
Endocannabinoid Metabolism and Traumatic Brain Injury

{kind=link}
{kind=link}
{kind=link}
Abstract
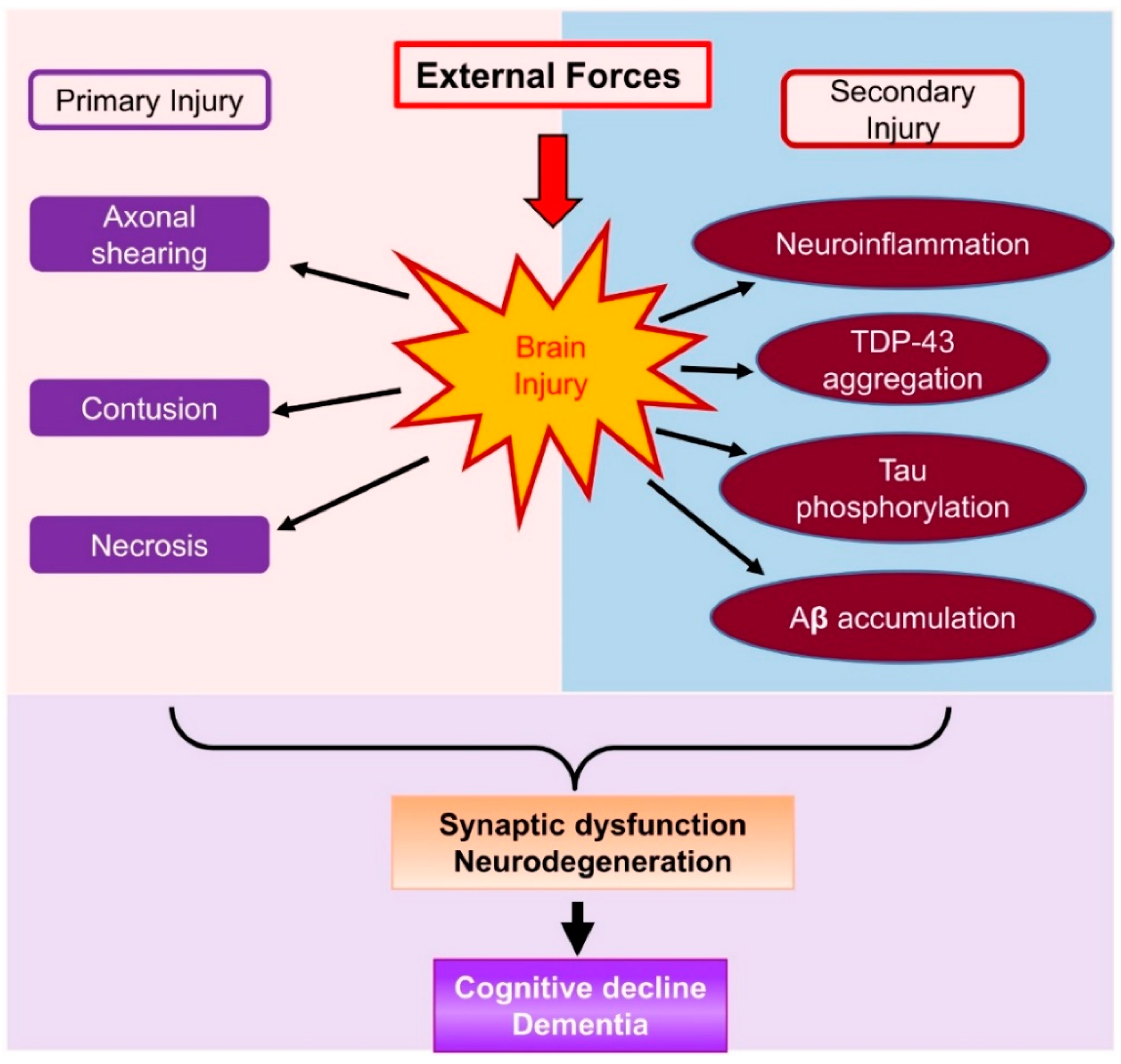
:1. Introduction
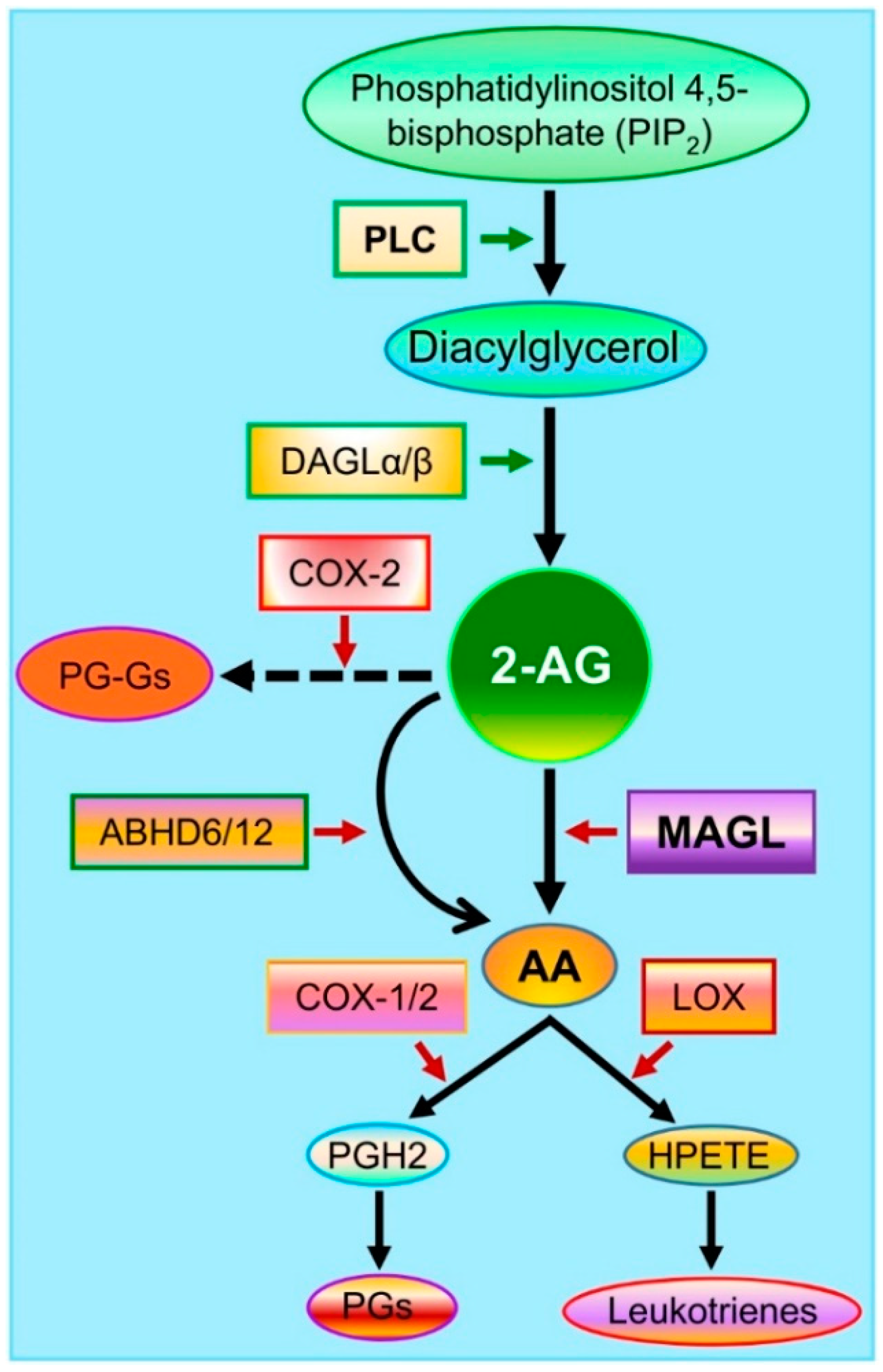
2. Endocannabinoid 2-AG Synthesis and Metabolism
3. Resolving Neuroinflammation and Maintaining the Integrity of the Blood–Brain Barrier by the Inhibition of 2-AG Metabolism in TBI
4. Alleviation of TBI-Induced Neuropathology by Inactivation of MAGL
5. Improvement of Synaptic and Cognitive Functions by Inactivation of MAGL in TBI
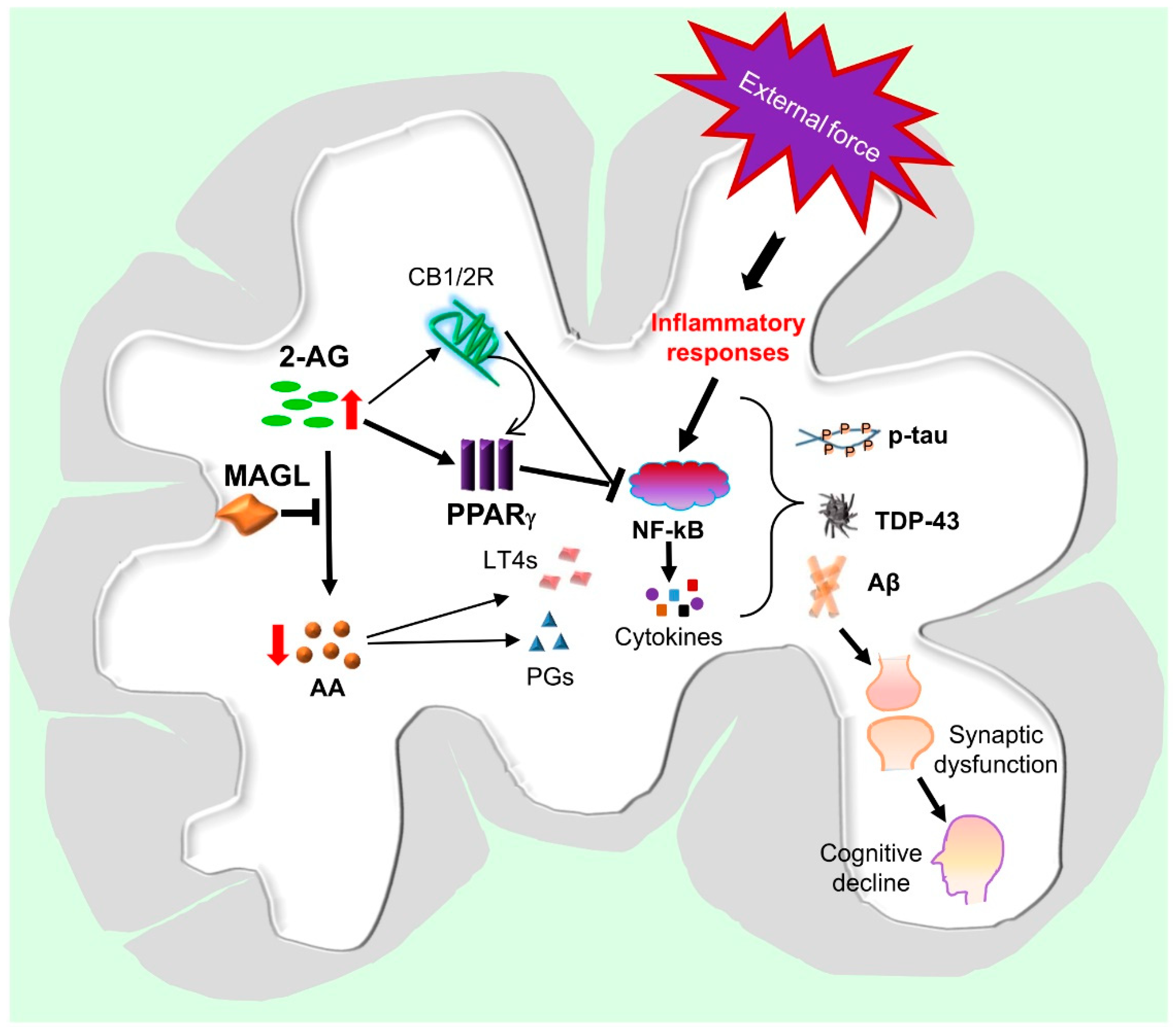
6. Potential Mechanisms Underlying the Neuroprotective Effects of MAGL Inactivation in TBI
7. Outlook on Potential Treatment Strategies for TBI
8. Summary
Funding
Conflicts of Interest
References
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.-C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2019, 130, 1080–1097. [Google Scholar] [CrossRef] [Green Version]
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness: A practical scale. Lancet 1974, 304, 81–84. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma 2015, 32, 1834–1848. [Google Scholar] [CrossRef]
- Bruns, J.; Hauser, W.A. The Epidemiology of Traumatic Brain Injury: A Review. Epilepsia 2003, 44, 2–10. [Google Scholar] [CrossRef]
- Andriessen, T.M.J.C.; Jacobs, B.; Vos, P.E. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J. Cell. Mol. Med. 2010, 14, 2381–2392. [Google Scholar] [CrossRef] [Green Version]
- Delic, V.; Beck, K.D.; Pang, K.C.H.; Citron, B.A. Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathol. Commun. 2020, 8, 45. [Google Scholar] [CrossRef]
- Fordington, S.; Manford, M. A review of seizures and epilepsy following traumatic brain injury. J. Neurol. 2020, 267, 3105–3111. [Google Scholar] [CrossRef]
- Al-Dahhak, R.; Khoury, R.; Qazi, E.; Grossberg, G.T. Traumatic Brain Injury, Chronic Traumatic Encephalopathy, and Alzheimer Disease. Clin. Geriatr. Med. 2018, 34, 617–635. [Google Scholar] [CrossRef]
- Dams-O’Connor, K.; Guetta, G.; Hahn-Ketter, A.E.; Fedor, A. Traumatic brain injury as a risk factor for Alzheimer’s disease: Current knowledge and future directions. Neurodegener. Dis. Manag. 2016, 6, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Fleminger, S. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef]
- Gardner, R.C.; Yaffe, K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol. Cell. Neurosci. 2015, 66, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-β pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortimer, J.A.; French, L.R.; Hutton, J.T.; Schuman, L.M. Head injury as a risk factor for Alzheimer’s disease. Neurology 1985, 35, 264. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Sivanandam, T.M.; Thakur, M.K. Traumatic brain injury: A risk factor for Alzheimer’s disease. Neurosci. Biobehav. Rev. 2012, 36, 1376–1381. [Google Scholar] [CrossRef]
- Saatman, K.E.; Duhaime, A.-C.; Bullock, R.; Maas, A.I.; Valadka, A.; Manley, G.T. Classification of Traumatic Brain Injury for Targeted Therapies. J. Neurotrauma 2008, 25, 719–738. [Google Scholar] [CrossRef] [Green Version]
- Panikashvili, D.; Mechoulam, R.; Beni, S.M.; Alexandrovich, A.; Shohami, E. CB1cannabinoid receptors are involved in neuroprotection via NF-κB inhibition. Br. J. Pharmacol. 2005, 25, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Panikashvili, D.; Shein, N.A.; Mechoulam, R.; Trembovler, V.; Kohen, R.; Alexandrovich, A.; Shohami, E. The endocannabinoid 2-AG protects the blood–brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol. Dis. 2006, 22, 257–264. [Google Scholar] [CrossRef]
- Panikashvili, D.; Simeonidou, C.; Ben-Shabat, S.; Hanus, L.O.; Breuer, A.; Mechoulam, R.; Shohami, E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nat. Cell Biol. 2001, 413, 527–531. [Google Scholar] [CrossRef]
- Shohami, E.; Cohen-Yeshurun, A.; Magid, L.; Algali, M.; Mechoulam, R. Endocannabinoids and traumatic brain injury. Br. J. Pharmacol. 2011, 163, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, J.; Chen, C. Endocannabinoid 2-arachidonoylglycerol protects neurons against β-amyloid insults. Neurosci. 2011, 178, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Chen, X.; Zhang, J.; Chen, C. Inhibition of COX-2 expression by endocannabinoid 2-arachidonoylglycerol is mediated via PPAR-γ. Br. J. Pharmacol. 2011, 163, 1533–1549. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, C. Endocannabinoid 2-Arachidonoylglycerol Protects Neurons by Limiting COX-2 Elevation. J. Biol. Chem. 2008, 283, 22601–22611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.-Y.; Chen, C. Endocannabinoids in Synaptic Plasticity and Neuroprotection. Neuroscientist 2015, 21, 152–168. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, T.; Kishimoto, S.; Oka, S.; Gokoh, M. Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog. Lipid Res. 2006, 45, 405–446. [Google Scholar] [CrossRef] [PubMed]
- Stella, N.; Schweitzer, P.J.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nat. Cell Biol. 1997, 388, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Viader, A.; Blankman, J.L.; Zhong, P.; Liu, X.; Schlosburg, J.E.; Joslyn, C.M.; Liu, Q.-S.; Tomarchio, A.J.; Lichtman, A.H.; Selley, D.E.; et al. Metabolic Interplay between Astrocytes and Neurons Regulates Endocannabinoid Action. Cell Rep. 2015, 12, 798–808. [Google Scholar] [CrossRef] [Green Version]
- Viader, A.; Ogasawara, D.; Joslyn, C.M.; Sanchez-Alavez, M.; Mori, S.; Nguyen, W.; Conti, B.; Cravatt, B.F. A chemical proteomic atlas of brain serine hydrolases identifies cell type-specific pathways regulating neuroinflammation. eLife 2016, 5, e12345. [Google Scholar] [CrossRef]
- Tornqvist, H.; Belfrage, P. Purification and some properties of a monoacylglycerol-hydrolyzing enzyme of rat adipose tissue. J. Biol. Chem. 1976, 251, 813–819. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [Green Version]
- Long, J.Z.; Nomura, D.K.; Cravatt, B.F. Characterization of Monoacylglycerol Lipase Inhibition Reveals Differences in Central and Peripheral Endocannabinoid Metabolism. Chem. Biol. 2009, 16, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, J.A.; Higgs, G.A. Prostaglandins and leukotrienes as inflammatory mediators. Br. Med Bull. 1987, 43, 285–296. [Google Scholar] [CrossRef]
- Serhan, C.N. Resolution Phase of Inflammation: Novel Endogenous Anti-Inflammatory and Proresolving Lipid Mediators and Pathways. Annu. Rev. Immunol. 2007, 25, 101–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Hamberg, M.; Samuelsson, B. Lipoxins: Novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. USA 1984, 81, 5335–5339. [Google Scholar] [CrossRef] [Green Version]
- Martini, A.C.; Forner, S.; Bento, A.F.; Rae, G.A. Neuroprotective Effects of Lipoxin A4 in Central Nervous System Pathologies. BioMed Res. Int. 2014, 2014, 316204. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.-H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef] [Green Version]
- Schaldach, C.M.; Riby, J.; Bjeldanes, L.F. Lipoxin A4: A New Class of Ligand for the Ah Receptor. Biochemistry 1999, 38, 7594–7600. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, J.; Wu, Y.; Wang, D.; Feng, G.; Tang, Y.-P.; Teng, Z.; Chen, C. Monoacylglycerol Lipase Is a Therapeutic Target for Alzheimer’s Disease. Cell Rep. 2012, 2, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Hashem, J.; Hu, M.; Zhang, J.; Gao, F.; Chen, C. Inhibition of 2-Arachidonoylglycerol Metabolism Alleviates Neuropathology and Improves Cognitive Function in a Tau Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 4122–4133. [Google Scholar] [CrossRef]
- Zhang, J.; Teng, Z.-Q.; Song, Y.; Hu, M.; Chen, C. Inhibition of Monoacylglycerol Lipase Prevents Chronic Traumatic Encephalopathy-like Neuropathology in a Mouse Model of Repetitive Mild Closed Head Injury. Br. J. Pharmacol. 2015, 35, 443–453. [Google Scholar] [CrossRef]
- Shi, K.; Zhang, J.; Dong, J.-F.; Shi, F.-D. Dissemination of brain inflammation in traumatic brain injury. Cell. Mol. Immunol. 2019, 16, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Suh, H.-S.; Zhao, M.-L.; Derico, L.; Choi, N.; Lee, S.C. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: Differential regulation by inflammatory mediators. J. Neuroinflamm. 2013, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Popiolek-Barczyk, K.; Ciechanowska, A.; Ciapała, K.; Pawlik, K.; Oggioni, M.; Mercurio, D.; De Simoni, M.-G.; Mika, J. The CCL2/CCL7/CCL12/CCR2 pathway is substantially and persistently upregulated in mice after traumatic brain injury, and CCL2 modulates the complement system in microglia. Mol. Cell. Probes 2020, 54, 101671. [Google Scholar] [CrossRef]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 2015, 173, 692–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, P.S.; Sulzer, J.K.; Impastato, R.A.; Teng, S.X.; Rogers, E.K.; Molina, P.E. Endocannabinoid Degradation Inhibition Improves Neurobehavioral Function, Blood–Brain Barrier Integrity, and Neuroinflammation following Mild Traumatic Brain Injury. J. Neurotrauma 2015, 32, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Mayeux, J.; Katz, P.; Edwards, S.; Middleton, J.W.; Molina, P.E. Inhibition of Endocannabinoid Degradation Improves Outcomes from Mild Traumatic Brain Injury: A Mechanistic Role for Synaptic Hyperexcitability. J. Neurotrauma 2017, 34, 436–443. [Google Scholar] [CrossRef]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.-L.; Li, Q.-Q.; Chen, X.-P.; Zhang, X.-M.; Li, L.; Li, B.-X.; Zhao, Z.-Q.; Tao, L.-Y. Lipoxin A4 attenuates brain damage and downregulates the production of pro-inflammatory cytokines and phosphorylated mitogen-activated protein kinases in a mouse model of traumatic brain injury. Brain Res. 2013, 1502, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, C. Endocannabinoid metabolism in neurodegenerative diseases. Neuroimmunol. Neuroinflamm. 2016, 3, 268–270. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, E.; Kelly, E.; Liu, Y.; Giordano, C.; Wallace, E.; Hynes, M.; Tiernan, S.; Meagher, A.; Greene, C.; Hughes, S.; et al. Dynamic Blood–Brain Barrier Regulation in Mild Traumatic Brain Injury. J. Neurotrauma 2020, 37, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Tchantchou, F.; Zhang, Y. Selective Inhibition of Alpha/Beta-Hydrolase Domain 6 Attenuates Neurodegeneration, Alleviates Blood Brain Barrier Breakdown, and Improves Functional Recovery in a Mouse Model of Traumatic Brain Injury. J. Neurotrauma 2013, 30, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hardy, J.; Zetterberg, H. The Neuropathology and Neurobiology of Traumatic Brain Injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.H.; Johnson, V.E.; Trojanowski, J.Q.; Stewart, W. Chronic traumatic encephalopathy—Confusion and controversies. Nat. Rev. Neurol. 2019, 15, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Kramer, E.-M.; Cardine, A.-M.; Schraven, B.; Brandt, R.; Trotter, J. Process Outgrowth of Oligodendrocytes Is Promoted by Interaction of Fyn Kinase with the Cytoskeletal Protein Tau. J. Neurosci. 2002, 22, 698–707. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, K.; Hasegawa, M.; Ihara, Y.; Iwatsubo, T. Somatodendritic localization of phosphorylated tau in neonatal and adult rat cerebral cortex. NeuroReport 1997, 8, 2797–2801. [Google Scholar] [CrossRef]
- Edwards, G.; Moreno-Gonzalez, I.; Soto, C. Amyloid-beta and tau pathology following repetitive mild traumatic brain injury. Biochem. Biophys. Res. Commun. 2017, 483, 1137–1142. [Google Scholar] [CrossRef]
- Edwards, G.; Zhao, J.; Dash, P.K.; Soto, C.; Moreno-Gonzalez, I. Traumatic Brain Injury Induces Tau Aggregation and Spreading. J. Neurotrauma 2020, 37, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Zanier, E.; Bertani, I.; Sammali, E.; Pischiutta, F.; Chiaravalloti, M.A.; Vegliante, G.; Masone, A.; Corbelli, A.; Smith, D.H.; Menon, D.K.; et al. Induction of a transmissible tau pathology by traumatic brain injury. Brain 2018, 141, 2685–2699. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen-Plotkin, A.S.; Lee, V.M.-Y.; Trojanowski, J.Q. TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geser, F.; Martinez-Lage, M.; Kwong, L.K.; Lee, V.M.-Y.; Trojanowski, J.Q. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: The TDP-43 diseases. J. Neurol. 2009, 256, 1205–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Kwong, L.K.; Sampathu, D.M.; Trojanowski, J.Q.; Lee, V.M.-Y. TDP-43 Proteinopathy in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Arch. Neurol. 2007, 64, 1388–1394. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.L.; Sun, M.; Brady, R.D.; Liu, S.J.; Llanos, R.; Cheung, S.; Wright, D.K.; Casillas-Espinosa, P.M.; Sashindranath, M.; O’Brien, T.J.; et al. Transactive Response DNA-Binding Protein 43 Abnormalities after Traumatic Brain Injury. J. Neurotrauma 2019, 36, 87–99. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Gavett, B.; Stern, R.; Nowinski, C.J.; Cantu, R.C.; Kowall, N.W.; Perl, D.P.; Hedley-Whyte, E.T.; Price, B.; Sullivan, C.; et al. TDP-43 Proteinopathy and Motor Neuron Disease in Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2010, 69, 918–929. [Google Scholar] [CrossRef]
- Yang, Z.; Lin, F.; Robertson, C.S.; Wang, K.K.W. Dual Vulnerability of TDP-43 to Calpain and Caspase-3 Proteolysis after Neurotoxic Conditions and Traumatic Brain Injury. Br. J. Pharmacol. 2014, 34, 1444–1452. [Google Scholar] [CrossRef] [Green Version]
- Roberts, G.W.; Gentleman, S.M.; Lynch, A.; Murray, L.; Landon, M.; Graham, D.I. Beta amyloid protein deposition in the brain after severe head injury: Implications for the pathogenesis of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1994, 57, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Uryu, K.; Chen, X.-H.; Martinez, D.; Browne, K.D.; Johnson, V.E.; Graham, D.I.; Lee, V.M.-Y.; Trojanowski, J.Q.; Smith, D.H. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 2007, 208, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Gerrow, K.; Triller, A. Synaptic stability and plasticity in a floating world. Curr. Opin. Neurobiol. 2010, 20, 631–639. [Google Scholar] [CrossRef]
- Miyazaki, S.; Katayama, Y.; Lyeth, B.; Jenkins, L.; DeWitt, D.; Goldberg, S.; Newlon, P.; Hayes, R. Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in rat. Brain Res. 1992, 585, 335–339. [Google Scholar] [CrossRef]
- Hernandez, A.; Tan, C.; Plattner, F.; Logsdon, A.F.; Pozo, K.; Yousuf, M.A.; Singh, T.; Turner, R.C.; Luke-Wold, B.P.; Huber, J.D.; et al. Exposure to mild blast forces induces neuropathological effects, neurophysiological deficits and biochemical changes. Mol. Brain 2018, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucich, E.A.; Stielper, Z.F.; Cancienne, H.L.; Edwards, S.; Gilpin, N.W.; Molina, P.E.; Middleton, J.W. Endocannabinoid degradation inhibitors ameliorate neuronal and synaptic alterations following traumatic brain injury. J. Neurophysiol. 2020, 123, 707–717. [Google Scholar] [CrossRef]
- Chauhan, N.B. Chronic neurodegenerative consequences of traumatic brain injury. Restor. Neurol. Neurosci. 2014, 32, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, J.; Sabbir, M.G.; Albensi, B.C. Traumatic Brain Injury as a Risk Factor for Alzheimer’s Disease: Is Inflammatory Signaling a Key Player? Curr. Alzheimer Res. 2016, 13, 730–738. [Google Scholar] [CrossRef]
- Smith, D.H.; Johnson, V.E.; Stewart, W. Chronic neuropathologies of single and repetitive TBI: Substrates of dementia? Nat. Rev. Neurol. 2013, 9, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Ordóñez, A.; Martin-Fontecha, M.; Ortega-Gutiérrez, S.; López-Rodríguez, M.L. Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem. Pharmacol. 2018, 157, 18–32. [Google Scholar] [CrossRef]
- Piro, J.; Benjamin, D.I.; Duerr, J.M.; Pi, Y.; Gonzales, C.; Wood, K.M.; Schwartz, J.W.; Nomura, D.K.; Samad, T.A. A Dysregulated Endocannabinoid-Eicosanoid Network Supports Pathogenesis in a Mouse Model of Alzheimer’s Disease. Cell Rep. 2012, 1, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, C. Alleviation of Neuropathology by Inhibition of Monoacylglycerol Lipase in APP Transgenic Mice Lacking CB2 Receptors. Mol. Neurobiol. 2017, 55, 4802–4810. [Google Scholar] [CrossRef]
- Dong, M.; Lu, Y.; Zou, Z.; Yang, H. Monoacylglycerol lipase inhibitor protects primary cultured neurons against homocysteine-induced impairments in rat caudate nucleus through COX-2 signaling. Life Sci. 2015, 138, 64–71. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Rockwell, C.; Snider, N.T.; Thompson, J.T.; Heuvel, J.P.V.; Kaminski, N.E. Interleukin-2 Suppression by 2-Arachidonyl Glycerol Is Mediated through Peroxisome Proliferator-Activated Receptor γ Independently of Cannabinoid Receptors 1 and 2. Mol. Pharmacol. 2006, 70, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chu, C.; Teng, Z.; Tang, Y.-P.; Chen, C. Synaptic and Cognitive Improvements by Inhibition of 2-AG Metabolism Are through Upregulation of MicroRNA-188-3p in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2014, 34, 14919–14933. [Google Scholar] [CrossRef] [Green Version]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nat. Cell Biol. 2008, 454, 470–477. [Google Scholar] [CrossRef]
- Sauerbeck, A.; Gao, J.; Readnower, R.; Liu, M.; Pauly, J.R.; Bing, G.; Sullivan, P.G. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp Neurol. 2011, 227, 128–135. [Google Scholar] [CrossRef] [Green Version]
- Cisar, J.S.; Weber, O.D.; Clapper, J.R.; Blankman, J.L.; Henry, C.L.; Simon, G.M.; Alexander, J.P.; Jones, T.K.; Ezekowitz, R.A.B.; O’Neill, G.P.; et al. Identification of ABX-1431, a Selective Inhibitor of Monoacylglycerol Lipase and Clinical Candidate for Treatment of Neurological Disorders. J. Med. Chem. 2018, 61, 9062–9084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.W.; Cognetta, I.A.B.; Niphakis, M.J.; Cravatt, B.F. Proteome-Wide Reactivity Profiling Identifies Diverse Carbamate Chemotypes Tuned for Serine Hydrolase Inhibition. ACS Chem. Biol. 2013, 8, 1590–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.Z.; Li, W.; Booker, L.; Burston, J.J.; Kinsey, S.G.; Schlosburg, J.E.; Pavón, F.J.; Serrano, A.M.; Selley, D.E.; Parsons, L.H.; et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009, 5, 37–44. [Google Scholar] [CrossRef] [Green Version]
- King, A.R.; Dotsey, E.Y.; Lodola, A.; Jung, K.-M.; Ghomian, A.; Qiu, Y.; Fu, J.; Mor, M.; Piomelli, D. Discovery of Potent and Reversible Monoacylglycerol Lipase Inhibitors. Chem. Biol. 2009, 16, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Torres, G.; Cipriano, M.; Hedén, E.; Björklund, E.; Canales, Á.; Zian, M.S.D.; Feliú, M.S.A.; Mecha, M.; Guaza, C.; Fowler, C.J.; et al. A Reversible and Selective Inhibitor of Monoacylglycerol Lipase Ameliorates Multiple Sclerosis. Angew. Chem. Int. Ed. 2014, 53, 13765–13770. [Google Scholar] [CrossRef]
- Tuccinardi, T.; Granchi, C.; Rizzolio, F.; Caligiuri, I.; Battistello, V.; Toffoli, G.; Minutolo, F.; Macchia, M.; Martinelli, A. Identification and characterization of a new reversible MAGL inhibitor. Bioorg. Med. Chem. 2014, 22, 3285–3291. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, G.M.; Kim, J.-K. Anti-Inflammatory Effect of Pristimerin on Lipopolysaccharide-Induced Inflammatory Responses in Murine Macrophages. Arch. Pharmacal. Res. 2013, 36, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Yuan, S.; Wang, X.; Lei, Y.; Zhang, X.; Huang, M.; Ouyang, H. Attenuation of pristimerin on TNF-α-induced endothelial inflammation. Int. Immunopharmacol. 2020, 82, 106326. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Bi, Y.; Zhong, J.; Ren, Z.; Liu, Y.; Jia, J.; Yu, M.; Tan, Y.; Zhang, Q.; Yu, X. Pristimerin suppresses colorectal cancer through inhibiting inflammatory responses and Wnt/β-catenin signaling. Toxicol. Appl. Pharmacol. 2020, 386, 114813. [Google Scholar] [CrossRef]
- Shaaban, A.A.; El-Kashef, D.H.; Hamed, M.; El-Agamy, D.S. Protective effect of pristimerin against LPS-induced acute lung injury in mice. Int. Immunopharmacol. 2018, 59, 31–39. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, D.; Gao, F.; Chen, C. Endocannabinoid Metabolism and Traumatic Brain Injury. Cells 2021, 10, 2979. https://doi.org/10.3390/cells10112979
Zhu D, Gao F, Chen C. Endocannabinoid Metabolism and Traumatic Brain Injury. Cells. 2021; 10(11):2979. https://doi.org/10.3390/cells10112979
Chicago/Turabian StyleZhu, Dexiao, Fei Gao, and Chu Chen. 2021. "Endocannabinoid Metabolism and Traumatic Brain Injury" Cells 10, no. 11: 2979. https://doi.org/10.3390/cells10112979
APA StyleZhu, D., Gao, F., & Chen, C. (2021). Endocannabinoid Metabolism and Traumatic Brain Injury. Cells, 10(11), 2979. https://doi.org/10.3390/cells10112979