaccination with a Paramyosin-Based Multi-Epitope Vaccine Elicits Significant Protective Immunity against Trichinella spiralis Infection in Mice. Front. Microbiol. 2017, 8, 1475. [Google Scholar] [CrossRef]
- Hasanzadeh, S.; Habibi, M.; Shokrgozar, M.A.; Ahangari Cohan, R.; Ahmadi, K.; Asadi Karam, M.R.; Bouzari, S. In silico analysis and in vivo assessment of a novel epitope-based vaccine candidate against uropathogenic Escherichia coli. Sci. Rep. 2020, 10, 16258. [Google Scholar] [CrossRef]
- Hassan, M.I.A.; Voigt, K. Pathogenicity patterns of mucormycosis: Epidemiology, interaction with immune cells and virulence factors. Med. Mycol. 2019, 57, S245–S256. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. Bioinformatic Approach for Identifying Parasite and Fungal Candidate Subunit Vaccines. Open Vaccine J. 2008, 1, 22–26. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; A Greenbaum, J.; Marcatili, P.; et al. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef]
- Weiskopf, D.; Angelo, M.A.; de Azeredo, E.L.; Sidney, J.; Greenbaum, J.A.; Fernando, A.N.; Broadwater, A.; Kolla, R.V.; De Silva, A.D.; de Silva, A.M.; et al. Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc. Natl. Acad. Sci. USA 2013, 110, E2046–E2053. [Google Scholar] [CrossRef]
- Greenbaum, J.; Sidney, J.; Chung, J.; Brander, C.; Peters, B.; Sette, A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics 2011, 63, 325–335. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Vir, P.; Raghava, G.P.S. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef]
- Larsen, J.E.P.; Lund, O.; Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, 2. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shen, Y.; Maupetit, J.; Derreumaux, P.; Tufféry, P. Improved PEP-FOLD Approach for Peptide and Miniprotein Structure Prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Franke, E.D.; Hoffman, S.L.; Sacci, J.B.; Wang, R.; Charoenvit, Y.; Appella, E.; Chesnut, R.; Alexander, J.; Del Guercio, M.-F.; Sette, A. Pan DR binding sequence provides T-cell help for induction of protective antibodies against Plasmodium yoelii sporozoites. Vaccine 1999, 17, 1201–1205. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. AlgPred: Prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S.; Open Source Drug Discovery Consortium. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed]
- Magnan, C.N.; Randall, A.Z.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: New York, NY, USA, 2005; pp. 571–607. [Google Scholar]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.B.; A Dombkowski, A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef]
- Monk, B.C.; Keniya, M.V. Roles for Structural Biology in the Discovery of Drugs and Agrochemicals Targeting Sterol 14α-Demethylases. J. Fungi 2021, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Nicolás, F.E.; Murcia, L.; Navarro, E.; Navarro-Mendoza, M.I.; Pérez-Arques, C.; Garre, V. Mucorales Species and Macrophages. J. Fungi 2020, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, H.; Voelz, K. Innate and adaptive immunity to mucorales. J. Fungi 2017, 3, 48. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Lopéz-Blanco, J.R.; Garzón, J.I.; Chacón, P. iMod: Multipurpose normal mode analysis in internal coordinates. Bioinformatics 2011, 27, 2843–2850. [Google Scholar] [CrossRef] [PubMed]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In International Conference on Exascale Applications and Software; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Elmaaty, A.A.; Darwish, K.M.; Khattab, M.; Elhady, S.S.; Salah, M.; Hamed, M.I.A.; Al-Karmalawy, A.A.; Saleh, M.M. In a search for potential drug candidates for combating COVID-19: Computational study revealed salvianolic acid B as a potential therapeutic targeting 3CLpro and spike proteins. J. Biomol. Struct. Dyn. 2021, 1–28. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the hACE2 Receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef]
- Saleh, A.H.; Abdelwaly, A.; Darwish, K.M.; Eissa, A.A.H.M.; Chittiboyina, A.; Helal, M.A. Deciphering the molecular basis of the kappa opioid receptor selectivity: A Molecular Dynamics study. J. Mol. Graph. Model. 2021, 106, 107940. [Google Scholar] [CrossRef]
- Zaki, A.A.; Ashour, A.; Elhady, S.S.; Darwish, K.M.; Al-Karmalawy, A.A. Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: Molecular docking, molecular dynamics, and SAR studies. J. Tradit. Complement. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tuble, S.C.; Anwar, J.; Gale, J.D. An Approach to Developing a Force Field for Molecular Simulation of Martensitic Phase Transitions between Phases with Subtle Differences in Energy and Structure. J. Am. Chem. Soc. 2004, 126, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. G-mmpbsa -A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef]
- Obaidullah, A.J.; Alanazi, M.M.; Alsaif, N.A.; Albassam, H.; Almehizia, A.A.; Alqahtani, A.M.; Mahmud, S.; Sami, S.A.; Bin Emran, T. Immunoinformatics-guided design of a multi-epitope vaccine based on the structural proteins of severe acute respiratory syndrome coronavirus. RSC Adv. 2021, 11, 18103–18121. [Google Scholar] [CrossRef]
- Azim, K.F.; Hasan, M.; Hossain, M.N.; Somana, S.R.; Hoque, S.F.; Bappy, M.N.I.; Chowdhury, A.T.; Lasker, T. Immunoinformatics approaches for designing a novel multi epitope peptide vaccine against human norovirus (Norwalk virus). Infect. Genet. Evol. 2019, 74, 103936. [Google Scholar] [CrossRef]
- Hasan, M.; Azim, K.F.; Imran, M.A.S.; Chowdhury, I.M.; Akhter Urme, S.R.; Parvez, M.S.A.; Uddin, M.B.; Uddin Ahmed, S.S. Comprehensive genome based analysis of Vibrio parahaemolyticus for identifying novel drug and vaccine molecules: Subtractive proteomics and vaccinomics approach. PLoS ONE 2020, 15, e0237181. [Google Scholar] [CrossRef]
- Hasan, M.; Azim, K.F.; Begum, A.; Khan, N.A.; Shammi, T.S.; Imran, A.S.; Chowdhury, I.M.; Urme, S.R.A. Vaccinomics strategy for developing a unique multi-epitope monovalent vaccine against Marburg marburgvirus. Infect. Genet. Evol. 2019, 70, 140–157. [Google Scholar] [CrossRef]
- Khan, J.; Sharma, P.K.; Mukhopadhaya, A. Vibrio cholerae porin OmpU mediates M1-polarization of macrophages/monocytes via TLR1/TLR2 activation. Immunobiology 2015, 220, 1199–1209. [Google Scholar] [CrossRef]
- Kardani, K.; Bolhassani, A.; Namvar, A. An overview of in silico vaccine design against different pathogens and cancer. Expert Rev. Vaccines 2020, 19, 699–726. [Google Scholar] [CrossRef] [PubMed]
- Raoufi, E.; Hemmati, M.; Eftekhari, S.; Khaksaran, K.; Mahmodi, Z.; Farajollahi, M.M.; Mohsenzadegan, M. Epitope Prediction by Novel Immunoinformatics Approach: A State-of-the-art Review. Int. J. Pept. Res. Ther. 2019, 26, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Liang, G.; Liu, W. Fungal Co-infections Associated with Global COVID-19 Pandemic: A Clinical and Diagnostic Perspective from China. Mycopathologia 2020, 185, 599–606. [Google Scholar] [CrossRef]
- Holanda, R.A.; Muñoz, J.E.; Dias, L.S.; Silva, L.B.R.; Santos, J.R.A.; Pagliari, S.; Vieira, É.L.M.; Paixão, T.A.; Taborda, C.P.; Santos, D.A.; et al. Recombinant vaccines of a CD4+ T-cell epitope promote efficient control of Paracoccidioides brasiliensis burden by restraining primary organ infection. PLOS Negl. Trop. Dis. 2017, 11, e0005927. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Ke, H.; Niu, X.; Li, S.; Lv, J.; Pan, L. Protection Against Helicobacter pylori Infection in BALB/c Mouse Model by Oral Administration of Multivalent Epitope-Based Vaccine of Cholera Toxin B Subunit-HUUC. Front. Immunol. 2018, 9, 1003. [Google Scholar] [CrossRef]
- Safavi, A.; Kefayat, A.; Sotoodehnejadnematalahi, F.; Salehi, M.; Modarressi, M.H. Production, purification, and in vivo evaluation of a novel multiepitope peptide vaccine consisted of immunodominant epitopes of SYCP1 and ACRBP antigens as a prophylactic melanoma vaccine. Int. Immunopharmacol. 2019, 76, 105872. [Google Scholar] [CrossRef]
- Champer, J.; Diaz-Arévalo, D.; Champer, M.; Hong, T.B.; Wong, M.; Shannahoff, M.; Ito, J.I.; Clemons, K.V.; Stevens, D.A.; Kalkum, M. Protein targets for broad-spectrum mycosis vaccines: Quantitative proteomic analysis of Aspergillus and Coccidioides and comparisons with other fungal pathogens. Ann. N. Y. Acad. Sci. 2012, 1273, 44–51. [Google Scholar] [CrossRef]
- Monod, M.; Capoccia, S.; Léchenne, B.; Zaugg, C.; Holdom, M.; Jousson, O. Secreted proteases from pathogenic fungi. Int. J. Med Microbiol. 2002, 292, 405–419. [Google Scholar] [CrossRef]
- Muszewska, A.; Stepniewska-Dziubinska, M.M.; Steczkiewicz, K.; Pawłowska, J.; Dziedzic, A.; Ginalski, K. Fungal lifestyle reflected in serine protease repertoire. Sci. Rep. 2017, 7, 9147. [Google Scholar] [CrossRef]
- Langner, T.; Göhre, V. Fungal chitinases: Function, regulation, and potential roles in plant/pathogen interactions. Curr. Genet. 2016, 62, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; Voelz, K. The mucormycete–host interface. Curr. Opin. Microbiol. 2017, 40, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Spellberg, B.; Phan, Q.T.; Fu, Y.; Fu, Y.; Lee, A.S.; Edwards, J.E.; Filler, S.G.; Ibrahim, A.S. The endothelial cell receptor GRP78 is required for mucormycosis pathogenesis in diabetic mice. J. Clin. Investig. 2010, 120, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.A.; Eweida, A.E.; El-Sayed, L.H. T-cell epitope mapping for the design of powerful vaccines. Vaccine Rep. 2016, 6, 13–22. [Google Scholar] [CrossRef]
- Thakur, R.; Shankar, J. In silico Identification of Potential Peptides or Allergen Shot Candidates Against Aspergillus fumigatus. BioResearch Open Access 2016, 5, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Joshi, A.; Kaushik, V.; Kumar, M.; Mannan, M.A.-U. In-silico design of a multivalent epitope-based vaccine against Candida auris. Microb. Pathog. 2021, 155, 104879. [Google Scholar] [CrossRef] [PubMed]
- Bär, E.; Gladiator, A.; Bastidas, S.; Roschitzki, B.; Acha-Orbea, H.; Oxenius, A.; LeibundGut-Landmann, S. A Novel Th Cell Epitope of Candida albicans Mediates Protection from Fungal Infection. J. Immunol. 2012, 188, 5636–5643. [Google Scholar] [CrossRef]
- Improving multi-epitope long peptide vaccine potency by using a strategy that enhances CD4+ T Help in BALB/c mice. PLoS ONE 2015, 10, e0142563. [CrossRef]
- Roilides, E.; Simitsopoulou, M. Immune responses to Mucorales growth forms: Do we know everything? Virulence 2017, 8, 1489–1491. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef]
- Chauhan, V.; Rungta, T.; Goyal, K.; Singh, M.P. Designing a multi-epitope based vaccine to combat Kaposi Sarcoma utilizing immunoinformatics approach. Sci. Rep. 2019, 9, 2517. [Google Scholar] [CrossRef]
- Sanches, R.C.O.; Tiwari, S.; Ferreira, L.C.G.; Oliveira, F.M.; Lopes, M.D.; Passos, M.J.F.; Maia, E.H.B.; Taranto, A.G.; Kato, R.; Azevedo, V.A.C.; et al. Immunoinformatics Design of Multi-Epitope Peptide-Based Vaccine against Schistosoma mansoni Using Transmembrane Proteins as a Target. Front. Immunol. 2021, 12, 621706. [Google Scholar] [CrossRef] [PubMed]
- Likić, V.A.; Gooley, P.R.; Speed, T.P.; Strehler, E.E. A statistical approach to the interpretation of molecular dynamics simulations of calmodulin equilibrium dynamics. Protein Sci. 2005, 14, 2955–2963. [Google Scholar] [CrossRef]
- Pirolli, D.; Sciandra, F.; Bozzi, M.; Giardina, B.; Brancaccio, A.; De Rosa, M.C. Insights from Molecular Dynamics Simulations: Structural Basis for the V567D Mutation-Induced Instability of Zebrafish Alpha-Dystroglycan and Comparison with the Murine Model. PLoS ONE 2014, 9, e103866. [Google Scholar] [CrossRef] [PubMed]
- Benson, N.C.; Daggett, V. A Comparison of Multiscale Methods for the Analysis of Molecular Dynamics Simulations. J. Phys. Chem. B 2012, 116, 8722–8731. [Google Scholar] [CrossRef]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.-G.; Lee, H.; Lee, J.-O. Crystal Structure of the TLR1-TLR2 Heterodimer Induced by Binding of a Tri-Acylated Lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Oh, S.C.; Lim, K.J.; Kasamatsu, J.; Heo, J.Y.; Park, B.S.; Lee, H.; Yoo, O.J.; Kasahara, M.; Lee, J.-O. Structural Diversity of the Hagfish Variable Lymphocyte Receptors. J. Biol. Chem. 2007, 282, 6726–6732. [Google Scholar] [CrossRef] [PubMed]
- Pancer, Z.; Cooper, M.D. The Evolution of adaptive immunity. Annu. Rev. Immunol. 2006, 24, 497–518. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Antigenicity Score | Surface Exposed or Secreted Protein |
|---|---|---|
| High-affinity iron permease | −0.03 | - |
| Spore coat protein | 0.80 | Yes |
| Serine protease | 0.86 | Yes |
| Calcium/calmodulin-dependent protein kinase | 0.55 | No |
| CotH | SP | ||||
|---|---|---|---|---|---|
| Epitope | Start–End | Antigenicity Score | Epitope | Start–End | Antigenicity Score |
| CATDPSYI | 38–44 | 1.14 | YAPVEAEAV | 36–44 | −0.11 |
| VFGNDQPGYKR | 109–119 | 2.04 | ETPNFKGYAGR | 96–106 | 2.04 |
| PTVKDYIEPRVN | 148–159 | 1.1 | NYDANTAGDG | 161–170 | 1.69 |
| QEYPSKSVSKDHT | 166–178 | 0.54 | IAGTKYGVAKKARP | 217–230 | 1.42 |
| LVPANEQKDADNSFK | 265–279 | −0.09 | SNGSGSMSD | 238–246 | 0.12 |
| KDKEQAQTEGKPFKG | 260–274 | 0.84 | |||
| NTATNTISGTSMASP | 364–378 | 1.5 | |||
| QSEPGVTPKEI | 390–400 | 0.35 | |||
| PNELTKIPKDT | 410–420 | −0.98 | |||
| CotH | SP | ||||
|---|---|---|---|---|---|
| Epitope | Antigenicity | Reacting Alleles Number | Epitope | Antigenicity | Reacting Alleles Number |
| GQNGRFIWL | 2.96 | 7 | TAGDGIKVY | 4.3 | 9 |
| RVFGNDQPGY | 2.63 | 11 | NDFGGRATW | 2.89 | 7 |
| ARASYVRLF | 2.26 | 6 | ISSRKALTL | 2.62 | 6 |
| RLIQIDVQW | 2.02 | 12 | ATWGKTIPA | 2.6 | 8 |
| TVNQSLSGF | 1.91 | 7 | HNDFGGRATW | 2.59 | 6 |
| YQDPGQNGRF | 1.91 | 14 | HVAGLAAYF | 2.4 | 12 |
| VQWDKQLQR | 1.68 | 9 | APGLDIQSIW | 2.19 | 6 |
| RIMQDYYDY | 1.63 | 8 | SSRKALTLR | 1.96 | 6 |
| HTMAPLVSF | 1.35 | 19 | VVLKDHLSM | 1.23 | 11 |
| SQLLQVDEF | 1.41 | 10 | KARPVAVKV | 1.11 | 11 |
| CotH | SP | ||||||
|---|---|---|---|---|---|---|---|
| Epitope | Antigenicity | IFN Epitope | Reacting Alleles | Epitope | Antigenicity | IFN Epitope | Reacting Alleles |
| AVGRLRLGANLGYLG | 1.66 | Yes | 4 | EAVRGSYIVVLKDHL | 1.77 | Yes | 9 |
| KIKFSLSGQTSRLFN | 1.73 | No | 6 | DGIKVYVIDTGINVS | 1.64 | Yes | 9 |
| VGRLRLGANLGYLGP | 1.8 | Yes | 4 | GIKVYVIDTGINVSH | 1.39 | Yes | 8 |
| DQFGLLNNIARRPLV | 0.99 | Yes | 8 | LARISSRKALTLRNF | 1.02 | Yes | 7 |
| DYLSTVNQSLSGFVL | 1.46 | Yes | 4 | DHAEWISSMVAAKAY | 0.71 | Yes | 13 |
| FGLLNNIARRPLVSQ | 1.07 | Yes | 4 | AITVGASTIADERAY | 0.69 | Yes | 5 |
| TDYLSTVNQSLSGFV | 1.68 | Yes | 6 | QDHAEWISSMVAAKA | 0.68 | Yes | 10 |
| GRLRLGANLGYLGPT | 1.56 | Yes | 4 | HAEWISSMVAAKAYN | 0.58 | Yes | 12 |
| ITDYLSTVNQSLSGF | 1.68 | Yes | 5 | GDGIKVYVIDTGINV | 2.63 | No | 9 |
| QFGLLNNIARRPLVS | 1.07 | Yes | 7 | GSYIVVLKDHLSMEQ | 2.21 | No | 8 |
| No. | Epitope | MHC-I Allele | Binding Energy (kcal/mol) | Epitope | MHC-II Allele | Binding Energy (kcal/mol) |
|---|---|---|---|---|---|---|
| 1 | GQNGRFIWL | −7.8 | AVGRLRLGANLGYLG | −8.1 | ||
| 2 | RLIQIDVQW | −7.4 | DQFGLLNNIARRPLV | −8.0 | ||
| 3 | HTMAPLVSF | HLA-A*11:01 | −8.3 | TDYLSTVNQSLSGFV | HLA-DRB1*04:01 | −7.6 |
| 4 | NDFGGRATW | −8.8 | EAVRGSYIVVLKDHL | −7.6 | ||
| 5 | ISSRKALTL | −8.0 | DGIKVYVIDTGINVS | −7.7 | ||
| 6 | HVAGLAAYF | −9.0 | DHAEWISSMVAAKAY | −8.4 |
| Percentage Identity (%) in | ||||||||
|---|---|---|---|---|---|---|---|---|
| Epitope Sequence | Epitope Type | Rhizopus microsporus | Rhizopus azygosporus | Rhizopus oryzae | Rhizopus delemar | Mucor lusitanicus | Apophysomyces sp. BC1015 | Lichtheimia corymbifera |
| GQNGRFIWL | CTL | 100 | 100 | 87.5 | 75 | 75 | 83.33 | 100 |
| RLIQIDVQW | CTL | 100 | 100 | 75 | 87.5 | 83.33 | 100 | 100 |
| HTMAPLVSF | CTL | 100 | 100 | 100 | 100 | 70 | 100 | 72.73 |
| NDFGGRATW | CTL | 100 | 75 | 87.5 | 87.5 | 87.5 | 87.5 | 77.78 |
| ISSRKALTL | CTL | 100 | 100 | 80 | 88.89 | 75 | 75 | 85.71 |
| HVAGLAAYF | CTL | 100 | 88.89 | 100 | 88.89 | 100 | 100 | 100 |
| AVGRLRLGANLGYLG | HTL | 100 | 100 | 86.76 | 86.76 | 73.33 | 87.50 | 88.89 |
| DQFGLLNNIARRPLV | HTL | 100 | 100 | 73.33 | 73.33 | 80 | 100 | 88.89 |
| TDYLSTVNQSLSGFV | HTL | 100 | 93.33 | 100 | 100 | 100 | 100 | 80 |
| EAVRGSYIVVLKDHL | HTL | 100 | 66.67 | 86.67 | 80 | 86.67 | 100 | 71.43 |
| DGIKVYVIDTGINVS | HTL | 100 | 78.57 | 92.86 | 92.86 | 85.71 | 92.86 | 91.67 |
| DHAEWISSMVAAKAY | HTL | 100 | 71.43 | 73.33 | 93.73 | 83.33 | 75 | 77.78 |
| VFGNDQPGYKR | BCL | 100 | 100 | 77.78 | 75 | 85.71 | 75 | 87.5 |
| PTVKDYIEPRVN | BCL | 100 | 91.67 | 69.23 | 70 | 75 | 87.5 | 80 |
| ETPNFKGYAGR | BCL | 100 | 72.73 | 81.82 | 81.82 | 81.82 | 87.5 | 87.5 |
| NYDANTAGDG | BCL | 100 | 75 | 80 | 70 | 80 | 87.5 | 75 |
| Physicochemical Characteristic | Molecular Weight | TheoreticalpI | Extinction Coefficient | GRAVY | Instability Index | Aliphatic Index |
|---|---|---|---|---|---|---|
| Score | 34.78 kDa | 10.03 | 51,255 M−1 cm−1 | −0.326 | 23.82 | 70.42 |
| Energy (kJ/mol ± SD) | Ligand–Receptor Complex |
|---|---|
| ΔGvan der Waal | −880.969 +/− 125.941 |
| ΔGElectrostatic | −2732.441 +/− 151.150 |
| ΔGSolvation; Polar | 2019.511 +/− 137.941 |
| ΔGSolvation; SASA | −119.052 +/− 9.876 |
| ΔGTotal binding | −1712.950 +/− 160.827 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soltan, M.A.; Eldeen, M.A.; Elbassiouny, N.; Kamel, H.L.; Abdelraheem, K.M.; El-Gayyed, H.A.; Gouda, A.M.; Sheha, M.F.; Fayad, E.; Ali, O.A.A.; et al. In Silico Designing of a Multitope Vaccine against Rhizopus microsporus with Potential Activity against Other Mucormycosis Causing Fungi. Cells 2021, 10, 3014. https://doi.org/10.3390/cells10113014
Soltan MA, Eldeen MA, Elbassiouny N, Kamel HL, Abdelraheem KM, El-Gayyed HA, Gouda AM, Sheha MF, Fayad E, Ali OAA, et al. In Silico Designing of a Multitope Vaccine against Rhizopus microsporus with Potential Activity against Other Mucormycosis Causing Fungi. Cells. 2021; 10(11):3014. https://doi.org/10.3390/cells10113014
Chicago/Turabian StyleSoltan, Mohamed A., Muhammad Alaa Eldeen, Nada Elbassiouny, Hasnaa L. Kamel, Kareem M. Abdelraheem, Hanaa Abd El-Gayyed, Ahmed M. Gouda, Mohammed F. Sheha, Eman Fayad, Ola A. Abu Ali, and et al. 2021. "In Silico Designing of a Multitope Vaccine against Rhizopus microsporus with Potential Activity against Other Mucormycosis Causing Fungi" Cells 10, no. 11: 3014. https://doi.org/10.3390/cells10113014
APA StyleSoltan, M. A., Eldeen, M. A., Elbassiouny, N., Kamel, H. L., Abdelraheem, K. M., El-Gayyed, H. A., Gouda, A. M., Sheha, M. F., Fayad, E., Ali, O. A. A., Ghany, K. A. E., El-damasy, D. A., Darwish, K. M., Elhady, S. S., & Sileem, A. E. (2021). In Silico Designing of a Multitope Vaccine against Rhizopus microsporus with Potential Activity against Other Mucormycosis Causing Fungi. Cells, 10(11), 3014. https://doi.org/10.3390/cells10113014