
DNMT3A and DNMT3B Targeting as an Effective Radiosensitizing Strategy in Embryonal Rhabdomyosarcoma

 , ,
, ,  ,
,  , ,
, ,  ,
,  ,
,  ,
, 
 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Compounds
2.2. Transient Transfection of Small Interfering RNA and Radiation Exposure
2.3. Cell Morphological Evaluation by Giemsa Staining
2.4. Cell Proliferation and MTT Assays
2.5. Trans-Well Migration Assay
2.6. RNA Extraction and Quantitative Real Time PCR (q-PCR)
2.7. Protein Extracts and Western Blot Analysis
2.8. Cell Cycle Analysis
2.9. Colony Formation Assay
2.10. Immunofluorescence
2.11. Statistical Analysis
3. Results
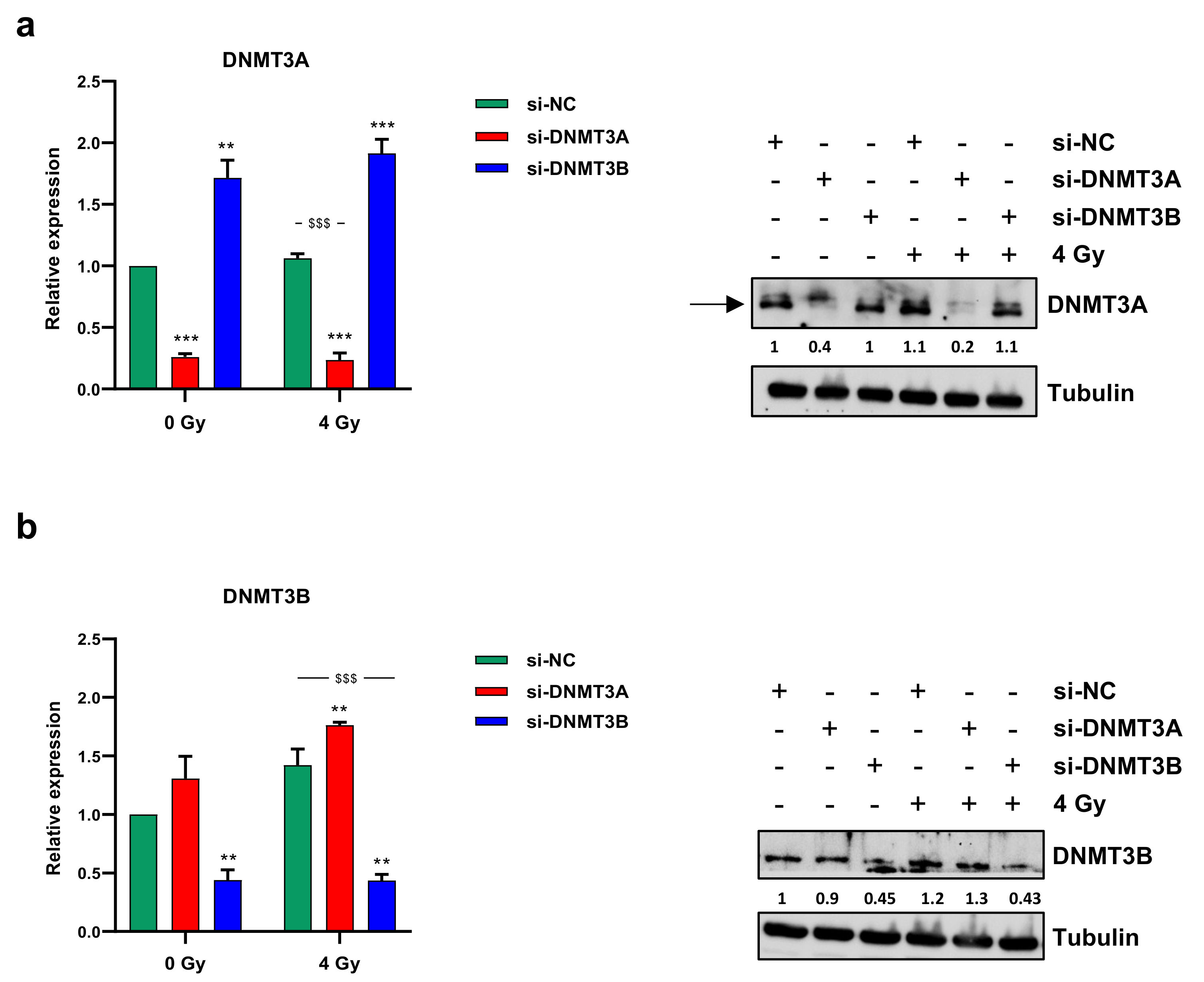
3.1. Radiotherapy Alone Does Not Decrease DNMT3A and DNMT3B Expression Levels
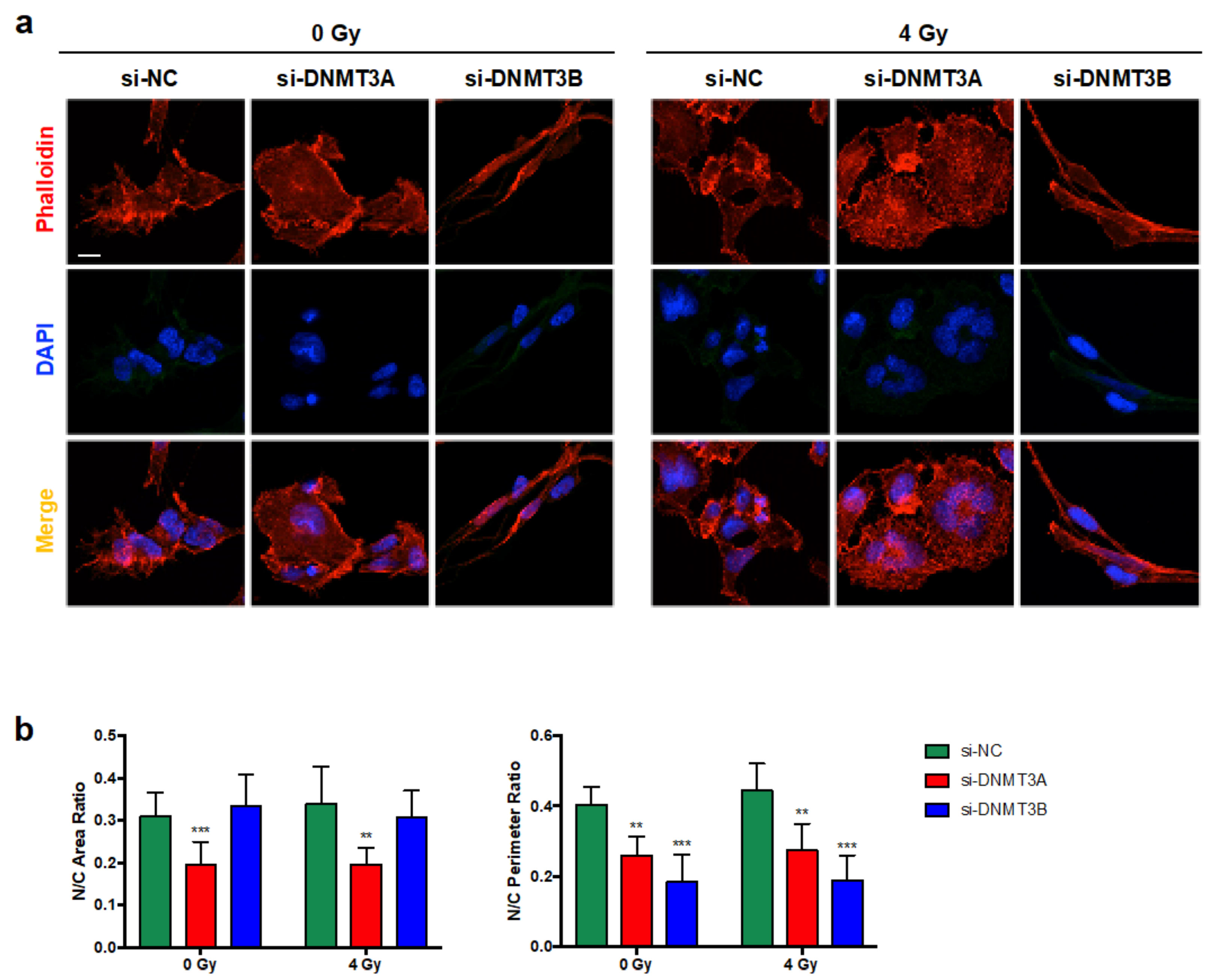
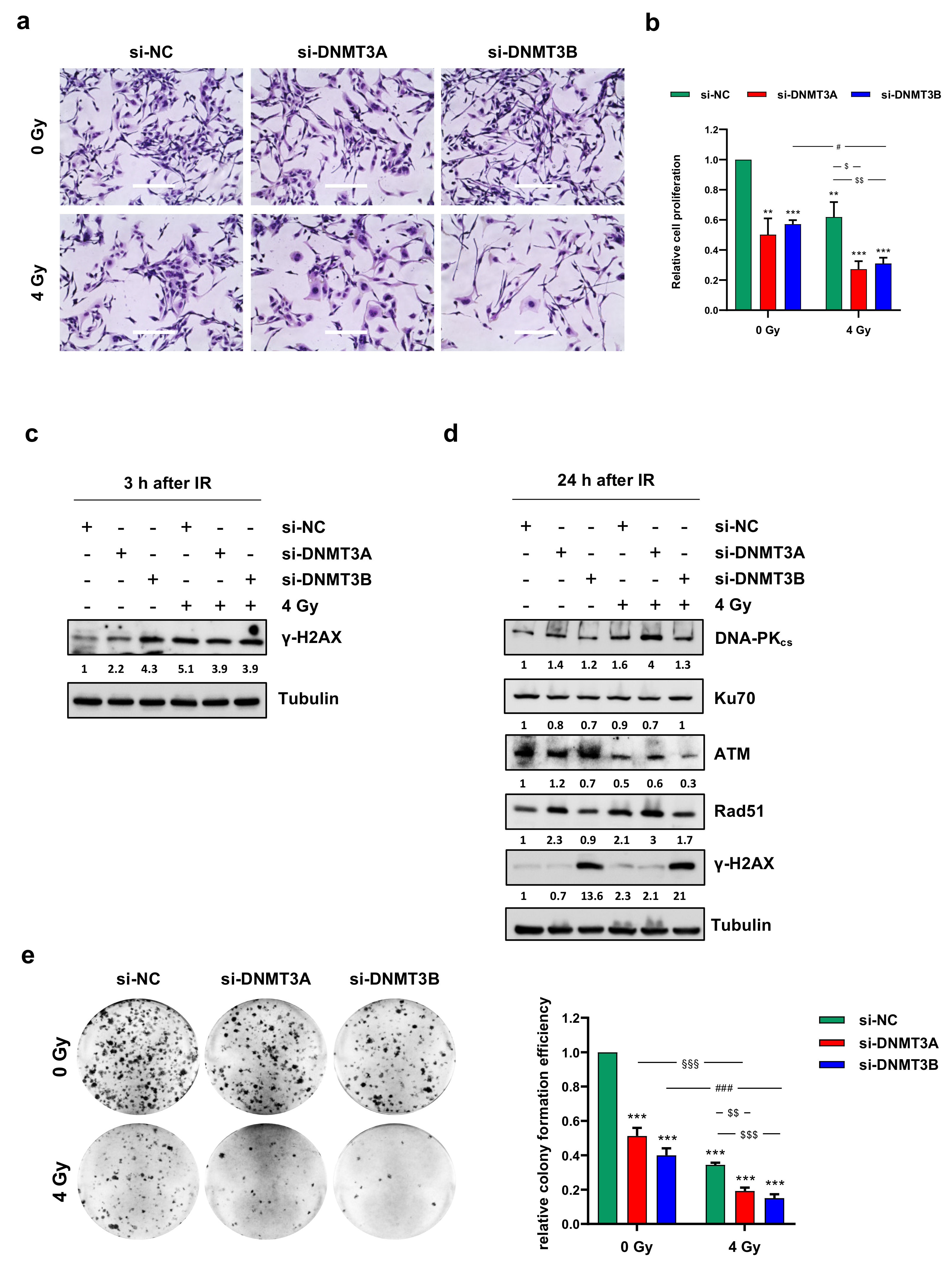
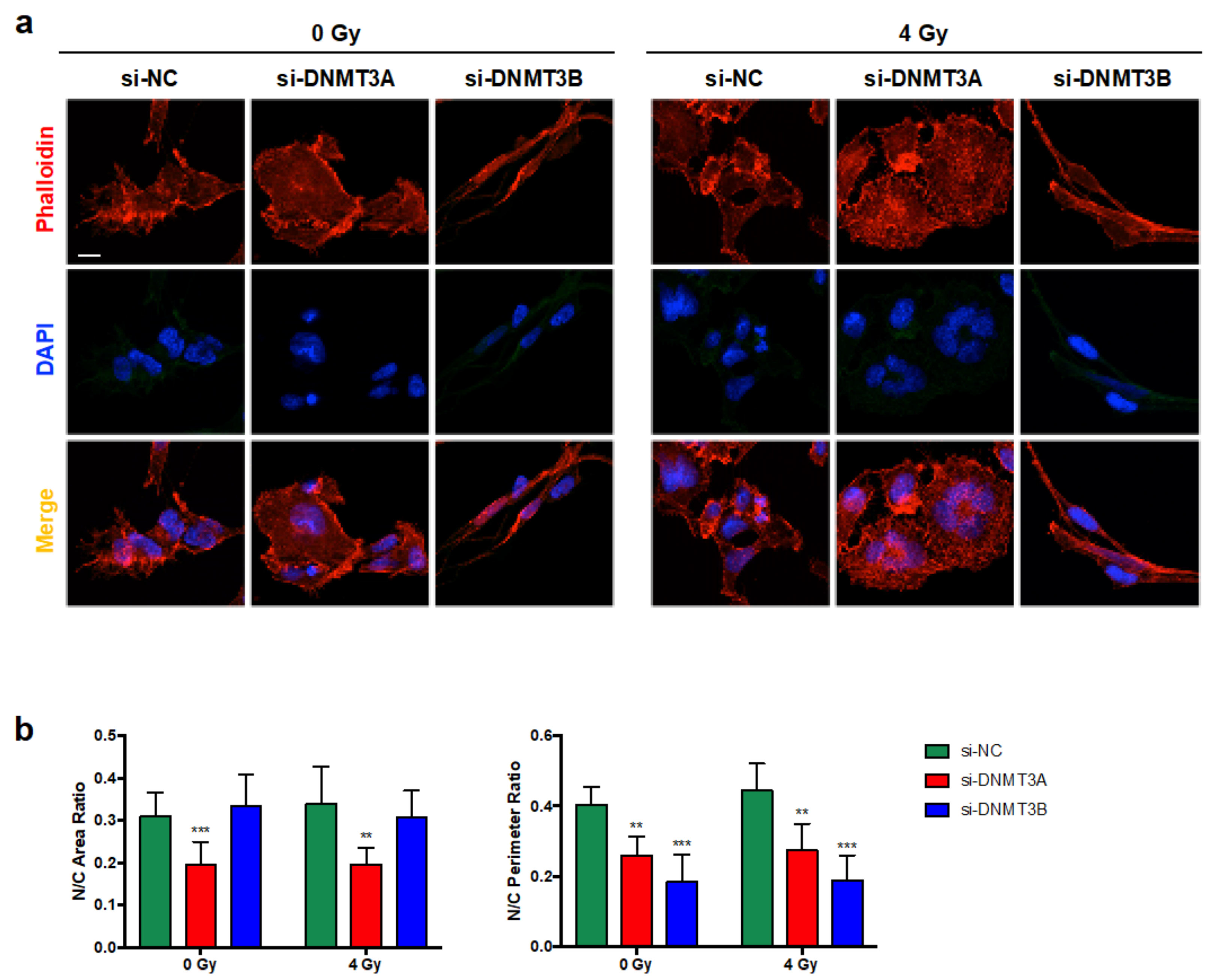
3.2. DNMT3A and DNMT3B Knocking down Enhance Radiotherapy-Induced Cell Proliferation Inhibition
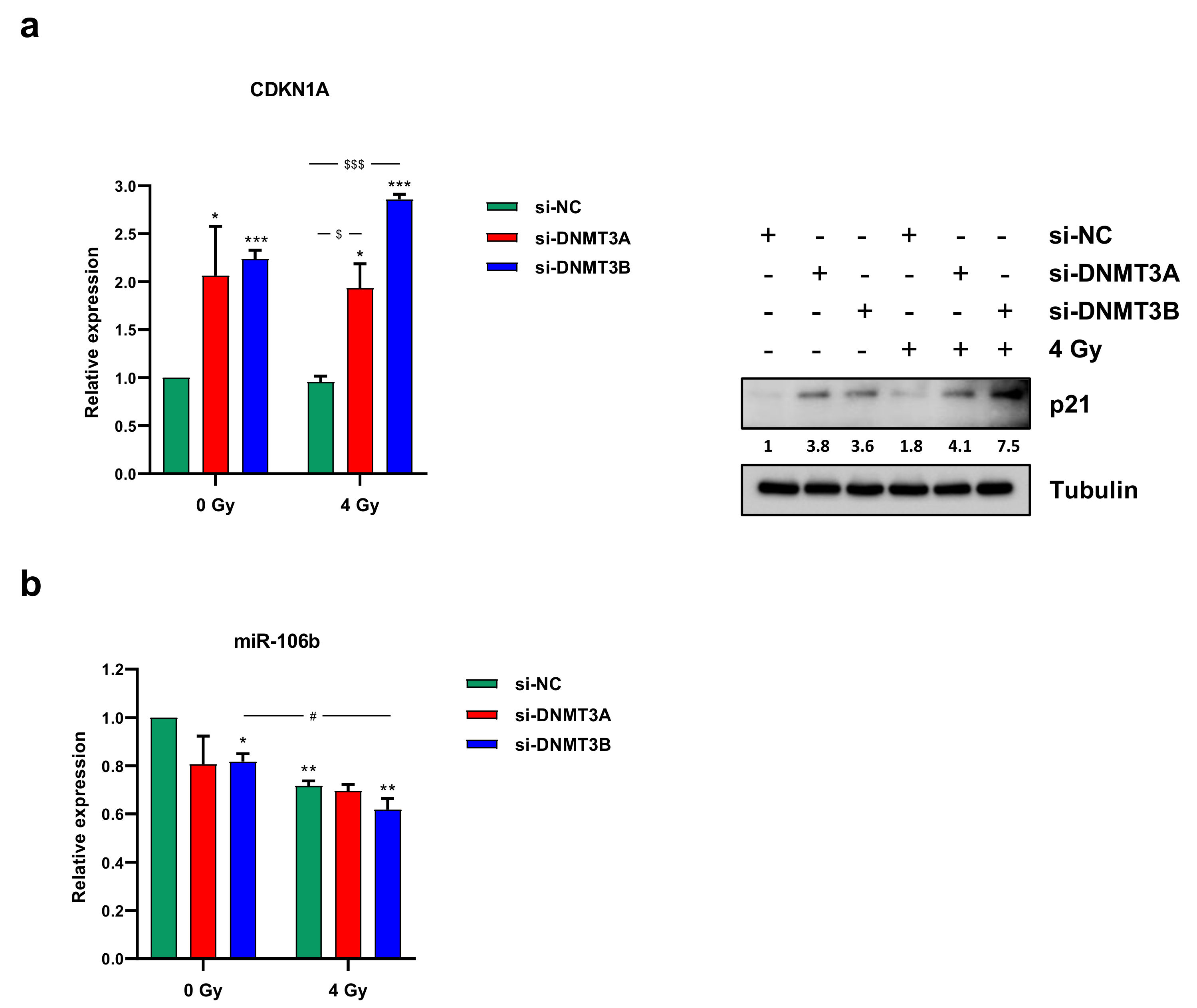
3.3. Cell Growth Inhibition Is Promoted by Sustained p21 Activation
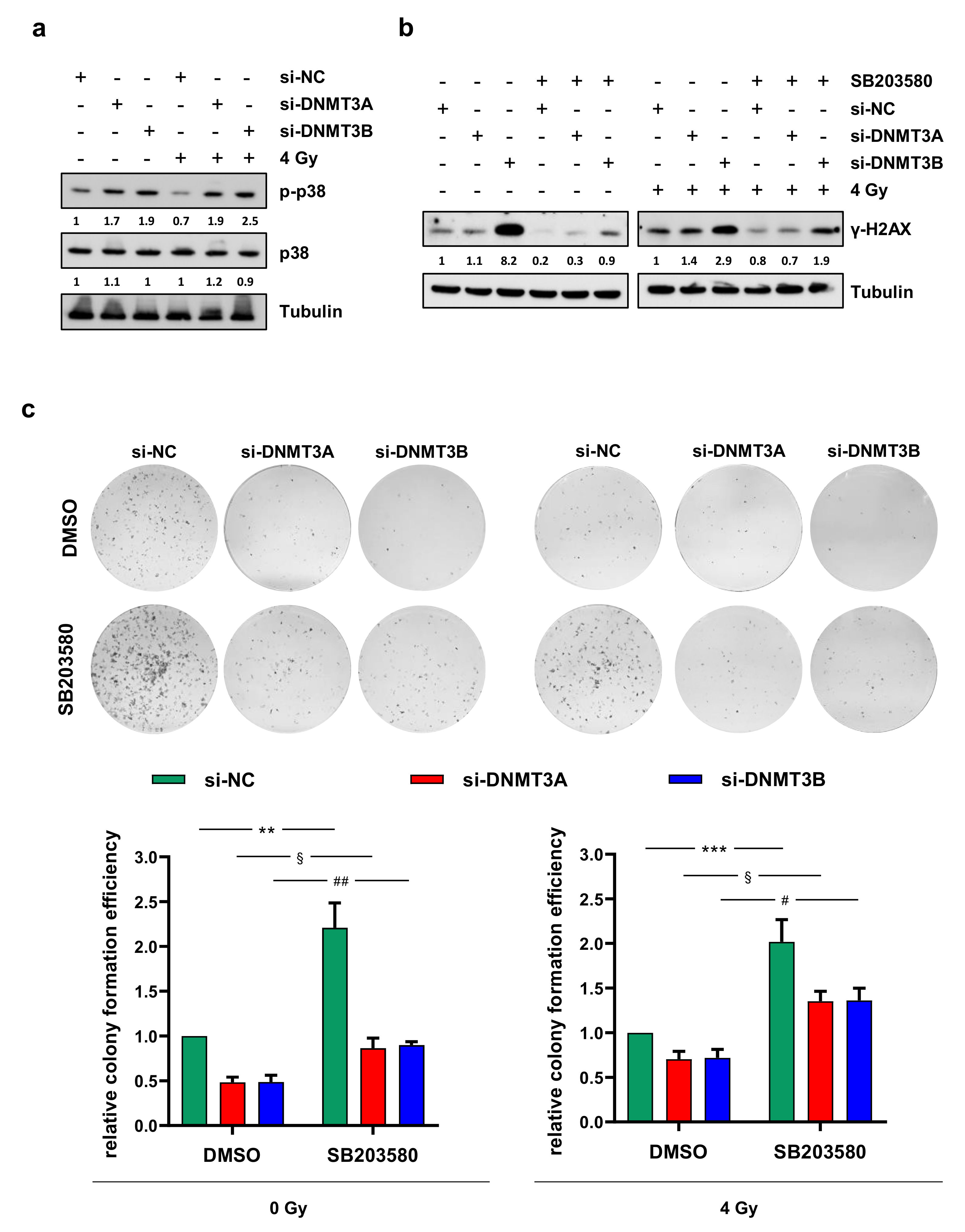
3.4. γ-H2AX Up-Regulation and the Reduced Clonogenic Capacity Induced by DNMT Knocking down Are Mediated by p38 Activation
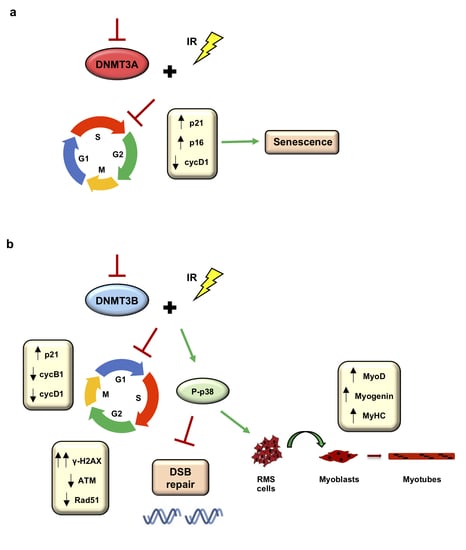
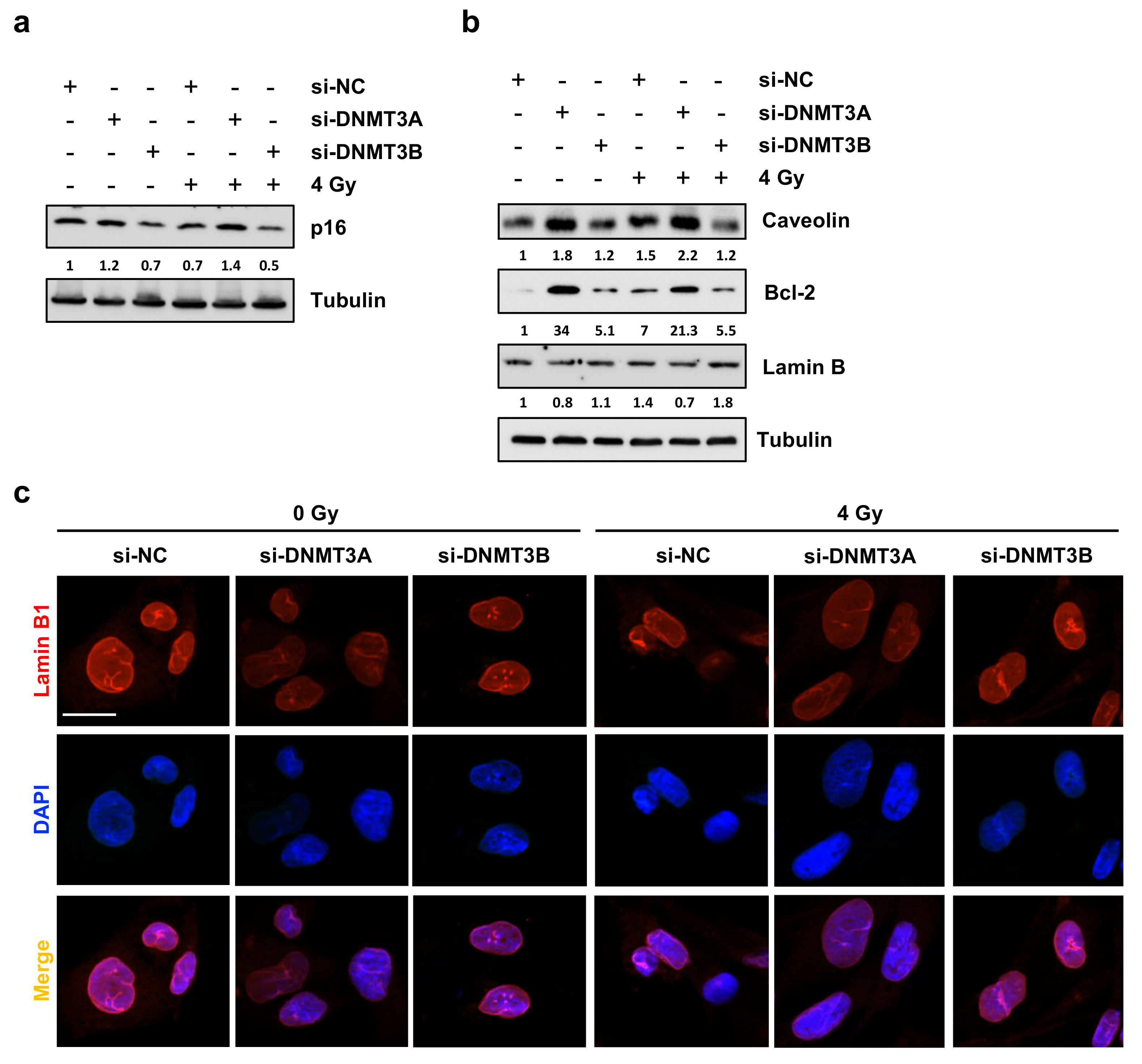
3.5. DNMT3A Depletion Stimulates Senescence in RMS Cells
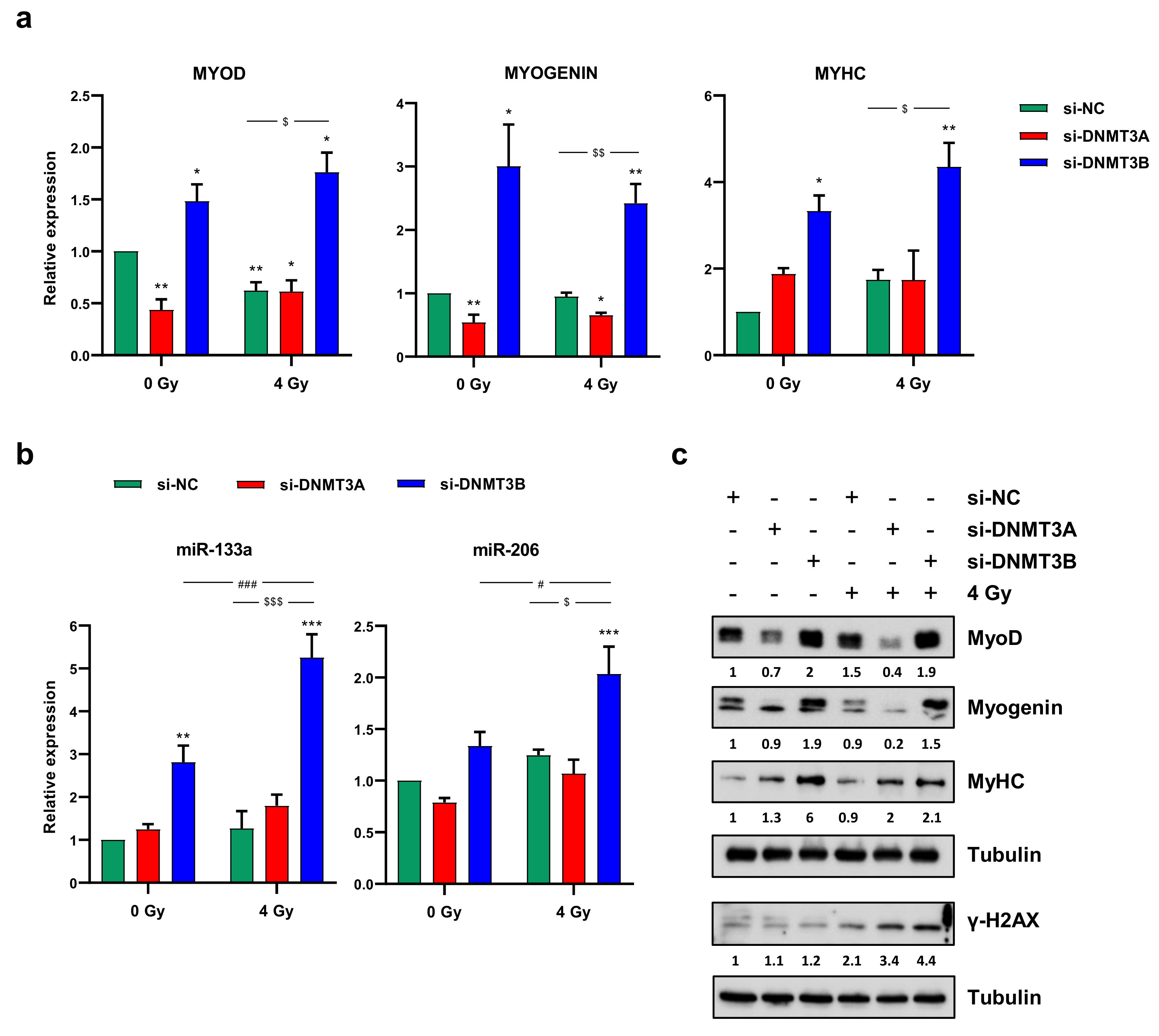
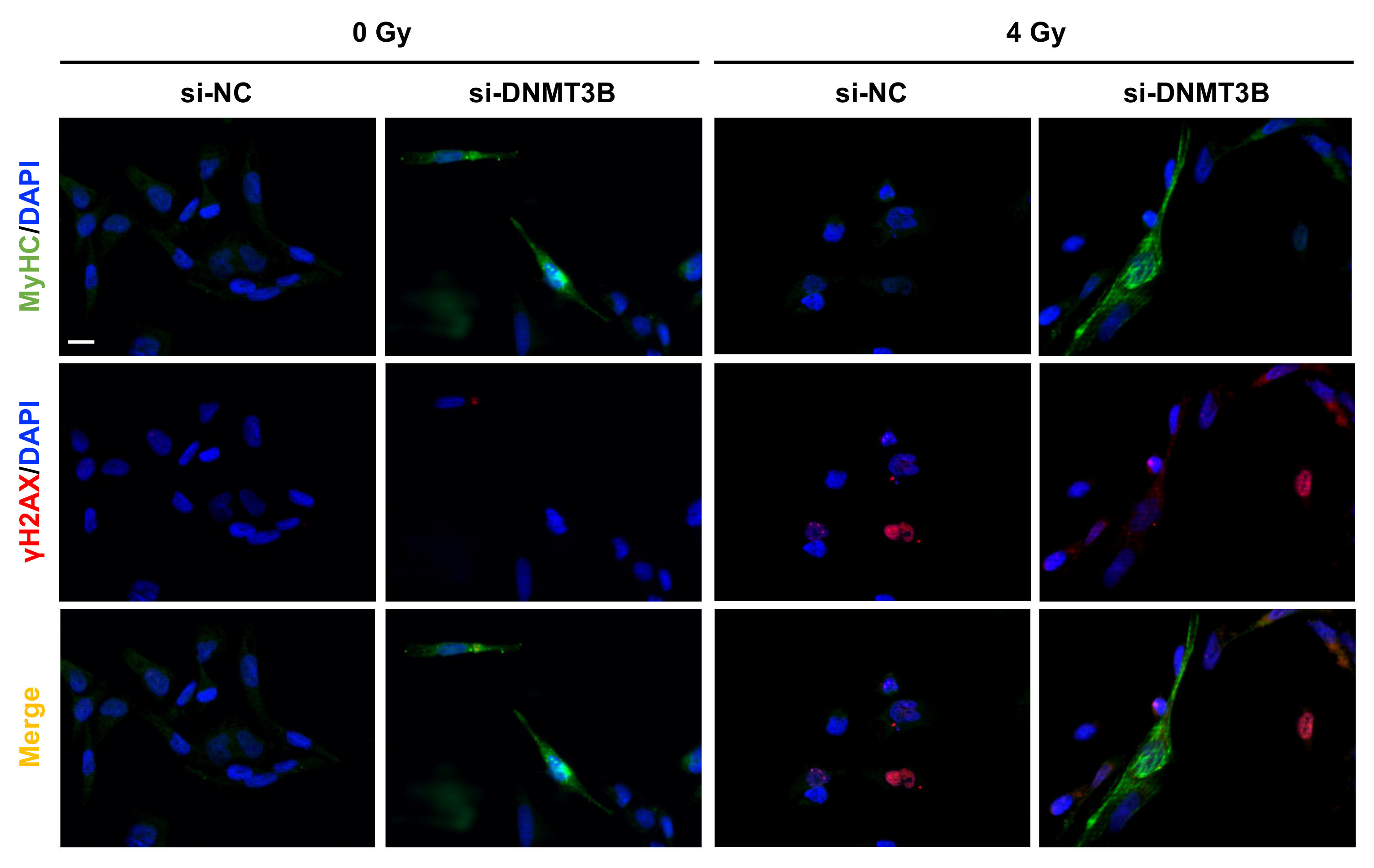
3.6. Radiation Treatment Preserves the Myogenic Program Triggered by DNMT3B Silencing and Induces Irreversible DNA Damage in Undifferentiated Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 2019, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Sultan, I.; Ferrari, A. Selecting multimodal therapy for rhabdomyosarcoma. Expert Rev. Anticancer Ther. 2010, 10, 1285–1301. [Google Scholar] [CrossRef] [PubMed]
- Parham, D.M.; Barr, F.G. Classification of Rhabdomyosarcoma and Its Molecular Basis. Adv. Anat. Pathol. 2013, 20, 387–397. [Google Scholar] [CrossRef]
- Chen, C.; Garcia, H.D.; Scheer, M.; Henssen, A.G. Current and Future Treatment Strategies for Rhabdomyosarcoma. Front. Oncol. 2019, 9, 1458. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Igarashi, K.; Tsuchiya, H. Recent advances and challenges in the treatment of rhabdomyosarcoma. Cancers 2020, 12, 1758. [Google Scholar] [CrossRef]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Hafiz, H.A.; Horwitz, K.B. Role of epigenetic modifications in luminal breast cancer. Epigenomics 2015, 7, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, V.; Soleimanian, A.; Ebrahimi, T.; Azargun, R.; Yazdani, P.; Eyvazi, S.; Tarhriz, V. Epigenetic modifications in gastric cancer: Focus on DNA methylation. Gene 2020, 742, 144577. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 49. [Google Scholar] [CrossRef]
- Megiorni, F.; Cialfi, S.; McDowell, H.P.; Felsani, A.; Camero, S.; Guffanti, A.; Pizer, B.; Clerico, A.; De Grazia, A.; Pizzuti, A.; et al. Deep Sequencing the microRNA profile in rhabdomyosarcoma reveals down-regulation of miR-378 family members. BMC Cancer 2014, 14, 880. [Google Scholar] [CrossRef] [Green Version]
- Camero, S.; Camicia, L.; Marampon, F.; Ceccarelli, S.; Shukla, R.; Mannarino, O.; Pizer, B.; Schiavetti, A.; Pizzuti, A.; Tombolini, V.; et al. BET inhibition therapy counteracts cancer cell survival, clonogenic potential and radioresistance mechanisms in rhabdomyosarcoma cells. Cancer Lett. 2020, 479, 71–88. [Google Scholar] [CrossRef]
- Hashimshony, T.; Zhang, J.; Keshet, I.; Bustin, M.; Cedar, H. The role of DNA methylation in setting up chromatin structure during development. Nat. Genet. 2003, 34, 187–192. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, S.Y.; Kim, K.H.; Kang, W.K.; Kim, J.I.; Oh, S.T.; Kim, J.S.; An, C.H. The combination effect of sodium butyrate and 5-Aza-2′-deoxycytidine on radiosensitivity in RKO colorectal cancer and MCF-7 breast cancer cell lines. World J. Surg. Oncol. 2009, 7, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dote, H.; Cerna, D.; Burgan, W.E.; Carter, D.J.; Cerra, M.A.; Hollingshead, M.G.; Camphausen, K.; Tofilon, P.J. Enhancement of in vitro and in vivo tumor cell radiosensitivity by the DNA methylation inhibitor zebularine. Clin. Cancer Res. 2005, 11, 4571–4579. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Shin, J.H.; Kim, I.H. Susceptibility and radiosensitization of human glioblastoma cells to trichostatin A, a histone deacetylase inhibitor. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.A.; Shin, J.H.; Kim, I.H.; Kim, J.H.; Kim, J.S.; Wu, H.G.; Chie, E.K.; Ha, S.W.; Park, C.; Kao, G.D. Histone deacetylase inhibitor-mediated radiosensitization of human cancer cells: Class differences and the potential influence of p53. Clin. Cancer Res. 2006, 12, 940–949. [Google Scholar] [CrossRef] [Green Version]
- Marampon, F.; Di Nisio, V.; Pietrantoni, I.; Petragnano, F.; Fasciani, I.; Scicchitano, B.M.; Ciccarelli, C.; Gravina, G.L.; Festuccia, C.; Del Fattore, A.; et al. Pro-differentiating and radiosensitizing effects of inhibiting HDACs by PXD-101 (Belinostat) in in vitro and in vivo models of human rhabdomyosarcoma cell lines. Cancer Lett. 2019, 461, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Ren, Z.; Chen, Y.; Zhu, J.; Du, Y.; Pan, D.; Li, X.; Hu, B. A feedback regulation between miR-145 and DNA methyltransferase 3b in prostate cancer cell and their responses to irradiation. Cancer Lett. 2015, 361, 121–127. [Google Scholar] [CrossRef]
- Wu, C.; Guo, E.; Ming, J.; Sun, W.; Nie, X.; Sun, L.; Peng, S.; Luo, M.; Liu, D.; Zhang, L.; et al. Radiation-Induced DNMT3B Promotes Radioresistance in Nasopharyngeal Carcinoma through Methylation of p53 and p21. Mol. Ther. Oncolytics 2020, 17, 306–319. [Google Scholar] [CrossRef]
- Chen, M.F.; Lu, M.S.; Lin, P.Y.; Chen, P.T.; Chen, W.C.; Lee, K. Der The role of DNA methyltransferase 3b in esophageal squamous cell carcinoma. Cancer 2012, 118, 4074–4089. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; Camero, S.; Ceccarelli, S.; McDowell, H.P.; Mannarino, O.; Marampon, F.; Pizer, B.; Shukla, R.; Pizzuti, A.; Marchese, C.; et al. DNMT3B in vitro knocking-down is able to reverse embryonal rhabdomyosarcoma cell phenotype through inhibition of proliferation and induction of myogenic differentiation. Oncotarget 2016, 7, 79342–79356. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Vescarelli, E.; Gerini, G.; Megiorni, F.; Anastasiadou, E.; Pontecorvi, P.; Solito, L.; De Vitis, C.; Camero, S.; Marchetti, C.; Mancini, R.; et al. MiR-200c sensitizes Olaparib-resistant ovarian cancer cells by targeting Neuropilin 1. J. Exp. Clin. Cancer Res. 2020, 39, 3. [Google Scholar] [CrossRef]
- Hinson, A.R.P.; Jones, R.; Lisa, L.E.; Belyea, B.C.; Barr, F.G.; Linardic, C.M. Human rhabdomyosarcoma cell lines for rhabdomyosarcoma research: Utility and pitfalls. Front. Oncol. 2013, 3, 183. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Roy, S.; Rana, A.; Akhter, Y.; Hande, M.P.; Banerjee, B. The role of p38 MAPK pathway in p53 compromised state and telomere mediated DNA damage response. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 836, 89–97. [Google Scholar] [CrossRef]
- Ohno-Iwashita, Y.; Shimada, Y.; Hayashi, M.; Inomata, M. Plasma membrane microdomains in aging and disease. Geriatr. Gerontol. Int. 2010, 10, S41–S52. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.J.; Oh, Y.S.; Park, S.C. Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 2007, 14, 1020–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [Green Version]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Choi, B.Y.; Lee, M.H.; Bode, A.M.; Dong, Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16INK4a) in Cancer. EBioMedicine 2016, 8, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Velicescu, M.; Weisenberger, D.J.; Gonzales, F.A.; Tsai, Y.C.T.; Nguyen, C.T.N.; Jones, P.A. Cell division is required for de Novo methylation of CpG islands in bladder cancer cells. Cancer Res. 2002, 62, 2378–2384. [Google Scholar] [PubMed]
- Pan, K.; Chen, Y.; Roth, M.; Wang, W.; Wang, S.; Yee, A.S.; Zhang, X. HBP1-Mediated Transcriptional Regulation of DNA Methyltransferase 1 and Its Impact on Cell Senescence. Mol. Cell. Biol. 2013, 33, 887–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Gong, Y.; Yan, G.; Wang, D.; Wang, Q.; Qiao, Y.; Hou, J.; Liu, B.; Tang, C. Atorvastatin treatment modulates p16 promoter methylation to regulate p16 expression. FEBS J. 2017, 284, 1868–1881. [Google Scholar] [CrossRef] [Green Version]
- Venturelli, S.; Berger, A.; Weiland, T.; Essmann, F.; Waibel, M.; Nuebling, T.; Häcker, S.; Schenk, M.; Schulze-Osthoff, K.; Salih, H.R.; et al. Differential induction of apoptosis and senescence by the DNA methyltransferase inhibitors 5-azacytidine and 5-aza-20-deoxycytidine in solid tumor cells. Mol. Cancer Ther. 2013, 12, 2226–2236. [Google Scholar] [CrossRef] [Green Version]
- Volonte, D.; Galbiati, F. Caveolin-1, a master regulator of cellular senescence. Cancer Metastasis Rev. 2020, 39, 397–414. [Google Scholar] [CrossRef]
- Dasari, A.; Bartholomew, J.N.; Volonte, D.; Galbiati, F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 2006, 66, 10805–10814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, A.; Patil, C.K.; Campisi, J. P38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gao, Y.; Zhang, G.; Huang, S.; Dong, Z.; Kong, C.; Su, D.; Du, J.; Zhu, S.; Liang, Q.; et al. DNMT3a plays a role in switches between doxorubicin-induced senescence and apoptosis of colorectal cancer cells. Int. J. Cancer 2011, 128, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Widodo, N.; Deocaris, C.C.; Kaur, K.; Hasan, K.; Yaguchi, T.; Yamasaki, K.; Sugihara, T.; Ishii, T.; Wadhwa, R.; Kaul, S.C. Stress chaperones, mortalin, and Pex19p mediate 5-Aza-2′ deoxycytidine-induced senescence of cancer cells by DNA methylation-independent pathway. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Amatori, S.; Bagaloni, I.; Viti, D.; Fanelli, M. Premature senescence induced by DNA demethylating agent (Decitabine) as therapeutic option for malignant pleural mesothelioma. Lung Cancer 2011, 71, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Putri, J.F.; Widodo, N.; Sakamoto, K.; Kaul, S.C.; Wadhwa, R. Induction of senescence in cancer cells by 5′-Aza-2′-deoxycytidine: Bioinformatics and experimental insights to its targets. Comput. Biol. Chem. 2017, 70, 49–55. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends in Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Gil, J. Senescence: A new weapon for cancer therapy. Trends Cell Biol. 2012, 22, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassen, P.; Primdahl, H.; Johansen, J.; Kristensen, C.A.; Andersen, E.; Andersen, L.J.; Evensen, J.F.; Eriksen, J.G.; Overgaard, J. Impact of HPV-associated p16-expression on radiotherapy outcome in advanced oropharynx and non-oropharynx cancer. Radiother. Oncol. 2014, 113, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Dikomey, E.; Dahm-Daphi, J.; Brammer, I.; Martensen, R.; Kaina, B. Correlation between cellular radiosensitivity and non-repaired double- strand breaks studied in nine mammalian cell lines. Int. J. Radiat. Biol. 1998, 73, 269–278. [Google Scholar] [CrossRef]
- Löbrich, M.; Shibata, A.; Beucher, A.; Fisher, A.; Ensminger, M.; Goodarzi, A.A.; Barton, O.; Jeggo, P.A. γH2AX foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimization. Cell Cycle 2010, 9, 662–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downey, M.; Durocher, D. γH2AX as a checkpoint maintenance signal. Cell Cycle 2006, 5, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Fillingham, J.; Keogh, M.C.; Krogan, N.J. γH2AX and its role in DNA double-strand break repair. Biochem. Cell Biol. 2006, 84, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; Kessinger, C.; Kobayashi, J.; Chen, B.P.C.; Chen, D.J.; Chatterjee, A.; Burma, S. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair 2006, 5, 575–590. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camero, S.; Vitali, G.; Pontecorvi, P.; Ceccarelli, S.; Anastasiadou, E.; Cicchetti, F.; Flex, E.; Pomella, S.; Cassandri, M.; Rota, R.; et al. DNMT3A and DNMT3B Targeting as an Effective Radiosensitizing Strategy in Embryonal Rhabdomyosarcoma. Cells 2021, 10, 2956. https://doi.org/10.3390/cells10112956
Camero S, Vitali G, Pontecorvi P, Ceccarelli S, Anastasiadou E, Cicchetti F, Flex E, Pomella S, Cassandri M, Rota R, et al. DNMT3A and DNMT3B Targeting as an Effective Radiosensitizing Strategy in Embryonal Rhabdomyosarcoma. Cells. 2021; 10(11):2956. https://doi.org/10.3390/cells10112956
Chicago/Turabian StyleCamero, Simona, Giulia Vitali, Paola Pontecorvi, Simona Ceccarelli, Eleni Anastasiadou, Francesca Cicchetti, Elisabetta Flex, Silvia Pomella, Matteo Cassandri, Rossella Rota, and et al. 2021. "DNMT3A and DNMT3B Targeting as an Effective Radiosensitizing Strategy in Embryonal Rhabdomyosarcoma" Cells 10, no. 11: 2956. https://doi.org/10.3390/cells10112956
APA StyleCamero, S., Vitali, G., Pontecorvi, P., Ceccarelli, S., Anastasiadou, E., Cicchetti, F., Flex, E., Pomella, S., Cassandri, M., Rota, R., Marampon, F., Marchese, C., Schiavetti, A., & Megiorni, F. (2021). DNMT3A and DNMT3B Targeting as an Effective Radiosensitizing Strategy in Embryonal Rhabdomyosarcoma. Cells, 10(11), 2956. https://doi.org/10.3390/cells10112956