
Lipid Metabolism Disorders in the Comorbid Course of Nonalcoholic Fatty Liver Disease and Chronic Obstructive Pulmonary Disease



{kind=link}
{kind=link}
{kind=link}
Abstract
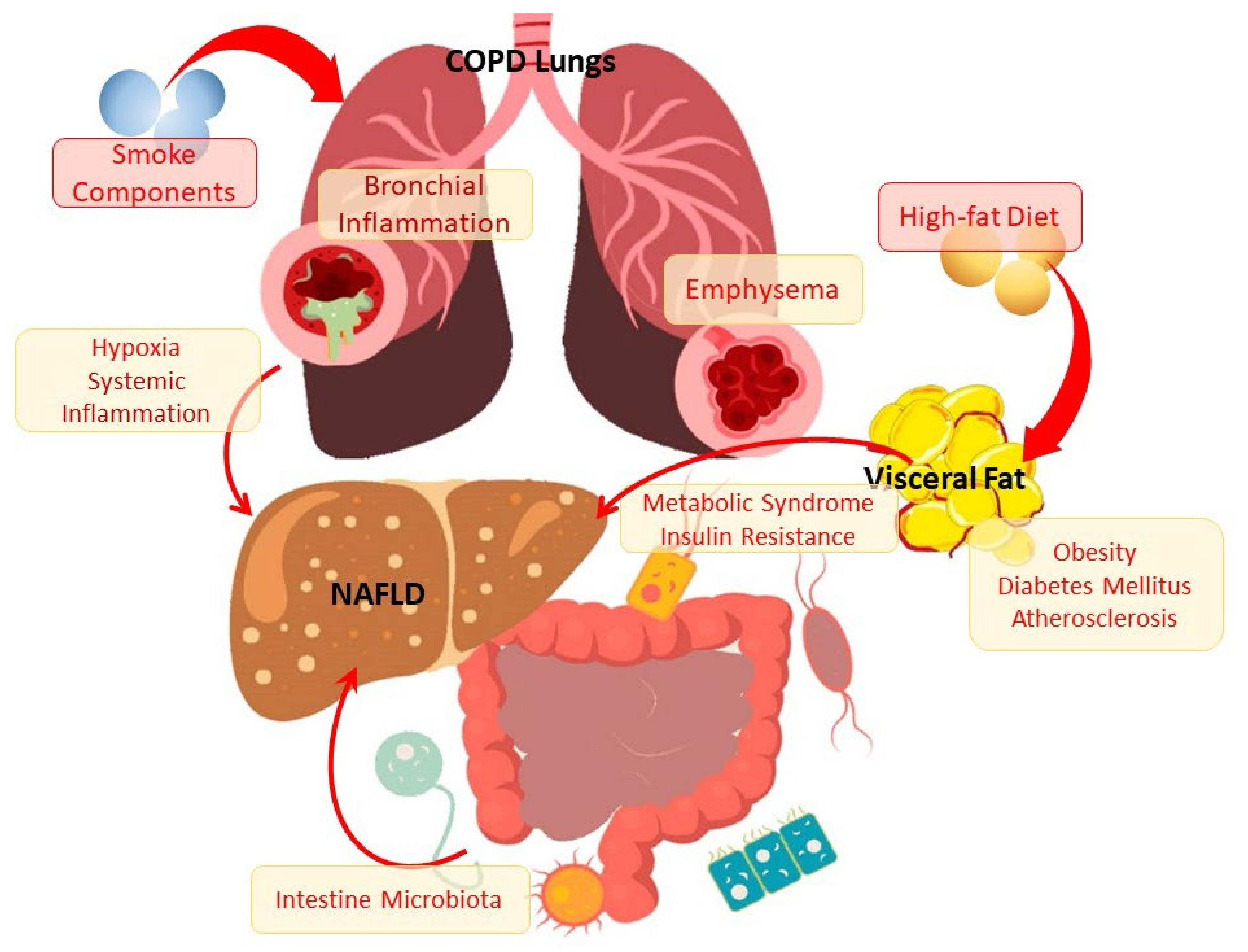
1. Introduction
2. Clinical, Morphological, and Pathogenetic Characteristics of NAFLD
3. Disorders of Lipid Metabolism in the Development and Progression of NAFLD
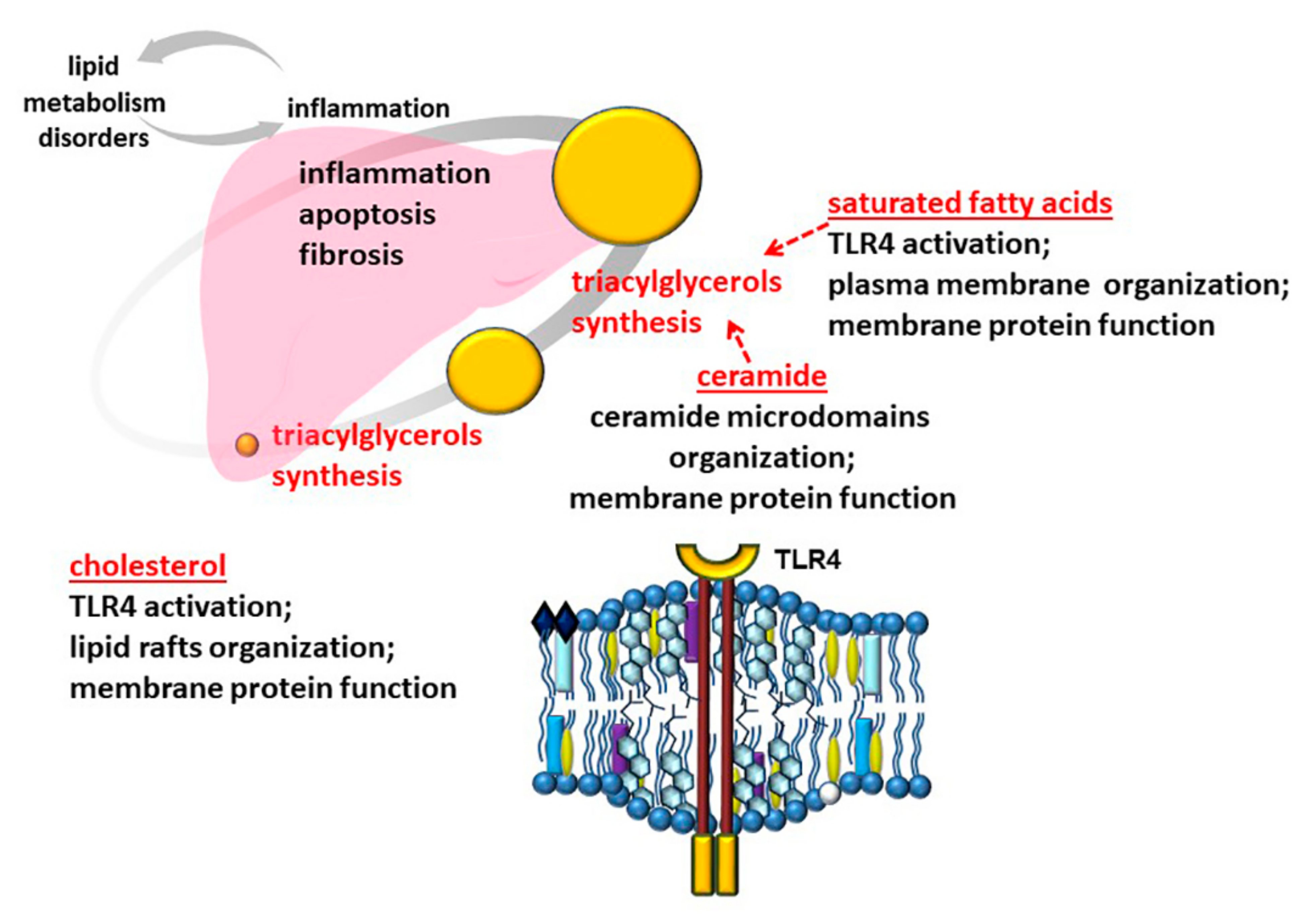
3.1. Role of Free Fatty Acids
3.2. Role of Cholesterol
3.3. Role of Ceramide
4. Lipid Metabolism Disorders in the Development and Progression of COPD
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, M.; Papadakis, S.; Linardakis, M.; Anyfantakis, D.; Symvoulakis, E.K.; Lionis, C.; Cretan Primary Care Research Group. Burden of metabolic syndrome among primary care patients in Crete, Greece: A descriptive study. Eur. J. Gen. Pract. 2020, 26, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Mehata, S.; Shrestha, N.; Mehta, R.K.; Bista, B.; Pandey, A.R.; Mishra, S.R. Prevalence of the Metabolic Syndrome and its determinants among Nepalese adults: Findings from a nationally representative cross-sectional study. Sci. Rep. 2018, 8, 14995. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef]
- Ge, X.; Zheng, L.; Wang, M.; Du, Y.; Jiang, J. Prevalence trends in non-alcoholic fatty liver disease at the global, regional and national levels, 1990-2017: A population-based observational study. BMJ Open 2020, 10, e036663. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; Miele, L.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef]
- Goldberg, D.; Ditah, I.C.; Saeian, K.; Lalehzari, M.; Aronsohn, A.; Gorospe, E.C.; Charlton, M. Changes in the Prevalence of Hepatitis C Virus Infection, Nonalcoholic Steatohepatitis, and Alcoholic Liver Disease among Patients with Cirrhosis or Liver Failure on the Waitlist for Liver Transplantation. Gastroenterology 2017, 152, 1090–1099.e1091. [Google Scholar] [CrossRef]
- Starley, B.Q.; Calcagno, C.J.; Harrison, S.A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: A weighty connection. Hepatology 2010, 51, 1820–1832. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Negro, F.; Hallaji, S.; Younossi, Y.; Lam, B.; Srishord, M. Nonalcoholic fatty liver disease in lean individuals in the United States. Medicine 2012, 91, 319–327. [Google Scholar] [CrossRef]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Andreoletti, M.; Colli, A.; Vanni, E.; Bertelli, C.; Fatta, E.; Bignamini, D.; Marchesini, G.; et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: A role for insulin resistance and diabetes. Hepatology 2008, 48, 792–798. [Google Scholar] [CrossRef] [PubMed]
- The Italian Association for the Study of the Liver (AISF). AISF position paper on nonalcoholic fatty liver disease (NAFLD): Updates and future directions. Dig. Liver Dis. 2017, 49, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Bellentani, S.; Cortez-Pinto, H.; Day, C.; Marchesini, G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol. 2010, 53, 372–384. [Google Scholar] [CrossRef]
- Bellentani, S.; Saccoccio, G.; Masutti, F.; Crocè, L.S.; Brandi, G.; Sasso, F.; Cristanini, G.; Tiribelli, C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann. Intern. Med. 2000, 132, 112–117. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J.; Panel, I.C. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Targher, G.; Loria, P. Diagnosis and management of cardiovascular risk in nonalcoholic fatty liver disease. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 629–650. [Google Scholar] [CrossRef]
- Viglino, D.; Jullian-Desayes, I.; Minoves, M.; Aron-Wisnewsky, J.; Leroy, V.; Zarski, J.P.; Tamisier, R.; Joyeux-Faure, M.; Pépin, J.L. Nonalcoholic fatty liver disease in chronic obstructive pulmonary disease. Eur. Respir. J. 2017, 49, 1601923. [Google Scholar] [CrossRef] [PubMed]
- Viglino, D.; Plazanet, A.; Bailly, S.; Benmerad, M.; Jullian-Desayes, I.; Tamisier, R.; Leroy, V.; Zarski, J.P.; Maignan, M.; Joyeux-Faure, M.; et al. Impact of Non-alcoholic Fatty Liver Disease on long-term cardiovascular events and death in Chronic Obstructive Pulmonary Disease. Sci. Rep. 2018, 8, 16559. [Google Scholar] [CrossRef] [PubMed]
- Song, J.U.; Jang, Y.; Lim, S.Y.; Ryu, S.; Song, W.J.; Byrne, C.D.; Sung, K.C. Decreased lung function is associated with risk of developing non-alcoholic fatty liver disease: A longitudinal cohort study. PLoS ONE 2019, 14, e0208736. [Google Scholar] [CrossRef]
- Jung, D.H.; Shim, J.Y.; Lee, H.R.; Moon, B.S.; Park, B.J.; Lee, Y.J. Relationship between non-alcoholic fatty liver disease and pulmonary function. Intern. Med. J. 2012, 42, 541–546. [Google Scholar] [CrossRef]
- Glass, L.M.; Hunt, C.M.; Fuchs, M.; Su, G.L. Comorbidities and Nonalcoholic Fatty Liver Disease: The Chicken, the Egg, or Both? Fed. Pract. 2019, 36, 64–71. [Google Scholar]
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Lonardo, A. The independent predictors of non-alcoholic steatohepatitis and its individual histological features.: Insulin resistance, serum uric acid, metabolic syndrome, alanine aminotransferase and serum total cholesterol are a clue to pathogenesis and candidate targets for treatment. Hepatol. Res. 2016, 46, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef]
- Tana, C.; Ballestri, S.; Ricci, F.; Di Vincenzo, A.; Ticinesi, A.; Gallina, S.; Giamberardino, M.A.; Cipollone, F.; Sutton, R.; Vettor, R.; et al. Cardiovascular Risk in Non-Alcoholic Fatty Liver Disease: Mechanisms and Therapeutic Implications. Int. J. Environ. Res. Public Health 2019, 16, 3104. [Google Scholar] [CrossRef] [PubMed]
- Misra, V.L.; Khashab, M.; Chalasani, N. Nonalcoholic fatty liver disease and cardiovascular risk. Curr. Gastroenterol. Rep. 2009, 11, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2021, 110, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Bang, K.B.; Cho, Y.K. Comorbidities and Metabolic Derangement of NAFLD. J. Lifestyle Med. 2015, 5, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Rosato, V.; Masarone, M.; Dallio, M.; Federico, A.; Aglitti, A.; Persico, M. NAFLD and Extra-Hepatic Comorbidities: Current Evidence on a Multi-Organ Metabolic Syndrome. Int. J. Environ. Res. Public Health 2019, 16, 3415. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Inflammatory mechanisms in the regulation of insulin resistance. Mol. Med. 2008, 14, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, E.; Krawczyk, M.; Stachowska, E.; Lammert, F.; Portincasa, P. Non-Alcoholic Fatty Liver Disease in Non-Obese Individuals: Prevalence, Pathogenesis and Treatment. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Tobari, M.; Hashimoto, E.; Taniai, M.; Ikarashi, Y.; Kodama, K.; Kogiso, T.; Tokushige, K.; Takayoshi, N.; Hashimoto, N. Characteristics of non-alcoholic steatohepatitis among lean patients in Japan: Not uncommon and not always benign. J. Gastroenterol. Hepatol. 2019, 34, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.C.; Lübbe, F.; Bressem, K.; Wagner, M.; Hamm, B.; Makowski, M.R. Non-alcoholic fatty liver disease in underweight patients with inflammatory bowel disease: A case-control study. PLoS ONE 2018, 13, e0206450. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, M.V.; Waddell, T.; Banerjee, R.; Guess, N. Nutrition and Nonalcoholic Fatty Liver Disease: Current Perspectives. Gastroenterol. Clin. N. Am. 2020, 49, 63–94. [Google Scholar] [CrossRef] [PubMed]
- Braunersreuther, V.; Viviani, G.L.; Mach, F.; Montecucco, F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.C.; Tai, M.S.; Chan, W.K.; Mahadeva, S. Association between non-alcoholic fatty liver disease evaluated by transient elastography with extracranial carotid atherosclerosis in a multiethnic Asian community. JGH Open 2019, 3, 117–125. [Google Scholar] [CrossRef]
- Das, K.; Mukherjee, P.S.; Ghosh, A.; Ghosh, S.; Mridha, A.R.; Dhibar, T.; Bhattacharya, B.; Bhattacharya, D.; Manna, B.; Dhali, G.K.; et al. Nonobese population in a developing country has a high prevalence of nonalcoholic fatty liver and significant liver disease. Hepatology 2010, 51, 1593–1602. [Google Scholar] [CrossRef]
- Chen, F.; Esmaili, S.; Rogers, G.B.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A Distinct Entity Shaped by Differential Metabolic Adaptation. Hepatology 2020, 71, 1213–1227. [Google Scholar] [CrossRef]
- Young, S.; Tariq, R.; Provenza, J.; Satapathy, S.K.; Faisal, K.; Choudhry, A.; Friedman, S.L.; Singal, A.K. Prevalence and Profile of Nonalcoholic Fatty Liver Disease in Lean Adults: Systematic Review and Meta-Analysis. Hepatol. Commun. 2020, 4, 953–972. [Google Scholar] [CrossRef]
- Kobyliak, N.; Abenavoli, L.; Mykhalchyshyn, G.; Kononenko, L.; Boccuto, L.; Kyriienko, D.; Dynnyk, O. A Multi-strain Probiotic Reduces the Fatty Liver Index, Cytokines and Aminotransferase levels in NAFLD Patients: Evidence from a Randomized Clinical Trial. J. Gastrointestin. Liver Dis. 2018, 27, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.G.; Kim, S.U.; Wong, V.W. New trends on obesity and NAFLD in Asia. J. Hepatol. 2017, 67, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.; Malik, V.; Jia, W.; Kadowaki, T.; Yajnik, C.S.; Yoon, K.H.; Hu, F.B. Diabetes in Asia: Epidemiology, risk factors, and pathophysiology. JAMA 2009, 301, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Uz-Zaman, M.H.; Rahman, A.; Yasmin, M. Epidemiology of Hepatitis B Virus Infection in Bangladesh: Prevalence among General Population, Risk Groups and Genotype Distribution. Genes 2018, 9, 541. [Google Scholar] [CrossRef]
- Rahman, M.M.; Kibria, M.G.; Begum, H.; Haque, M.; Sultana, N.; Akhter, M.; Rowshon, A.H.M.; Ahmed, F.; Hasan, M. Prevalence, risk factors and metabolic profile of the non-obese and obese non-alcoholic fatty liver disease in a rural community of South Asia. BMJ Open Gastroenterol. 2020, 7, e000535. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, W.; Joo, S.K.; Kim, J.H.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Predictors of nonalcoholic steatohepatitis and significant fibrosis in non-obese nonalcoholic fatty liver disease. Liver Int. 2019, 39, 332–341. [Google Scholar] [CrossRef]
- Chang, Y.; Ryu, S.; Sung, K.C.; Cho, Y.K.; Sung, E.; Kim, H.N.; Jung, H.S.; Yun, K.E.; Ahn, J.; Shin, H.; et al. Alcoholic and non-alcoholic fatty liver disease and associations with coronary artery calcification: Evidence from the Kangbuk Samsung Health Study. Gut 2019, 68, 1667–1675. [Google Scholar] [CrossRef]
- Yoshitaka, H.; Hamaguchi, M.; Kojima, T.; Fukuda, T.; Ohbora, A.; Fukui, M. Nonoverweight nonalcoholic fatty liver disease and incident cardiovascular disease: A post hoc analysis of a cohort study. Medicine 2017, 96, e6712. [Google Scholar] [CrossRef]
- Qureshi, K.; Abrams, G.A. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 3540–3553. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Liu, Y.L. Non-alcoholic fatty liver disease: The problems we are facing. Hepatobiliary Pancreat. Dis. Int. 2003, 2, 334–337. [Google Scholar] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tripathi, M.; Sinha, R.A.; Singh, B.K.; Yen, P.M. Gut microbiota and their metabolites in the progression of non-alcoholic fatty liver disease. Hepatoma Res. 2021, 7, 11. [Google Scholar] [CrossRef]
- Poss, A.M.; Summers, S.A. Too Much of a Good Thing? An Evolutionary Theory to Explain the Role of Ceramides in NAFLD. Front. Endocrinol. 2020, 11, 505. [Google Scholar] [CrossRef]
- Tamura, S.; Shimomura, I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1139–1142. [Google Scholar] [CrossRef]
- Sahini, N.; Borlak, J. Recent insights into the molecular pathophysiology of lipid droplet formation in hepatocytes. Prog. Lipid Res. 2014, 54, 86–112. [Google Scholar] [CrossRef]
- Tauchi-Sato, K.; Ozeki, S.; Houjou, T.; Taguchi, R.; Fujimoto, T. The surface of lipid droplets is a phospholipid monolayer with a unique Fatty Acid composition. J. Biol. Chem. 2002, 277, 44507–44512. [Google Scholar] [CrossRef] [PubMed]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef]
- Wang, H.; Quiroga, A.D.; Lehner, R. Analysis of lipid droplets in hepatocytes. Methods Cell Biol. 2013, 116, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Onal, G.; Kutlu, O.; Gozuacik, D.; Dokmeci Emre, S. Lipid Droplets in Health and Disease. Lipids Health Dis. 2017, 16, 128. [Google Scholar] [CrossRef] [PubMed]
- Farese, R.V.; Walther, T.C. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 2009, 139, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.S.; Seow, C.J.; Goh, V.J.; Silver, D.L. Recent advances in understanding proteins involved in lipid droplet formation, growth and fusion. J. Genet. Genom. 2014, 41, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G. Hepatic lipid droplets: A balancing act between energy storage and metabolic dysfunction in NAFLD. Mol. Metab. 2021, 50, 101115. [Google Scholar] [CrossRef]
- Schott, M.B.; Weller, S.G.; Schulze, R.J.; Krueger, E.W.; Drizyte-Miller, K.; Casey, C.A.; McNiven, M.A. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 2019, 218, 3320–3335. [Google Scholar] [CrossRef]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V.; et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef]
- McClain, C.J.; Barve, S.; Deaciuc, I. Good fat/bad fat. Hepatology 2007, 45, 1343–1346. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 445–451. [Google Scholar] [CrossRef]
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, Y.; Xu, C.; Hong, Y.; Lu, H.; Wu, J.; Chen, Y. Association between serum free fatty acid levels and nonalcoholic fatty liver disease: A cross-sectional study. Sci. Rep. 2014, 4, 5832. [Google Scholar] [CrossRef]
- Gambino, R.; Bugianesi, E.; Rosso, C.; Mezzabotta, L.; Pinach, S.; Alemanno, N.; Saba, F.; Cassader, M. Different Serum Free Fatty Acid Profiles in NAFLD Subjects and Healthy Controls after Oral Fat Load. Int. J. Mol. Sci. 2016, 17, 479. [Google Scholar] [CrossRef]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: Role of stearoyl-CoA desaturase. J. Biol. Chem. 2009, 284, 5637–5644. [Google Scholar] [CrossRef] [PubMed]
- Fernández Gianotti, T.; Burgueño, A.; Gonzales Mansilla, N.; Pirola, C.J.; Sookoian, S. Fatty liver is associated with transcriptional downregulation of stearoyl-CoA desaturase and impaired protein dimerization. PLoS ONE 2013, 8, e76912. [Google Scholar] [CrossRef]
- Silbernagel, G.; Kovarova, M.; Cegan, A.; Machann, J.; Schick, F.; Lehmann, R.; Häring, H.U.; Stefan, N.; Schleicher, E.; Fritsche, A.; et al. High hepatic SCD1 activity is associated with low liver fat content in healthy subjects under a lipogenic diet. J. Clin. Endocrinol. Metab. 2012, 97, E2288–E2292. [Google Scholar] [CrossRef]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110.e1095. [Google Scholar] [CrossRef]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Obesity and free fatty acids. Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.H.; Kim, J.A.; Lee, J.Y. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur. J. Pharmacol. 2016, 785, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef]
- Kang, H.H.; Kim, I.K.; Lee, H.I.; Joo, H.; Lim, J.U.; Lee, J.; Lee, S.H.; Moon, H.S. Chronic intermittent hypoxia induces liver fibrosis in mice with diet-induced obesity via TLR4/MyD88/MAPK/NF-kB signaling pathways. Biochem. Biophys. Res. Commun. 2017, 490, 349–355. [Google Scholar] [CrossRef]
- Heyens, L.J.M.; Busschots, D.; Koek, G.H.; Robaeys, G.; Francque, S. Liver Fibrosis in Non-alcoholic Fatty Liver Disease: From Liver Biopsy to Non-invasive Biomarkers in Diagnosis and Treatment. Front. Med. 2021, 8, 615978. [Google Scholar] [CrossRef]
- Yong, S.H.; Leem, A.Y.; Kim, Y.S.; Park, M.S.; Chang, J.; Kim, S.U.; Jung, J.Y. Hepatic Fibrosis Assessed Using Fibrosis-4 Index Is Predictive of All-Cause Mortality in Patients with Chronic Obstructive Pulmonary Disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 831–839. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Ther. 2017, 179, 142–157. [Google Scholar] [CrossRef]
- Burns, K.A.; Vanden Heuvel, J.P. Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta 2007, 1771, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.; Siersbæk, M.; Mandrup, S. PPARs: Fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef]
- Sozen, E.; Ozer, N.K. Impact of high cholesterol and endoplasmic reticulum stress on metabolic diseases: An updated mini-review. Redox Biol. 2017, 12, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef]
- Puri, P.; Wiest, M.M.; Cheung, O.; Mirshahi, F.; Sargeant, C.; Min, H.K.; Contos, M.J.; Sterling, R.K.; Fuchs, M.; Zhou, H.; et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1827–1838. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; De Lacy, A.M.; Fernández-Checa, J.C.; Caballería, J.; García-Ruiz, C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Haigh, W.G.; Thorning, D.; Savard, C. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J. Lipid Res. 2013, 54, 1326–1334. [Google Scholar] [CrossRef]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164.e110. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, P.; Gill, R.K.; Saksena, S.; Alrefai, W.A. Disturbances in Cholesterol Homeostasis and Non-alcoholic Fatty Liver Diseases. Front. Med. 2020, 7, 467. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Pathak, P.; Boehme, S.; Chiang, J.L. Cholesterol 7α-hydroxylase protects the liver from inflammation and fibrosis by maintaining cholesterol homeostasis. J. Lipid Res. 2016, 57, 1831–1844. [Google Scholar] [CrossRef]
- Min, H.K.; Kapoor, A.; Fuchs, M.; Mirshahi, F.; Zhou, H.; Maher, J.; Kellum, J.; Warnick, R.; Contos, M.J.; Sanyal, A.J. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012, 15, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Vega-Badillo, J.; Gutiérrez-Vidal, R.; Hernández-Pérez, H.A.; Villamil-Ramírez, H.; León-Mimila, P.; Sánchez-Muñoz, F.; Morán-Ramos, S.; Larrieta-Carrasco, E.; Fernández-Silva, I.; Méndez-Sánchez, N.; et al. Hepatic miR-33a/miR-144 and their target gene ABCA1 are associated with steatohepatitis in morbidly obese subjects. Liver Int. 2016, 36, 1383–1391. [Google Scholar] [CrossRef]
- Hussein, M.A.; Shrestha, E.; Ouimet, M.; Barrett, T.J.; Leone, S.; Moore, K.J.; Hérault, Y.; Fisher, E.A.; Garabedian, M.J. LXR-Mediated ABCA1 Expression and Function Are Modulated by High Glucose and PRMT2. PLoS ONE 2015, 10, e0135218. [Google Scholar] [CrossRef]
- Chawla, A.; Boisvert, W.A.; Lee, C.H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Pan, H.; Zheng, Y.; Pan, Q.; Chen, H.; Chen, F.; Wu, J.; Di, D. Expression of LXR-β, ABCA1 and ABCG1 in human triple-negative breast cancer tissues. Oncol Rep. 2019, 42, 1869–1877. [Google Scholar] [CrossRef]
- Kotlyarov, S. Participation of ABCA1 Transporter in Pathogenesis of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 3334. [Google Scholar] [CrossRef]
- Soumian, S.; Albrecht, C.; Davies, A.H.; Gibbs, R.G. ABCA1 and atherosclerosis. Vasc. Med. 2005, 10, 109–119. [Google Scholar] [CrossRef]
- Yokoyama, S. ABCA1 and biogenesis of HDL. J. Atheroscler. Thromb. 2006, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.B.; Ammit, A.J.; Gelissen, I.C. Examining the role of ABC lipid transporters in pulmonary lipid homeostasis and inflammation. Respir. Res. 2017, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.; Kruijt, J.K.; Van Eck, M.; Van Berkel, T.J. Specific gene expression of ATP-binding cassette transporters and nuclear hormone receptors in rat liver parenchymal, endothelial, and Kupffer cells. J. Biol. Chem. 2003, 278, 25448–25453. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, B.; Zhao, J.; Feng, Q.; Peng, J.; Hu, Y.; Zhao, Y. Biological mechanisms and related natural modulators of liver X receptor in nonalcoholic fatty liver disease. Biomed. Pharmacother. 2019, 113, 108778. [Google Scholar] [CrossRef]
- Wang, B.; Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Bełtowski, J. Liver X receptors (LXR) as therapeutic targets in dyslipidemia. Cardiovasc. Ther. 2008, 26, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Duval, C.; Touche, V.; Tailleux, A.; Fruchart, J.C.; Fievet, C.; Clavey, V.; Staels, B.; Lestavel, S. Niemann-Pick C1 like 1 gene expression is down-regulated by LXR activators in the intestine. Biochem. Biophys. Res. Commun. 2006, 340, 1259–1263. [Google Scholar] [CrossRef]
- Bonamassa, B.; Moschetta, A. Atherosclerosis: Lessons from LXR and the intestine. Trends Endocrinol. Metab. 2013, 24, 120–128. [Google Scholar] [CrossRef]
- Cannon, M.V.; van Gilst, W.H.; de Boer, R.A. Emerging role of liver X receptors in cardiac pathophysiology and heart failure. Basic Res. Cardiol. 2016, 111, 3. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, W.; Yang, X.; Liu, Y.; Liu, L.; Feng, K.; Zhang, X.; Yang, S.; Sun, L.; Yu, M.; et al. Functional interplay between liver X receptor and AMP-activated protein kinase α inhibits atherosclerosis in apolipoprotein E-deficient mice—A new anti-atherogenic strategy. Br. J. Pharmacol. 2018, 175, 1486–1503. [Google Scholar] [CrossRef]
- Gage, M.C.; Bécares, N.; Louie, R.; Waddington, K.E.; Zhang, Y.; Tittanegro, T.H.; Rodríguez-Lorenzo, S.; Jathanna, A.; Pourcet, B.; Pello, O.M.; et al. Disrupting LXRα phosphorylation promotes FoxM1 expression and modulates atherosclerosis by inducing macrophage proliferation. Proc. Natl. Acad. Sci. USA 2018, 115, E6556–E6565. [Google Scholar] [CrossRef] [PubMed]
- Valledor, A.F. The innate immune response under the control of the LXR pathway. Immunobiology 2005, 210, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; Andreu-Agullo, C.; Derbyshire, M.L.; Posada, J.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840.e818. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.J.; Sheu, M.Y.; Schmuth, M.; Kao, J.; Fluhr, J.W.; Rhein, L.; Collins, J.L.; Willson, T.M.; Mangelsdorf, D.J.; Elias, P.M.; et al. Liver X receptor activators display anti-inflammatory activity in irritant and allergic contact dermatitis models: Liver-X-receptor-specific inhibition of inflammation and primary cytokine production. J. Investig. Dermatol. 2003, 120, 246–255. [Google Scholar] [CrossRef]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Magida, J.A.; Evans, R.M. Rational application of macrophage-specific LXR agonists avoids the pitfalls of SREBP-induced lipogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 5051–5053. [Google Scholar] [CrossRef]
- Field, F.J.; Born, E.; Mathur, S.N. LXR/RXR ligand activation enhances basolateral efflux of beta-sitosterol in CaCo-2 cells. J. Lipid Res. 2004, 45, 905–913. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Bakke, S.S.; Smith, R.; Ravna, A.W.; Sylte, I.; Rustan, A.C.; Thoresen, G.H.; Kase, E.T. The liver X receptor modulator 22(S)-hydroxycholesterol exerts cell-type specific effects on lipid and glucose metabolism. J. Steroid Biochem. Mol. Biol. 2012, 128, 154–164. [Google Scholar] [CrossRef]
- Kuang, Y.L.; Paulson, K.E.; Lichtenstein, A.H.; Lamon-Fava, S. Regulation of the expression of key genes involved in HDL metabolism by unsaturated fatty acids. Br. J. Nutr. 2012, 108, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Korach-André, M.; Archer, A.; Gabbi, C.; Barros, R.P.; Pedrelli, M.; Steffensen, K.R.; Pettersson, A.T.; Laurencikiene, J.; Parini, P.; Gustafsson, J. Liver X receptors regulate de novo lipogenesis in a tissue-specific manner in C57BL/6 female mice. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E210–E222. [Google Scholar] [CrossRef]
- Dixon, E.D.; Nardo, A.D.; Claudel, T.; Trauner, M. The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD. Genes 2021, 12, 645. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, N.; Kato, M.; Shundo, Y.; Tajiri, H.; Tanaka, M.; Yamashita, N.; Kohjima, M.; Kotoh, K.; Nakamuta, M.; Takayanagi, R.; et al. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol. Res. 2008, 38, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.Y.; Repa, J.J. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 2007, 282, 743–751. [Google Scholar] [CrossRef]
- Ai, Z.L.; Chen, D.F. The significance and effects of liver X receptor alpha in nonalcoholic fatty liver disease in rats. Zhonghua Gan Zang Bing Za Zhi 2007, 15, 127–130. [Google Scholar] [PubMed]
- Ito, A.; Hong, C.; Rong, X.; Zhu, X.; Tarling, E.J.; Hedde, P.N.; Gratton, E.; Parks, J.; Tontonoz, P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. eLife 2015, 4, e08009. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Han, X.; Bian, Z.; Peng, Y.; You, Z.; Wang, Q.; Chen, X.; Qiu, D.; Ma, X. Activation of liver X receptors attenuates endotoxin-induced liver injury in mice with nonalcoholic fatty liver disease. Dig. Dis. Sci. 2012, 57, 390–398. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Sot, J.; Aranda, F.J.; Collado, M.I.; Goñi, F.M.; Alonso, A. Different effects of long- and short-chain ceramides on the gel-fluid and lamellar-hexagonal transitions of phospholipids: A calorimetric, NMR, and x-ray diffraction study. Biophys. J. 2005, 88, 3368–3380. [Google Scholar] [CrossRef]
- Sot, J.; Goñi, F.M.; Alonso, A. Molecular associations and surface-active properties of short- and long-N-acyl chain ceramides. Biochim. Biophys. Acta 2005, 1711, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Pullmannová, P.; Pavlíková, L.; Kováčik, A.; Sochorová, M.; Školová, B.; Slepička, P.; Maixner, J.; Zbytovská, J.; Vávrová, K. Permeability and microstructure of model stratum corneum lipid membranes containing ceramides with long (C16) and very long (C24) acyl chains. Biophys. Chem. 2017, 224, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Goñi, F.M.; Contreras, F.X.; Montes, L.R.; Sot, J.; Alonso, A. Biophysics (and sociology) of ceramides. Biochem. Soc. Symp. 2005, 72, 177–188. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Becker, K.A.; Gulbins, E. Ceramide-enriched membrane domains--structure and function. Biochim. Biophys. Acta 2009, 1788, 178–183. [Google Scholar] [CrossRef]
- Megha; London, E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): Implications for lipid raft structure and function. J. Biol. Chem. 2004, 279, 9997–10004. [Google Scholar] [CrossRef]
- Stancevic, B.; Kolesnick, R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010, 584, 1728–1740. [Google Scholar] [CrossRef]
- Siskind, L.J. Mitochondrial ceramide and the induction of apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, J.W.; Donato, L.J.; Bryant, S.C.; Baudhuin, L.M.; Berger, P.B.; Jaffe, A.S. Plasma Ceramides. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1933–1939. [Google Scholar] [CrossRef]
- Tippetts, T.S.; Holland, W.L.; Summers, S.A. The ceramide ratio: A predictor of cardiometabolic risk. J. Lipid Res. 2018, 59, 1549–1550. [Google Scholar] [CrossRef] [PubMed]
- Hilvo, M.; Meikle, P.J.; Pedersen, E.R.; Tell, G.S.; Dhar, I.; Brenner, H.; Schöttker, B.; Lääperi, M.; Kauhanen, D.; Koistinen, K.M.; et al. Development and validation of a ceramide- and phospholipid-based cardiovascular risk estimation score for coronary artery disease patients. Eur. Heart J. 2020, 41, 371–380. [Google Scholar] [CrossRef]
- Wigger, L.; Cruciani-Guglielmacci, C.; Nicolas, A.; Denom, J.; Fernandez, N.; Fumeron, F.; Marques-Vidal, P.; Ktorza, A.; Kramer, W.; Schulte, A.; et al. Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Rep. 2017, 18, 2269–2279. [Google Scholar] [CrossRef]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Brönneke, H.S.; et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef]
- Brozinick, J.T.; Hawkins, E.; Hoang Bui, H.; Kuo, M.S.; Tan, B.; Kievit, P.; Grove, K. Plasma sphingolipids are biomarkers of metabolic syndrome in non-human primates maintained on a Western-style diet. Int. J. Obes. 2013, 37, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Tonks, K.T.; Coster, A.C.; Christopher, M.J.; Chaudhuri, R.; Xu, A.; Gagnon-Bartsch, J.; Chisholm, D.J.; James, D.E.; Meikle, P.J.; Greenfield, J.R.; et al. Skeletal muscle and plasma lipidomic signatures of insulin resistance and overweight/obesity in humans. Obesity 2016, 24, 908–916. [Google Scholar] [CrossRef]
- Mah, M.; Febbraio, M.; Turpin-Nolan, S. Circulating Ceramides- Are Origins Important for Sphingolipid Biomarkers and Treatments? Front. Endocrinol. 2021, 12, 684448. [Google Scholar] [CrossRef]
- Xia, J.Y.; Holland, W.L.; Kusminski, C.M.; Sun, K.; Sharma, A.X.; Pearson, M.J.; Sifuentes, A.J.; McDonald, J.G.; Gordillo, R.; Scherer, P.E. Targeted Induction of Ceramide Degradation Leads to Improved Systemic Metabolism and Reduced Hepatic Steatosis. Cell Metab. 2015, 22, 266–278. [Google Scholar] [CrossRef]
- Merrill, A.H.; Lingrell, S.; Wang, E.; Nikolova-Karakashian, M.; Vales, T.R.; Vance, D.E. Sphingolipid biosynthesis de novo by rat hepatocytes in culture. Ceramide and sphingomyelin are associated with, but not required for, very low density lipoprotein secretion. J. Biol. Chem. 1995, 270, 13834–13841. [Google Scholar] [CrossRef]
- Carlier, A.; Phan, F.; Szpigel, A.; Hajduch, E.; Salem, J.E.; Gautheron, J.; Le Goff, W.; Guérin, M.; Lachkar, F.; Ratziu, V.; et al. Dihydroceramides in Triglyceride-Enriched VLDL Are Associated with Nonalcoholic Fatty Liver Disease Severity in Type 2 Diabetes. Cell Rep. Med. 2020, 1, 100154. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab. 2012, 23, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A.; Chaurasia, B.; Holland, W.L. Metabolic Messengers: Ceramides. Nat. Metab. 2019, 1, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, B.; Tippetts, T.S.; Mayoral Monibas, R.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 2019, 365, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef]
- Xu, S.; Jay, A.; Brunaldi, K.; Huang, N.; Hamilton, J.A. CD36 enhances fatty acid uptake by increasing the rate of intracellular esterification but not transport across the plasma membrane. Biochemistry 2013, 52, 7254–7261. [Google Scholar] [CrossRef]
- Jay, A.G.; Hamilton, J.A. The enigmatic membrane fatty acid transporter CD36: New insights into fatty acid binding and their effects on uptake of oxidized LDL. Prostaglandins Leukot. Essent. Fatty Acids 2018, 138, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef]
- Frangioudakis, G.; Garrard, J.; Raddatz, K.; Nadler, J.L.; Mitchell, T.W.; Schmitz-Peiffer, C. Saturated- and n-6 polyunsaturated-fat diets each induce ceramide accumulation in mouse skeletal muscle: Reversal and improvement of glucose tolerance by lipid metabolism inhibitors. Endocrinology 2010, 151, 4187–4196. [Google Scholar] [CrossRef]
- Hyde, R.; Hajduch, E.; Powell, D.J.; Taylor, P.M.; Hundal, H.S. Ceramide down-regulates System A amino acid transport and protein synthesis in rat skeletal muscle cells. FASEB J. 2005, 19, 461–463. [Google Scholar] [CrossRef]
- Finicle, B.T.; Ramirez, M.U.; Liu, G.; Selwan, E.M.; McCracken, A.N.; Yu, J.; Joo, Y.; Nguyen, J.; Ou, K.; Roy, S.G.; et al. Sphingolipids inhibit endosomal recycling of nutrient transporters by inactivating ARF6. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Guenther, G.G.; Peralta, E.R.; Rosales, K.R.; Wong, S.Y.; Siskind, L.J.; Edinger, A.L. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 17402–17407. [Google Scholar] [CrossRef] [PubMed]
- Zigdon, H.; Kogot-Levin, A.; Park, J.W.; Goldschmidt, R.; Kelly, S.; Merrill, A.H.; Scherz, A.; Pewzner-Jung, Y.; Saada, A.; Futerman, A.H. Ablation of ceramide synthase 2 causes chronic oxidative stress due to disruption of the mitochondrial respiratory chain. J. Biol. Chem. 2013, 288, 4947–4956. [Google Scholar] [CrossRef] [PubMed]
- Hajduch, E.; Turban, S.; Le Liepvre, X.; Le Lay, S.; Lipina, C.; Dimopoulos, N.; Dugail, I.; Hundal, H.S. Targeting of PKCzeta and PKB to caveolin-enriched microdomains represents a crucial step underpinning the disruption in PKB-directed signalling by ceramide. Biochem. J. 2008, 410, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Muñoz, A.; Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Trueba, M.; Ordoñez, M. Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Prog. Lipid Res. 2016, 61, 51–62. [Google Scholar] [CrossRef]
- Gomez-Muñoz, A.; Gangoiti, P.; Arana, L.; Ouro, A.; Rivera, I.G.; Ordoñez, M.; Trueba, M. New insights on the role of ceramide 1-phosphate in inflammation. Biochim. Biophys. Acta 2013, 1831, 1060–1066. [Google Scholar] [CrossRef]
- Hait, N.C.; Maiti, A. The Role of Sphingosine-1-Phosphate and Ceramide-1-Phosphate in Inflammation and Cancer. Mediat. Inflamm. 2017, 2017, 4806541. [Google Scholar] [CrossRef]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, C.W.; Samaha, G.; Garcia-Arcos, I. Alveolar lipids in pulmonary disease. A review. Lipids Health Dis. 2020, 19, 122. [Google Scholar] [CrossRef]
- Morissette, M.C.; Shen, P.; Thayaparan, D.; Stämpfli, M.R. Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. Eur. Respir. J. 2015, 46, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, C.W.; Kumley, B.K.; Area-Gomez, E.; Xu, Y.; Dabo, A.J.; Geraghty, P.; Campos, M.; Foronjy, R.; Garcia-Arcos, I. Decreased surfactant lipids correlate with lung function in chronic obstructive pulmonary disease (COPD). PLoS ONE 2020, 15, e0228279. [Google Scholar] [CrossRef]
- Lugg, S.T.; Scott, A.; Parekh, D.; Naidu, B.; Thickett, D.R. Cigarette smoke exposure and alveolar macrophages: Mechanisms for lung disease. Thorax 2021. [Google Scholar] [CrossRef]
- Mirza, S.; Benzo, R. Chronic Obstructive Pulmonary Disease Phenotypes: Implications for Care. Mayo Clin. Proc. 2017, 92, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Corlateanu, A.; Mendez, Y.; Wang, Y.; Garnica, R.J.A.; Botnaru, V.; Siafakas, N. Chronic obstructive pulmonary disease and phenotypes: A state-of-the-art. Pulmonology 2020, 26, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, E.; André, S.; Boleo-Tomé, J.P.; Areias, V.; Munhá, J.; Cardoso, J.; GI DPOC-Grupo de Interesse na Doença Pulmonar Obstrutiva Crónica. Understanding COPD: A vision on phenotypes, comorbidities and treatment approach. Rev. Port. Pneumol. 2016, 22, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Naik, D.; Joshi, A.; Paul, T.V.; Thomas, N. Chronic obstructive pulmonary disease and the metabolic syndrome: Consequences of a dual threat. Indian J. Endocrinol. Metab. 2014, 18, 608–616. [Google Scholar] [CrossRef]
- Snider, G.L. Chronic obstructive pulmonary disease: A definition and implications of structural determinants of airflow obstruction for epidemiology. Am. Rev. Respir. Dis. 1989, 140, S3–S8. [Google Scholar] [CrossRef]
- Ogawa, E.; Nakano, Y.; Ohara, T.; Muro, S.; Hirai, T.; Sato, S.; Sakai, H.; Tsukino, M.; Kinose, D.; Nishioka, M.; et al. Body mass index in male patients with COPD: Correlation with low attenuation areas on CT. Thorax 2009, 64, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Coxson, H.O.; Chan, I.H.; Mayo, J.R.; Hlynsky, J.; Nakano, Y.; Birmingham, C.L. Early emphysema in patients with anorexia nervosa. Am. J. Respir. Crit. Care Med. 2004, 170, 748–752. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.N.; Wouters, E.F.M.; Rutten, E.; Casaburi, R.; Rennard, S.I.; Lomas, D.A.; Bamman, M.; Celli, B.; Agusti, A.; Tal-Singer, R.; et al. It’s more than low BMI: Prevalence of cachexia and associated mortality in COPD. Respir. Res. 2019, 20, 100. [Google Scholar] [CrossRef]
- Brigham, E.P.; Anderson, J.A.; Brook, R.D.; Calverley, P.M.A.; Celli, B.R.; Cowans, N.J.; Crim, C.; Diserens, J.E.; Martinez, F.J.; McCormack, M.C.; et al. Challenging the obesity paradox: Extreme obesity and COPD mortality in the SUMMIT trial. ERJ Open Res. 2021, 7, 00902–2020. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Hasegawa, W.; Yasunaga, H.; Sunohara, M.; Jo, T.; Takami, K.; Matsui, H.; Fushimi, K.; Nagase, T. Paradoxical association between body mass index and in-hospital mortality in elderly patients with chronic obstructive pulmonary disease in Japan. Int. J. Chron. Obstruct. Pulmon. Dis. 2014, 9, 1337–1346. [Google Scholar] [CrossRef]
- Hickson, D.A.; Burchfiel, C.M.; Liu, J.; Petrini, M.F.; Harrison, K.; White, W.B.; Sarpong, D.F. Diabetes, impaired glucose tolerance, and metabolic biomarkers in individuals with normal glucose tolerance are inversely associated with lung function: The Jackson Heart Study. Lung 2011, 189, 311–321. [Google Scholar] [CrossRef][Green Version]
- Ubhi, B.K.; Riley, J.H.; Shaw, P.A.; Lomas, D.A.; Tal-Singer, R.; MacNee, W.; Griffin, J.L.; Connor, S.C. Metabolic profiling detects biomarkers of protein degradation in COPD patients. Eur. Respir. J. 2012, 40, 345–355. [Google Scholar] [CrossRef]
- Walter, R.E.; Beiser, A.; Givelber, R.J.; O’Connor, G.T.; Gottlieb, D.J. Association between glycemic state and lung function: The Framingham Heart Study. Am. J. Respir. Crit. Care Med. 2003, 167, 911–916. [Google Scholar] [CrossRef] [PubMed]
- McKeever, T.M.; Weston, P.J.; Hubbard, R.; Fogarty, A. Lung function and glucose metabolism: An analysis of data from the Third National Health and Nutrition Examination Survey. Am. J. Epidemiol. 2005, 161, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Davis, W.A.; Knuiman, M.; Kendall, P.; Grange, V.; Davis, T.M.; Study, F.D. Glycemic exposure is associated with reduced pulmonary function in type 2 diabetes: The Fremantle Diabetes Study. Diabetes Care 2004, 27, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Ebata, M.; Saito, M. The relationship between low vital capacity and impaired glucose metabolism in men. Diabet. Med. 2010, 27, 1460–1461. [Google Scholar] [CrossRef]
- Klein, O.L.; Krishnan, J.A.; Glick, S.; Smith, L.J. Systematic review of the association between lung function and Type 2 diabetes mellitus. Diabet. Med. 2010, 27, 977–987. [Google Scholar] [CrossRef]
- Van den Borst, B.; Gosker, H.R.; Zeegers, M.P.; Schols, A.M. Pulmonary function in diabetes: A metaanalysis. Chest 2010, 138, 393–406. [Google Scholar] [CrossRef]
- Heianza, Y.; Arase, Y.; Tsuji, H.; Saito, K.; Amakawa, K.; Hsieh, S.D.; Kodama, S.; Shimano, H.; Yamada, N.; Hara, S.; et al. Low lung function and risk of type 2 diabetes in Japanese men: The Toranomon Hospital Health Management Center Study 9 (TOPICS 9). Mayo Clin. Proc. 2012, 87, 853–861. [Google Scholar] [CrossRef]
- Kwon, C.H.; Rhee, E.J.; Song, J.U.; Kim, J.T.; Kwag, H.J.; Sung, K.C. Reduced lung function is independently associated with increased risk of type 2 diabetes in Korean men. Cardiovasc. Diabetol. 2012, 11, 38. [Google Scholar] [CrossRef][Green Version]
- Engström, G.; Janzon, L. Risk of developing diabetes is inversely related to lung function: A population-based cohort study. Diabet. Med. 2002, 19, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Rana, J.S.; Mittleman, M.A.; Sheikh, J.; Hu, F.B.; Manson, J.E.; Colditz, G.A.; Speizer, F.E.; Barr, R.G.; Camargo, C.A. Chronic obstructive pulmonary disease, asthma, and risk of type 2 diabetes in women. Diabetes Care 2004, 27, 2478–2484. [Google Scholar] [CrossRef]
- Yeh, H.C.; Punjabi, N.M.; Wang, N.Y.; Pankow, J.S.; Duncan, B.B.; Cox, C.E.; Selvin, E.; Brancati, F.L. Cross-sectional and prospective study of lung function in adults with type 2 diabetes: The Atherosclerosis Risk in Communities (ARIC) study. Diabetes Care 2008, 31, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Wannamethee, S.G.; Shaper, A.G.; Rumley, A.; Sattar, N.; Whincup, P.H.; Thomas, M.C.; Lowe, G.D. Lung function and risk of type 2 diabetes and fatal and nonfatal major coronary heart disease events: Possible associations with inflammation. Diabetes Care 2010, 33, 1990–1996. [Google Scholar] [CrossRef] [PubMed]
- Engström, G.; Lind, P.; Hedblad, B.; Wollmer, P.; Stavenow, L.; Janzon, L.; Lindgärde, F. Lung function and cardiovascular risk: Relationship with inflammation-sensitive plasma proteins. Circulation 2002, 106, 2555–2560. [Google Scholar] [CrossRef] [PubMed]
- Fujii, W.; Kapellos, T.S.; Baßler, K.; Händler, K.; Holsten, L.; Knoll, R.; Warnat-Herresthal, S.; Oestreich, M.; Hinkley, E.R.; Hasenauer, J.; et al. Alveolar macrophage transcriptomic profiling in COPD shows major lipid metabolism changes. ERJ Open Res. 2021, 7, 00915-2020. [Google Scholar] [CrossRef]
- Draper, D.W.; Madenspacher, J.H.; Dixon, D.; King, D.H.; Remaley, A.T.; Fessler, M.B. ATP-binding cassette transporter G1 deficiency dysregulates host defense in the lung. Am. J. Respir. Crit. Care Med. 2010, 182, 404–412. [Google Scholar] [CrossRef]
- Wojcik, A.J.; Skaflen, M.D.; Srinivasan, S.; Hedrick, C.C. A critical role for ABCG1 in macrophage inflammation and lung homeostasis. J. Immunol. 2008, 180, 4273–4282. [Google Scholar] [CrossRef]
- Baldán, A.; Gomes, A.V.; Ping, P.; Edwards, P.A. Loss of ABCG1 results in chronic pulmonary inflammation. J. Immunol. 2008, 180, 3560–3568. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.R.; Tao, J.Q.; Collins, H.L.; Francone, O.L.; Rothblat, G.H. Pulmonary abnormalities due to ABCA1 deficiency in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L980–L989. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.M.; Nair, P.; Hargreave, F.E.; Efthimiadis, A.E.; Anvari, M.; Allen, C.J.; ELVIS Research Study Group. Lipid and smoker’s inclusions in sputum macrophages in patients with airway diseases. Respir. Med. 2011, 105, 1691–1695. [Google Scholar] [CrossRef]
- Basset-Léobon, C.; Lacoste-Collin, L.; Aziza, J.; Bes, J.C.; Jozan, S.; Courtade-Saïdi, M. Cut-off values and significance of Oil Red O-positive cells in bronchoalveolar lavage fluid. Cytopathology 2010, 21, 245–250. [Google Scholar] [CrossRef]
- Chakinala, R.C.; Khatri, A.; Gupta, K.; Koike, K.; Epelbaum, O. Sphingolipids in COPD. Eur. Respir. Rev. 2019, 28. [Google Scholar] [CrossRef]
- Scarpa, M.C.; Baraldo, S.; Marian, E.; Turato, G.; Calabrese, F.; Saetta, M.; Maestrelli, P. Ceramide expression and cell homeostasis in chronic obstructive pulmonary disease. Respiration 2013, 85, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Khan, E.; Careaga, M.; Goldkorn, T. Neutral sphingomyelinase 2 is activated by cigarette smoke to augment ceramide-induced apoptosis in lung cell death. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L125–L133. [Google Scholar] [CrossRef] [PubMed]
- Petrache, I.; Natarajan, V.; Zhen, L.; Medler, T.R.; Richter, A.T.; Cho, C.; Hubbard, W.C.; Berdyshev, E.V.; Tuder, R.M. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat. Med. 2005, 11, 491–498. [Google Scholar] [CrossRef]
- Petrache, I.; Petrusca, D.N. The involvement of sphingolipids in chronic obstructive pulmonary diseases. Handb. Exp. Pharmacol. 2013, 216, 247–264. [Google Scholar] [CrossRef]
- Bowler, R.P.; Jacobson, S.; Cruickshank, C.; Hughes, G.J.; Siska, C.; Ory, D.S.; Petrache, I.; Schaffer, J.E.; Reisdorph, N.; Kechris, K. Plasma sphingolipids associated with chronic obstructive pulmonary disease phenotypes. Am. J. Respir. Crit. Care Med. 2015, 191, 275–284. [Google Scholar] [CrossRef]
- Serban, K.A.; Rezania, S.; Petrusca, D.N.; Poirier, C.; Cao, D.; Justice, M.J.; Patel, M.; Tsvetkova, I.; Kamocki, K.; Mikosz, A.; et al. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci. Rep. 2016, 6, 31596. [Google Scholar] [CrossRef]
- Koike, K.; Berdyshev, E.V.; Bowler, R.P.; Scruggs, A.K.; Cao, D.; Schweitzer, K.S.; Serban, K.A.; Petrache, I. Bioactive Sphingolipids in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2018, 15, S249–S252. [Google Scholar] [CrossRef]
- Paudel, K.R.; Panth, N.; Kim, D.W. Circulating Endothelial Microparticles: A Key Hallmark of Atherosclerosis Progression. Scientifica 2016, 2016, 8514056. [Google Scholar] [CrossRef] [PubMed]
- França, C.N.; Izar, M.C.; Amaral, J.B.; Tegani, D.M.; Fonseca, F.A. Microparticles as potential biomarkers of cardiovascular disease. Arq. Bras. Cardiol. 2015, 104, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A. Leukocyte-derived microparticles in vascular homeostasis. Circ. Res. 2012, 110, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Agarwal, S.; Clauss, M.; Britt, N.S.; Dhillon, N.K. Extracellular vesicles: Novel communicators in lung diseases. Respir. Res. 2020, 21, 175. [Google Scholar] [CrossRef]
- Liang, J.; Gu, S.; Mao, X.; Tan, Y.; Wang, H.; Li, S.; Zhou, Y. Endothelial Cell Morphology Regulates Inflammatory Cells Through MicroRNA Transferred by Extracellular Vesicles. Front. Bioeng. Biotechnol. 2020, 8, 369. [Google Scholar] [CrossRef] [PubMed]
- Petrusca, D.N.; Gu, Y.; Adamowicz, J.J.; Rush, N.I.; Hubbard, W.C.; Smith, P.A.; Berdyshev, E.V.; Birukov, K.G.; Lee, C.H.; Tuder, R.M.; et al. Sphingolipid-mediated inhibition of apoptotic cell clearance by alveolar macrophages. J. Biol. Chem. 2010, 285, 40322–40332. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.S.; Kim, M.C.; Ahn, J.H. Sarcopenia Is an Independent Risk Factor for NAFLD in COPD: A Nationwide Survey (KNHANES 2008–2011). Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Lee, D.H.; Lee, J.K.; Lee, S.; Koo, B.K.; Joo, S.K.; Heo, E.Y.; Jung, Y.J.; Kim, W.; Kim, D.K. Pulmonary function is associated with fibrosis severity in patients with biopsy-proven nonalcoholic fatty liver disease. Liver Int. 2020, 40, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Ou, Z.; Ruan, X.; Gong, J. Role of liver X receptors in cholesterol efflux and inflammatory signaling (review). Mol. Med. Rep. 2012, 5, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Higham, A.; Lea, S.; Plumb, J.; Maschera, B.; Simpson, K.; Ray, D.; Singh, D. The role of the liver X receptor in chronic obstructive pulmonary disease. Respir. Res. 2013, 14, 106. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, H.; Koarai, A.; Ichikawa, T.; Minakata, Y.; Matsunaga, K.; Hirano, T.; Akamatsu, K.; Yanagisawa, S.; Furusawa, M.; Uno, Y.; et al. Increased 25-hydroxycholesterol concentrations in the lungs of patients with chronic obstructive pulmonary disease. Respirology 2012, 17, 533–540. [Google Scholar] [CrossRef]
- Kikuchi, T.; Sugiura, H.; Koarai, A.; Ichikawa, T.; Minakata, Y.; Matsunaga, K.; Nakanishi, M.; Hirano, T.; Akamatsu, K.; Yanagisawa, S.; et al. Increase of 27-hydroxycholesterol in the airways of patients with COPD: Possible role of 27-hydroxycholesterol in tissue fibrosis. Chest 2012, 142, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Kidani, Y.; Bensinger, S.J. Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunol. Rev. 2012, 249, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Giguère, V. Orphan nuclear receptors: From gene to function. Endocr. Rev. 1999, 20, 689–725. [Google Scholar] [CrossRef]
- Castrillo, A.; Tontonoz, P. Nuclear receptors in macrophage biology: At the crossroads of lipid metabolism and inflammation. Annu. Rev. Cell Dev. Biol. 2004, 20, 455–480. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications--a review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef]
- Lakshmi, S.P.; Reddy, A.T.; Zhang, Y.; Sciurba, F.C.; Mallampalli, R.K.; Duncan, S.R.; Reddy, R.C. Down-regulated peroxisome proliferator-activated receptor γ (PPARγ) in lung epithelial cells promotes a PPARγ agonist-reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD). J. Biol. Chem. 2014, 289, 6383–6393. [Google Scholar] [CrossRef]
- Shan, M.; You, R.; Yuan, X.; Frazier, M.V.; Porter, P.; Seryshev, A.; Hong, J.S.; Song, L.Z.; Zhang, Y.; Hilsenbeck, S.; et al. Agonistic induction of PPARγ reverses cigarette smoke-induced emphysema. J. Clin. Investig. 2014, 124, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Solleti, S.K.; Simon, D.M.; Srisuma, S.; Arikan, M.C.; Bhattacharya, S.; Rangasamy, T.; Bijli, K.M.; Rahman, A.; Crossno, J.T.; Shapiro, S.D.; et al. Airway epithelial cell PPARγ modulates cigarette smoke-induced chemokine expression and emphysema susceptibility in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L293–L304. [Google Scholar] [CrossRef]
- Remels, A.H.; Gosker, H.R.; Schrauwen, P.; Langen, R.C.; Schols, A.M. Peroxisome proliferator-activated receptors: A therapeutic target in COPD? Eur. Respir. J. 2008, 31, 502–508. [Google Scholar] [CrossRef]
- Morissette, M.C.; Shen, P.; Thayaparan, D.; Stämpfli, M.R. Impacts of peroxisome proliferator-activated receptor-γ activation on cigarette smoke-induced exacerbated response to bacteria. Eur. Respir. J. 2015, 45, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Lestavel, S.; Bocher, V.; Remaley, A.T.; Neve, B.; Torra, I.P.; Teissier, E.; Minnich, A.; Jaye, M.; Duverger, N.; et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 2001, 7, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, H.; Ayaori, M.; Iizuka, M.; Terao, Y.; Uto-Kondo, H.; Yakushiji, E.; Takiguchi, S.; Nakaya, K.; Hisada, T.; Uehara, Y.; et al. Pioglitazone enhances cholesterol efflux from macrophages by increasing ABCA1/ABCG1 expressions via PPARγ/LXRα pathway: Findings from in vitro and ex vivo studies. Atherosclerosis 2011, 219, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Sticozzi, C.; Pecorelli, A.; Belmonte, G.; Valacchi, G. Cigarette smoke affects ABCAl expression via liver X receptor nuclear translocation in human keratinocytes. Int. J. Mol. Sci. 2010, 11, 3375–3386. [Google Scholar] [CrossRef]
- Smoak, K.; Madenspacher, J.; Jeyaseelan, S.; Williams, B.; Dixon, D.; Poch, K.R.; Nick, J.A.; Worthen, G.S.; Fessler, M.B. Effects of liver X receptor agonist treatment on pulmonary inflammation and host defense. J. Immunol. 2008, 180, 3305–3312. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Hiroshima, A.; Ariga, A.; Honzumi, S.; Koieyama, T.; Inaba, T.; Fujiwara, T. Liver X receptor agonists inhibit tissue factor expression in macrophages. FEBS J. 2005, 272, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef]
- Birrell, M.A.; Catley, M.C.; Hardaker, E.; Wong, S.; Willson, T.M.; McCluskie, K.; Leonard, T.; Farrow, S.N.; Collins, J.L.; Haj-Yahia, S.; et al. Novel role for the liver X nuclear receptor in the suppression of lung inflammatory responses. J. Biol. Chem. 2007, 282, 31882–31890. [Google Scholar] [CrossRef]
- Beuther, D.A.; Sutherland, E.R. Overweight, obesity, and incident asthma: A meta-analysis of prospective epidemiologic studies. Am. J. Respir. Crit. Care Med. 2007, 175, 661–666. [Google Scholar] [CrossRef]
- Wada, H.; Goto, H.; Saitoh, E.; Ieki, R.; Okamura, T.; Ota, T.; Hagiwara, S.; Kodaka, T.; Yamamoto, Y. Reduction in plasma free fatty acid in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 171, 1465. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Knudsen, N.H.; Wang, G.; Qiu, W.; Naing, Z.Z.C.; Bai, Y.; Ai, X.; Lee, C.H.; Zhou, X. Genetic Control of Fatty Acid β-Oxidation in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell. Mol. Biol. 2017, 56, 738–748. [Google Scholar] [CrossRef]
- Cornell, K.; Alam, M.; Lyden, E.; Wood, L.; LeVan, T.D.; Nordgren, T.M.; Bailey, K.; Hanson, C. Saturated Fat Intake Is Associated with Lung Function in Individuals with Airflow Obstruction: Results from NHANES 2007–2012. Nutrients 2019, 11, 317. [Google Scholar] [CrossRef]
- Ceco, E.; Celli, D.; Weinberg, S.; Shigemura, M.; Welch, L.C.; Volpe, L.; Chandel, N.S.; Bharat, A.; Lecuona, E.; Sznajder, J.I. Elevated CO. Front. Physiol. 2020, 11, 630910. [Google Scholar] [CrossRef]
- Gong, J.; Zhao, H.; Liu, T.; Li, L.; Cheng, E.; Zhi, S.; Kong, L.; Yao, H.W.; Li, J. Cigarette Smoke Reduces Fatty Acid Catabolism, Leading to Apoptosis in Lung Endothelial Cells: Implication for Pathogenesis of COPD. Front. Pharmacol. 2019, 10, 941. [Google Scholar] [CrossRef] [PubMed]
- Van der Does, A.M.; Heijink, M.; Mayboroda, O.A.; Persson, L.J.; Aanerud, M.; Bakke, P.; Eagan, T.M.; Hiemstra, P.S.; Giera, M. Dynamic differences in dietary polyunsaturated fatty acid metabolism in sputum of COPD patients and controls. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2019, 1864, 224–233. [Google Scholar] [CrossRef]
- Rutting, S.; Papanicolaou, M.; Xenaki, D.; Wood, L.G.; Mullin, A.M.; Hansbro, P.M.; Oliver, B.G. Dietary ω-6 polyunsaturated fatty acid arachidonic acid increases inflammation, but inhibits ECM protein expression in COPD. Respir. Res. 2018, 19, 211. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef]
- Kotlyarov, S.; Kotlyarova, A. Molecular Mechanisms of Lipid Metabolism Disorders in Infectious Exacerbations of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 7634. [Google Scholar] [CrossRef] [PubMed]
- Rovina, N.; Koutsoukou, A.; Koulouris, N.G. Inflammation and immune response in COPD: Where do we stand? Mediat. Inflamm. 2013, 2013, 413735. [Google Scholar] [CrossRef]
- Shabalala, S.C.; Dludla, P.V.; Mabasa, L.; Kappo, A.P.; Basson, A.K.; Pheiffer, C.; Johnson, R. The effect of adiponectin in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) and the potential role of polyphenols in the modulation of adiponectin signaling. Biomed. Pharmacother. 2020, 131, 110785. [Google Scholar] [CrossRef] [PubMed]
- Rotundo, L.; Persaud, A.; Feurdean, M.; Ahlawat, S.; Kim, H.S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin. Mol. Hepatol. 2018, 24, 392–401. [Google Scholar] [CrossRef]
- Whitehead, J.P.; Richards, A.A.; Hickman, I.J.; Macdonald, G.A.; Prins, J.B. Adiponectin—A key adipokine in the metabolic syndrome. Diabetes Obes. Metab. 2006, 8, 264–280. [Google Scholar] [CrossRef] [PubMed]
- Halpin, D.M.G.; Criner, G.J.; Papi, A.; Singh, D.; Anzueto, A.; Martinez, F.J.; Agusti, A.A.; Vogelmeier, C.F. Global Initiative for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease. The 2020 GOLD Science Committee Report on COVID-19 and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 203, 24–36. [Google Scholar] [CrossRef]
- Rochlani, Y.; Pothineni, N.V.; Kovelamudi, S.; Mehta, J.L. Metabolic syndrome: Pathophysiology, management, and modulation by natural compounds. Ther. Adv. Cardiovasc. Dis. 2017, 11, 215–225. [Google Scholar] [CrossRef]
- Naseem, S.; Baneen, U. Systemic inflammation in patients of chronic obstructive pulmonary disease with metabolic syndrome. J. Family Med. Prim. Care 2019, 8, 3393–3398. [Google Scholar] [CrossRef]
- Hu, C.J.; Wang, L.Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhang, F. Role of the HIF-1 signaling pathway in chronic obstructive pulmonary disease. Exp. Ther. Med. 2018, 16, 4553–4561. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Walters, E.H.; Simpson, J.L.; Keely, S.; Wark, P.A.B.; O’Toole, R.F.; Hansbro, P.M. Hypoxia-inducible factor and bacterial infections in chronic obstructive pulmonary disease. Respirology 2020, 25, 53–63. [Google Scholar] [CrossRef]
- Foglia, B.; Novo, E.; Protopapa, F.; Maggiora, M.; Bocca, C.; Cannito, S.; Parola, M. Hypoxia, Hypoxia-Inducible Factors and Liver Fibrosis. Cells 2021, 10, 1764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, C.; Li, X.; Shangguan, Z.; Wei, W.; Liu, S.; Yang, S.; Liu, Y. HFD and HFD-provoked hepatic hypoxia act as reciprocal causation for NAFLD via HIF-independent signaling. BMC Gastroenterol. 2020, 20, 366. [Google Scholar] [CrossRef] [PubMed]
- Morello, E.; Sutti, S.; Foglia, B.; Novo, E.; Cannito, S.; Bocca, C.; Rajsky, M.; Bruzzì, S.; Abate, M.L.; Rosso, C.; et al. Hypoxia-inducible factor 2α drives nonalcoholic fatty liver progression by triggering hepatocyte release of histidine-rich glycoprotein. Hepatology 2018, 67, 2196–2214. [Google Scholar] [CrossRef]
- Chen, J.; Fu, H.; Li, Y.; Wang, L.; Luo, S.; Lu, H. Hypoxia exacerbates nonalcoholic fatty liver disease via the HIF-2α/PPARα pathway. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E710–E722. [Google Scholar] [CrossRef]
- Han, J.; He, Y.; Zhao, H.; Xu, X. Hypoxia inducible factor-1 promotes liver fibrosis in nonalcoholic fatty liver disease by activating PTEN/p65 signaling pathway. J. Cell. Biochem. 2019, 120, 14735–14744. [Google Scholar] [CrossRef] [PubMed]
- Mesarwi, O.A.; Shin, M.K.; Bevans-Fonti, S.; Schlesinger, C.; Shaw, J.; Polotsky, V.Y. Hepatocyte Hypoxia Inducible Factor-1 Mediates the Development of Liver Fibrosis in a Mouse Model of Nonalcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0168572. [Google Scholar] [CrossRef]
- Zhan, L.; Huang, C.; Meng, X.M.; Song, Y.; Wu, X.Q.; Yang, Y.; Li, J. Hypoxia-inducible factor-1alpha in hepatic fibrosis: A promising therapeutic target. Biochimie 2015, 108, 1–7. [Google Scholar] [CrossRef]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schödel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, O. Hypoxic Signaling and Cholesterol Lipotoxicity in Fatty Liver Disease Progression. Oxid. Med. Cell. Longev. 2018, 2018, 2548154. [Google Scholar] [CrossRef] [PubMed]
- Pasupneti, S.; Tian, W.; Tu, A.B.; Dahms, P.; Granucci, E.; Gandjeva, A.; Xiang, M.; Butcher, E.C.; Semenza, G.L.; Tuder, R.M.; et al. Endothelial HIF-2α as a Key Endogenous Mediator Preventing Emphysema. Am. J. Respir. Crit. Care Med. 2020, 202, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Takikawa, S.; Geraghty, P.; Argmann, C.; Campbell, J.; Lin, L.; Huang, T.; Tu, Z.; Foronjy, R.F.; Feronjy, R.; et al. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet. 2015, 11, e1004898. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotlyarov, S.; Bulgakov, A. Lipid Metabolism Disorders in the Comorbid Course of Nonalcoholic Fatty Liver Disease and Chronic Obstructive Pulmonary Disease. Cells 2021, 10, 2978. https://doi.org/10.3390/cells10112978
Kotlyarov S, Bulgakov A. Lipid Metabolism Disorders in the Comorbid Course of Nonalcoholic Fatty Liver Disease and Chronic Obstructive Pulmonary Disease. Cells. 2021; 10(11):2978. https://doi.org/10.3390/cells10112978
Chicago/Turabian StyleKotlyarov, Stanislav, and Aleksei Bulgakov. 2021. "Lipid Metabolism Disorders in the Comorbid Course of Nonalcoholic Fatty Liver Disease and Chronic Obstructive Pulmonary Disease" Cells 10, no. 11: 2978. https://doi.org/10.3390/cells10112978
APA StyleKotlyarov, S., & Bulgakov, A. (2021). Lipid Metabolism Disorders in the Comorbid Course of Nonalcoholic Fatty Liver Disease and Chronic Obstructive Pulmonary Disease. Cells, 10(11), 2978. https://doi.org/10.3390/cells10112978