
Cardiac Conduction Velocity, Remodeling and Arrhythmogenesis


{kind=link}
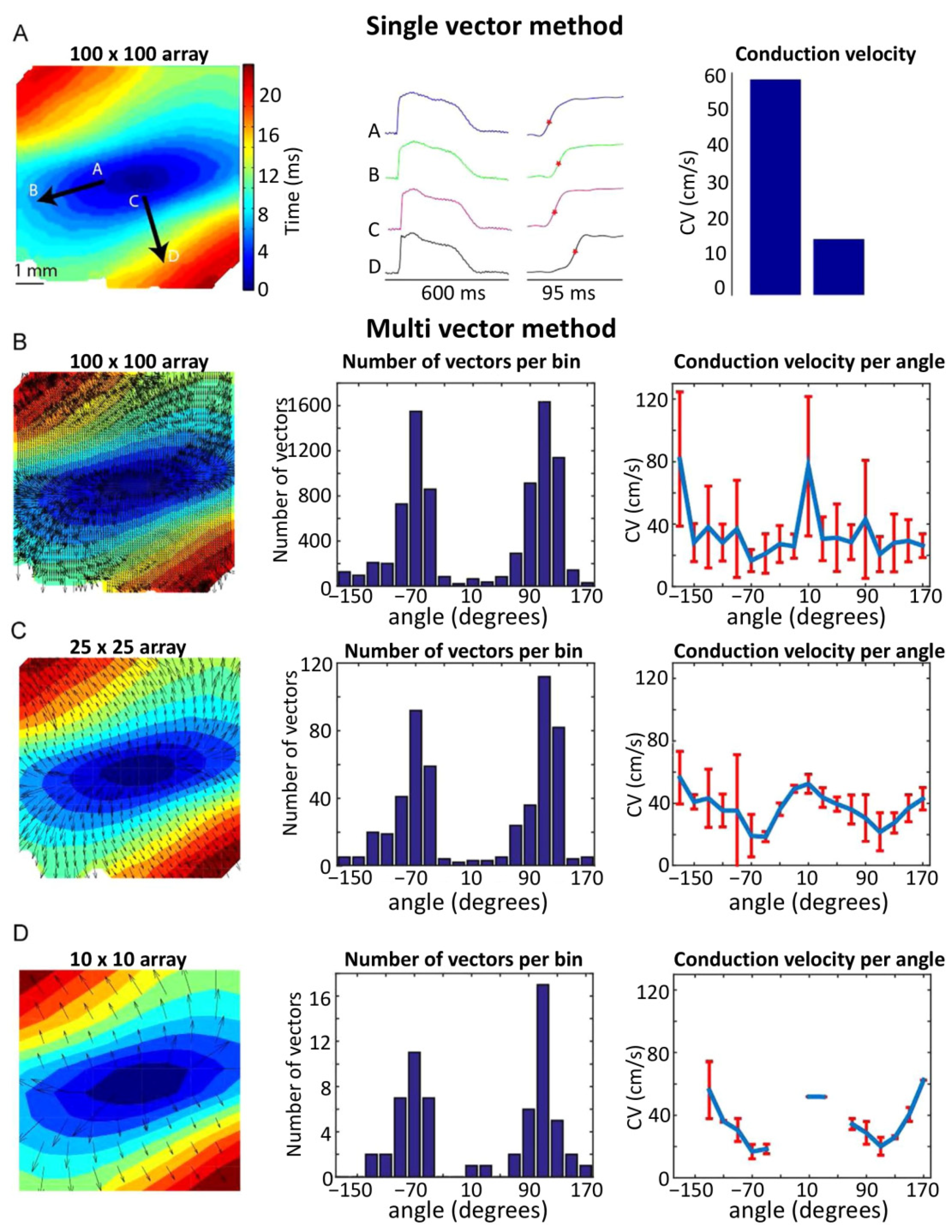{kind=link}
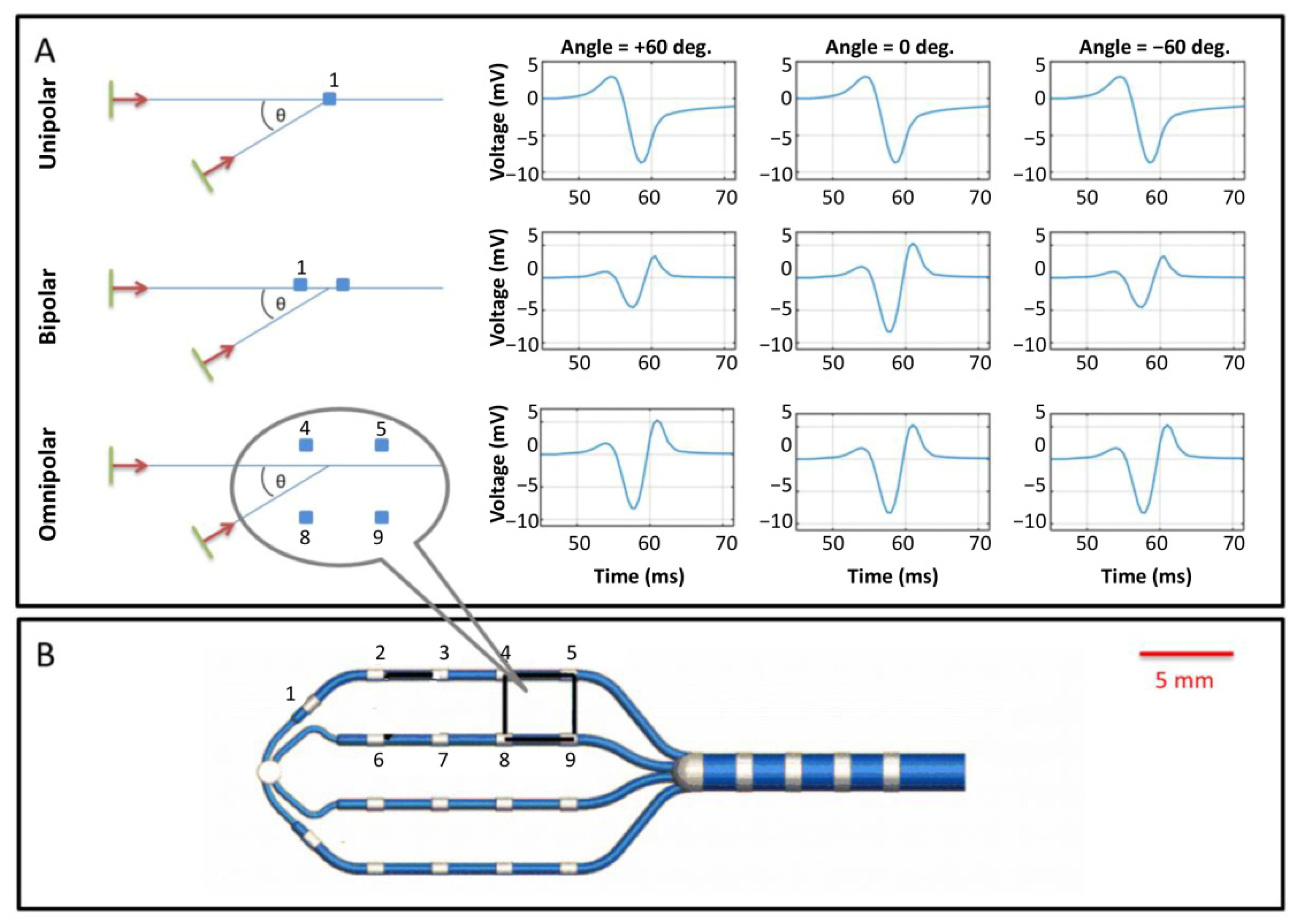{kind=link}
{kind=link}
{kind=link}
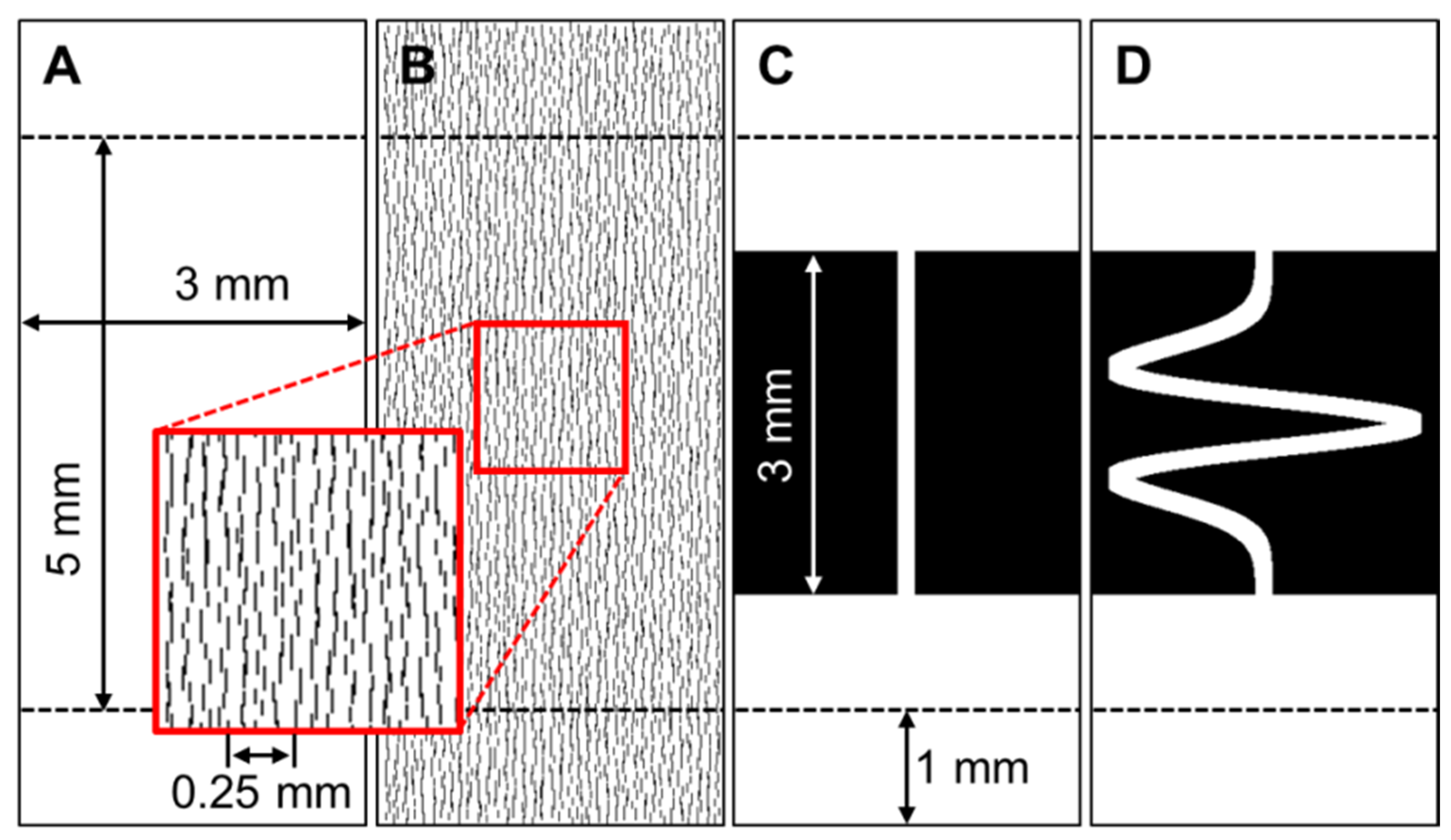{kind=link}
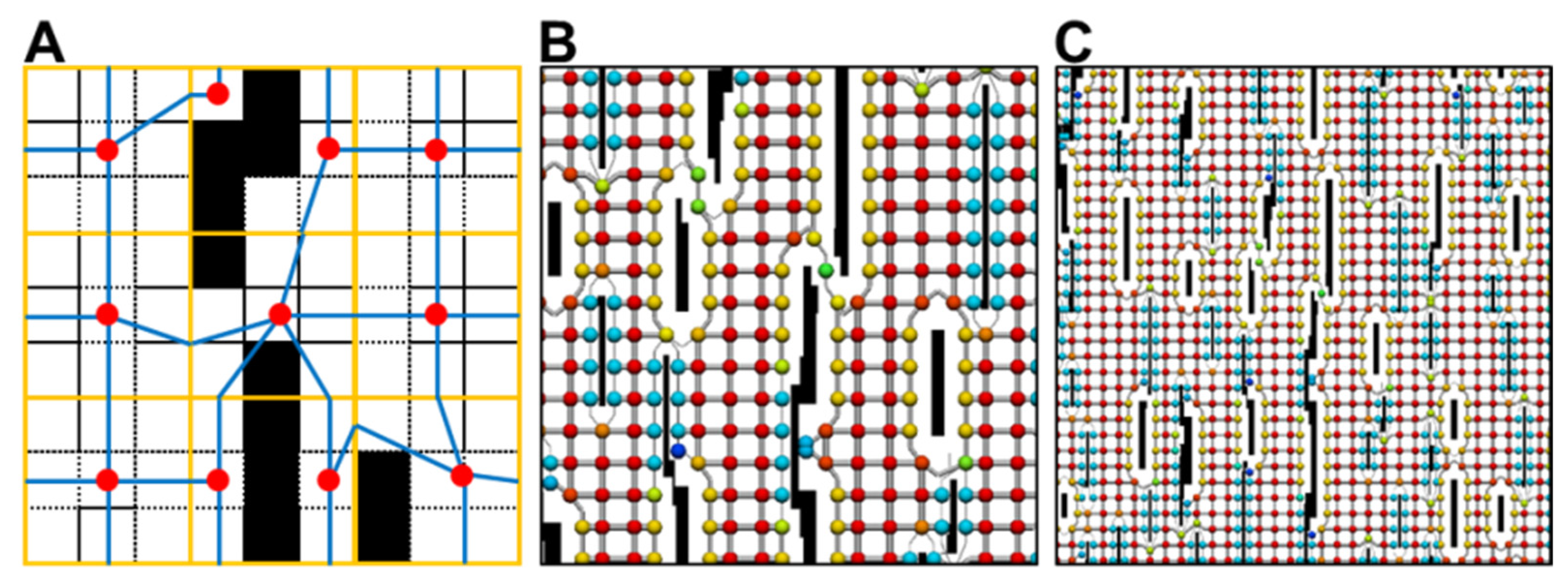{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods of CV Measurement
2.1. Optical Mapping
2.2. Multi-Electrode Arrays
2.3. Intracardiac (Catheter) Electrograms
2.4. Electrocardiographic Imaging
2.5. Limitations of Current CV Measuring Techniques
3. Physiological Factors Affecting CV
3.1. Geometric and Intra-/Extracellular Factors
3.2. Gap Junctions and Connexins
3.3. Mechanical Loading
4. Pathological Factors Affecting CV
4.1. Aging-Associated Changes
4.2. Heart Failure
4.3. Hypertrophy
4.4. Ischemia
5. Compounds Affecting CV
5.1. Experimental Compounds
5.2. Clinical Compounds
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Introduction
Appendix A.2. Model Domains


Appendix A.3. Modeling Electrical Activity in the Networks

References
- Taniguchi, N.; Miyasaka, Y.; Suwa, Y.; Harada, S.; Nakai, E.; Shiojima, I. Heart failure in atrial fibrillation: An update on clinical and echocardiographic implications. Circ. J. 2020, 84, 1212–1217. [Google Scholar] [CrossRef]
- Magnussen, C.; Niiranen, T.J.; Ojeda, F.M.; Gianfagna, F.; Blankenberg, S.; Njølstad, I.; Vartiainen, E.; Sans, S.; Pasterkamp, G.; Hughes, M.; et al. Sex differences and similarities in atrial fibrillation epidemiology, risk factors, and mortality in community cohorts: Results from the BiomarCaRE consortium (Biomarker for cardiovascular risk assessment in Europe). Circulation 2017, 136, 1588–1597. [Google Scholar] [CrossRef]
- Aiba, T.; Tomaselli, G.F. Electrical remodeling in the failing heart. Curr. Opin. Cardiol. 2010, 25, 29–36. [Google Scholar] [CrossRef]
- Hernández-Romero, I.; Guillem, M.S.; Figuera, C.; Atienza, F.; Fernández-Avilés, F.; Climent, A.M. Optical imaging of voltage and calcium in isolated hearts: Linking spatiotemporal heterogeneities and ventricular fibrillation initiation. PLoS ONE 2019, 14, e0215951. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-R.; Salama, G. Simultaneous maps of optical action potentials and calcium transients in guinea-pig hearts: Mechanisms underlying concordant alternans. J. Physiol. 2000, 529, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Mironov, S.; Jalife, J.; Tolkacheva, E.G. Role of conduction velocity restitution and short-term memory in the development of action potential duration alternans in isolated rabbit hearts. Circulation 2008, 118, 17–25. [Google Scholar] [CrossRef]
- Roney, C.H.; Whitaker, J.; Sim, I.; O’Neill, L.; Mukherjee, R.K.; Razeghi, O.; Vigmond, E.J.; Wright, M.; O’Neill, M.D.; Williams, S.E.; et al. A technique for measuring anisotropy in atrial conduction to estimate conduction velocity and atrial fibre direction. Comput. Biol. Med. 2019, 104, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Mines, G.R. On dynamic equilibrium in the heart. J. Physiol. 1913, 46, 349–383. [Google Scholar] [CrossRef]
- de Jong, S.; van Veen, T.A.B.; van Rijen, H.V.M.; de Bakker, J.M.T. Fibrosis and cardiac arrhythmias. J. Cardiovasc. Pharmacol. 2011, 57, 630–638. [Google Scholar] [CrossRef]
- LeGrice, I.J.; Smaill, B.H.; Chai, L.Z.; Edgar, S.G.; Gavin, J.B.; Hunter, P.J. Laminar structure of the heart: Ventricular myocyte arrangement and connective tissue architecture in the dog. Am. J. Physiol. Heart Circ. Physiol. 1995, 269, H571–582. [Google Scholar] [CrossRef]
- Fedorov, V.V.; Lozinsky, I.T.; Sosunov, E.A.; Anyukhovsky, E.P.; Rosen, M.R.; Balke, C.W.; Efimov, I.R. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm 2007, 4, 619–626. [Google Scholar] [CrossRef]
- Lou, Q.; Li, W.; Efimov, I.R. Multiparametric optical mapping of the langendorff-perfused rabbit heart. J. Vis. Exp. 2011, 55, 3160. [Google Scholar] [CrossRef] [PubMed]
- Sabeh, M.K.; Kekhia, H.; MacRae, C.A. Optical mapping in the developing zebrafish heart. Pediatr. Cardiol. 2012, 33, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Banville, I.; Gray, R.A.; Ideker, R.E.; Smith, W.M. Shock-induced figure-of-eight reentry in the isolated rabbit heart. Circ. Res. 1999, 85, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Banville, I.; Gray, R.A. Effect of action potential duration and conduction velocity restitution and their spatial dispersion on alternans and the stability of arrhythmias. J. Cardiovasc. Electrophysiol. 2002, 13, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Gutbrod, S.R.; Walton, R.; Gilbert, S.; Meillet, V.; Jaïs, P.; Hocini, M.; Haïssaguerre, M.; Dubois, R.; Bernus, O.; Efimov, I.R. Quantification of the transmural dynamics of atrial fibrillation by simultaneous endocardial and epicardial optical mapping in an acute sheep model. Circ. Arrhythmia Electrophysiol. 2015, 8, 456–465. [Google Scholar] [CrossRef]
- Baxter, W.T.; Mironov, S.F.; Zaitsev, A.V.; Jalife, J.; Pertsov, A.M. Visualizing excitation waves inside cardiac muscle using transillumination. Biophys. J. 2001, 80, 516–530. [Google Scholar] [CrossRef]
- Bray, M.A.; Lin, S.F.; Wikswo, J.P., Jr. Three-dimensional surface reconstruction and flourescent visualization of cardiac activation. IEEE Trans. Biomed. Eng. 2000, 47, 1382–1391. [Google Scholar] [CrossRef]
- Qu, F.; Ripplinger, C.M.; Nikolski, V.P.; Grimm, C.; Efimov, I.R. Three-dimensional panoramic imaging of cardiac arrhythmias in rabbit heart. J. Biomed. Opt. 2007, 12, 044019. [Google Scholar] [CrossRef]
- Kay, M.W.; Walcott, G.P.; Gladden, J.D.; Melnick, S.B.; Rogers, J.M. Lifetimes of epicardial rotors in panoramic optical maps of fibrillating swine ventricles. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1935–H1941. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ripplinger, C.M.; Lou, Q.; Li, W.; Hadley, J.; Efimov, I.R. Panoramic imaging reveals basic mechanisms of induction and termination of ventricular tachycardia in rabbit heart with chronic infarction: Implications for low voltage cardioversion. Heart Rhythm 2009, 6, 87–97. [Google Scholar] [CrossRef]
- Dura, M.; Schröder-Schetelig, J.; Luther, S.; Lehnart, S.E. Towards panoramic in situ mapping of action potential propagation in transgenic hearts to investigate initiation and therapeutic control of arrhythmias. Front. Physiol. 2014, 5, 337. [Google Scholar] [CrossRef] [PubMed]
- Linnenbank, A.C.; de Bakker, J.M.T.; Coronel, R. How to measure propagation velocity in cardiac tissue: A simulation study. Front. Physiol. 2014, 5, 267. [Google Scholar] [CrossRef]
- Doshi, A.N.; Walton, R.D.; Krul, S.P.; de Groot, J.R.; Bernus, O.; Efimov, I.R.; Boukens, B.J.; Coronel, R. Feasibility of a semi-automated method for cardiac conduction velocity analysis of high-resolution activation maps. Comput. Biol. Med. 2015, 65, 177–183. [Google Scholar] [CrossRef]
- Bayly, P.V.; Kenknight, B.H.; Rogers, J.M.; Hillsley, R.E.; Ideker, R.E.; Smith, W.M. Estimation of conduction velocity vector fields from epicardial mapping data. IEEE Trans. Biomed. Eng. 1998, 45, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Swift, L.M.; Asfour, H.; Posnack, N.G.; Arutunyan, A.; Kay, M.W.; Sarvazyan, N. Properties of blebbistatin for cardiac optical mapping and other imaging applications. Pflugers Arch. Eur. J. Physiol. 2012, 464, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Brack, K.E.; Narang, R.; Winter, J.; Ng, G.A. The mechanical uncoupler blebbistatin is associated with significant electrophysiological effects in the isolated rabbit heart. Exp. Physiol. 2013, 98, 1009–1027. [Google Scholar] [CrossRef] [PubMed]
- Kappadan, V.; Telele, S.; Uzelac, I.; Fenton, F.; Parlitz, U.; Luther, S.; Christoph, J. High-resolution optical measurement of cardiac restitution, contraction, and fibrillation dynamics in beating vs. blebbistatin-uncoupled isolated rabbit hearts. Front. Physiol. 2020, 11, 464. [Google Scholar] [CrossRef]
- Lee, P.; Quintanilla, J.G.; Alfonso-Almazán, J.M.; Galán-Arriola, C.; Yan, P.; Sánchez-González, J.; Pérez-Castellano, N.; Pérez-Villacastín, J.; Ibañez, B.; Loew, L.M.; et al. In vivo ratiometric optical mapping enables high-resolution cardiac electrophysiology in pig models. Cardiovasc. Res. 2019, 115, 1659–1671. [Google Scholar] [CrossRef]
- Zhang, H.; Iijima, K.; Huang, J.; Walcott, G.P.; Rogers, J.M. Optical mapping of membrane potential and epicardial deformation in beating hearts. Biophys. J. 2016, 111, 438–451. [Google Scholar] [CrossRef]
- Christoph, J.; Luther, S. Marker-free tracking for motion artifact compensation and deformation measurements in optical mapping videos of contracting hearts. Front. Physiol. 2018, 9, 1483. [Google Scholar] [CrossRef] [PubMed]
- Pogwizd, S.M.; Corr, P.B. Electrophysiologic mechanisms underlying arrhythmias due to reperfusion of ischemic myocardium. Circulation 1987, 76, 404–426. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.; Ideker, R.E.; Smith, W.M.; Klein, G.J.; Kasell, J.; Wallace, A.G.; Gallagher, J.J. The sock electrode array: A tool for determining global epicardial activation during unstable arrhythmias. Pacing Clin. Electrophysiol. 1980, 3, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Faris, O.P.; Evans, F.J.; Ennis, D.B.; Helm, P.A.; Taylor, J.L.; Chesnick, A.S.; Guttman, M.A.; Ozturk, C.; McVeigh, E.R. Novel technique for cardiac electromechanical mapping with magnetic resonance imaging tagging and an epicardial electrode sock. Ann. Biomed. Eng. 2003, 31, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Usui, M.; Callihan, R.L.; Walker, R.G.; Walcott, G.P.; Rollins, D.L.; Wolf, P.D.; Smith, W.M.; Ideker, R.E. Epicardial sock mapping following monophasic and biphasic shocks of equal voltage with an endocardial lead system. J. Cardiovasc. Electrophysiol. 1996, 7, 322–334. [Google Scholar] [CrossRef]
- Orini, M.; Taggart, P.; Bhuva, A.; Roberts, N.; Di Salvo, C.; Yates, M.; Badiani, S.; Van Duijvenboden, S.; Lloyd, G.; Smith, A.; et al. Direct in vivo assessment of global and regional mechanoelectric feedback in the intact human heart. Heart Rhythm 2021, 18, 1406–1413. [Google Scholar] [CrossRef]
- Eckstein, J.; Maesen, B.; Linz, D.; Zeemering, S.; van Hunnik, A.; Verheule, S.; Allessie, M.; Schotten, U. Time course and mechanisms of endo-epicardial electrical dissociation during atrial fibrillation in the goat. Cardiovasc. Res. 2011, 89, 816–824. [Google Scholar] [CrossRef]
- Xu, L.; Gutbrod, S.R.; Bonifas, A.P.; Su, Y.; Sulkin, M.S.; Lu, N.; Chung, H.-J.; Jang, K.-I.; Liu, Z.; Ying, M.; et al. 3D multifunctional integumentary membranes for spatiotemporal cardiac measurements and stimulation across the entire epicardium. Nat. Commun. 2014, 5, 3329. [Google Scholar] [CrossRef]
- Kim, D.-H.; Ghaffari, R.; Lu, N.; Wang, S.; Lee, S.P.; Keum, H.; D’Angelo, R.; Klinker, L.; Su, Y.; Lu, C.; et al. Electronic sensor and actuator webs for large-area complex geometry cardiac mapping and therapy. Proc. Natl. Acad. Sci. USA 2012, 109, 19910–19915. [Google Scholar] [CrossRef]
- Shariat, M.H.; Redfearn, D.P. Cardiac conduction velocity estimation during wavefront collision. Proc. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. EMBS 2018, 2018, 4840–4843. [Google Scholar] [CrossRef]
- Tedrow, U.B.; Stevenson, W.G. Recording and interpreting unipolar electrograms to guide catheter ablation. Heart Rhythm 2011, 8, 791–796. [Google Scholar] [CrossRef]
- Stevenson, W.G.; Soejima, K. Recording techniques for clinical electrophysiology. J. Cardiovasc. Electrophysiol. 2005, 16, 1017–1022. [Google Scholar] [CrossRef]
- Blanchard, S.M.; Damiano, R.J., Jr.; Asano, T.; Smith, W.M.; Ideker, R.E.; Lowe, J.E. The effects of distant cardiac electrical events on local activation in unipolar epicardial electrograms. IEEE Trans. Biomed. Eng. 1987, 34, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Anter, E.; Josephson, M.E. Bipolar voltage amplitude: What does it really mean? Heart Rhythm 2016, 13, 326–327. [Google Scholar] [CrossRef] [PubMed]
- Josephson, M.E.; Anter, E. Substrate mapping for ventricular tachycardia assumptions and misconceptions. JACC Clin. Electrophysiol. 2015, 1, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, M.; Magtibay, K.; Massé, S.; Porta-Sanchez, A.; Haldar, S.; Bhaskaran, A.; Nayyar, S.; Glover, B.; Deno, D.C.; Vigmond, E.J.; et al. Determinants of atrial bipolar voltage: Inter electrode distance and wavefront angle. Comput. Biol. Med. 2018, 102, 449–457. [Google Scholar] [CrossRef]
- Takigawa, M.; Relan, J.; Martin, R.; Kim, S.; Kitamura, T.; Frontera, A.; Cheniti, G.; Vlachos, K.; Massoullié, G.; Martin, C.A.; et al. Effect of bipolar electrode orientation on local electrogram properties. Heart Rhythm 2018, 15, 1853–1861. [Google Scholar] [CrossRef]
- Takigawa, M.; Relan, J.; Martin, R.; Kim, S.; Kitamura, T.; Cheniti, G.; Vlachos, K.; Pillois, X.; Frontera, A.; Massoullié, G.; et al. Detailed analysis of the relation between bipolar electrode spacing and far- and near-field electrograms. JACC Clin. Electrophysiol. 2019, 5, 66–77. [Google Scholar] [CrossRef]
- Cantwell, C.D.; Roney, C.H.; Ng, F.S.; Siggers, J.H.; Sherwin, S.J.; Peters, N.S. Techniques for automated local activation time annotation and conduction velocity estimation in cardiac mapping. Comput. Biol. Med. 2015, 65, 229–242. [Google Scholar] [CrossRef]
- D Pooter, J.; El Haddad, M.; Wolf, M.; Phlips, T.; van Heuverswyn, F.; Timmers, L.; Tavernier, R.; Knecht, S.; Vandekerckhove, Y.; Duytschaever, M. Clinical assessment and comparison of annotation algorithms in high-density mapping of regular atrial tachycardias. J. Cardiovasc. Electrophysiol. 2018, 29, 177–185. [Google Scholar] [CrossRef]
- Weiss, C.; Willems, S.; Rueppel, R.; Hoffmann, M.; Meinertz, T. Electroanatomical Mapping (CARTO) of ectopic atrial tachycardia: Impact of bipolar and unipolar local electrogram annotation for localization the focal origin. J. Interv. Card. Electrophysiol. 2001, 5, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Deno, D.C.; Balachandran, R.; Morgan, D.; Ahmad, F.; Masse, S.; Nanthakumar, K. Orientation-independent catheter-based characterization of myocardial activation. IEEE Trans. Biomed. Eng. 2017, 64, 1067–1177. [Google Scholar] [CrossRef]
- Magtibay, K.; Porta-Sánchez, A.; Haldar, S.K.; Deno, D.C.; Massé, S.; Nanthakumar, K. Reinserting physiology into cardiac mapping using omnipolar electrograms. Card. Electrophysiol. Clin. 2019, 11, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, C.; Ghanem, R.N.; Jia, P.; Ryu, K.; Rudy, Y. Noninvasive electrocardiographic imaging for cardiac electrophysiology and arrhythmia. Nat. Med. 2004, 10, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, C.; Jia, P.; Ghanem, R.; Ryu, K.; Rudy, Y. Activation and repolarization of the normal human heart under complete physiological conditions. Proc. Natl. Acad. Sci. USA 2006, 103, 6309–6314. [Google Scholar] [CrossRef]
- Duchateau, J.; Sacher, F.; Pambrun, T.; Derval, N.; Chamorro-Servent, J.; Denis, A.; Ploux, S.; Hocini, M.; Jaïs, P.; Bernus, O.; et al. Performance and limitations of noninvasive cardiac activation mapping. Heart Rhythm 2019, 16, 435–442. [Google Scholar] [CrossRef]
- Bear, L.R.; Bouhamama, O.; Cluitmans, M.; Duchateau, J.; Walton, R.D.; Abell, E.; Belterman, C.; Haissaguerre, M.; Bernus, O.; Coronel, R.; et al. Advantages and pitfalls of noninvasive electrocardiographic imaging. J. Electrocardiol. 2019, 57S, S15–S20. [Google Scholar] [CrossRef]
- Bear, L.R.; Walton, R.D.; Abell, E.; Coudière, Y.; Haissaguerre, M.; Bernus, O.; Dubois, R. Optical imaging of ventricular action potentials in a torso tank: A new platform for non-invasive electrocardiographic imaging validation. Front. Physiol. 2019, 10, 146. [Google Scholar] [CrossRef]
- Aras, K.K.; Faye, N.R.; Cathey, B.; Efimov, I.R. Critical volume of human myocardium necessary to maintain ventricular fibrillation. Circ. Arrhythmia Electrophysiol. 2018, 11, ee006692. [Google Scholar] [CrossRef]
- Thomas, S.P.; Kucera, J.P.; Bircher-Lehmann, L.; Rudy, Y.; Saffitz, J.E.; Kléber, A.G. Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ. Res. 2003, 92, 1209–1216. [Google Scholar] [CrossRef]
- Spach, M.S.; Heidlage, J.F.; Dolber, P.C.; Barr, R.C. Electrophysiological effects of remodeling cardiac gap junctions and cell size. Experimental and model studies of normal cardiac growth. Circ. Res. 2000, 86, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Joyner, R.W.; Ramza, B.M.; Tan, R.C.; Matsuda, J.; Do, T.T. Effects of tissue geometry on initiation of a cardiac action potential. Am. J. Physiol. Heart Circ. Physiol. 1989, 256, 391–403. [Google Scholar] [CrossRef]
- Jack, J.; Noble, D.; Tsien, R. Electric Current Flow in Excitable Cells; Oxford University Press: Oxford, UK, 1983. [Google Scholar]
- Roberts, D.E.; Scher, A.M. Effect of tissue anisotropy on extracellular potential fields in canine myocardium in situ. Circ. Res. 1982, 50, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Stinstra, J.G.; Hopenfeld, B.; MacLeod, R.S. On the passive cardiac conductivity. Ann. Biomed. Eng. 2005, 33, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Kohl, P.; Camelliti, P.; Burton, F.L.; Smith, G.L. Electrical coupling of fibroblasts and myocytes: Relevance for cardiac propagation. J. Electrocardiol. 2005, 38, 45–50. [Google Scholar] [CrossRef]
- Camelliti, P.; Green, C.R.; LeGrice, I.; Kohl, P. Fibroblast network in rabbit sinoatrial node: Structural and functional identification of homogeneous and heterogeneous cell coupling. Circ. Res. 2004, 94, 828–835. [Google Scholar] [CrossRef]
- Walker, N.L.; Burton, F.L.; Kettlewell, S.; Smith, G.L.; Cobbe, S.M. Mapping of epicardial activation in a rabbit model of chronic myocardial infarction. J. Cardiovasc. Electrophysiol. 2007, 18, 862–868. [Google Scholar] [CrossRef]
- Quinn, T.A.; Camelliti, P.; Rog-Zielinska, E.A.; Siedlecka, U.; Poggioli, T.; O’Toole, E.T.; Knöpfel, T.; Kohl, P. Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc. Natl. Acad. Sci. USA 2016, 113, 14852–14857. [Google Scholar] [CrossRef]
- Rohr, S. Arrhythmogenic implications of fibroblast-myocyte interactions. Circ. Arrhythmia Electrophysiol. 2012, 5, 442–452. [Google Scholar] [CrossRef]
- Spach, M.S.; Heidlage, J.F.; Barr, R.C.; Dolber, P.C. Cell size and communication: Role in structural and electrical development and remodeling of the heart. Heart Rhythm 2004, 1, 500–515. [Google Scholar] [CrossRef]
- Rutherford, S.L.; Trew, M.L.; Sands, G.B.; Legrice, I.J.; Smaill, B.H. High-resolution 3-dimensional reconstruction of the infarct border zone: Impact of structural remodeling on electrical activation. Circ. Res. 2012, 111, 301–311. [Google Scholar] [CrossRef]
- Cabo, C.; Pertsov, A.M.; Baxter, W.T.; Davidenko, J.M.; Gray, R.A.; Jalife, J. Wave-front curvature as a cause of slow conduction and block in isolated cardiac muscle. Circ. Res. 1994, 75, 1014–1028. [Google Scholar] [CrossRef] [PubMed]
- Trew, M.L.; Engelman, Z.J.; Caldwell, B.J.; Lever, N.A.; LeGrice, I.J.; Smaill, B.H. Cardiac intramural electrical mapping reveals focal delays but no conduction velocity slowing in the peri-infarct region. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H743–H753. [Google Scholar] [CrossRef]
- de Bakker, J.M.; Coronel, R.; Tasseron, S.; Wilde, A.A.; Opthof, T.; Janse, M.J.; van Capelle, F.J.; Becker, A.E.; Jambroes, G. Ventricular tachycardia in the infarcted, Langendorff-perfused human heart: Role of the arrangement of surviving cardiac fibers. J. Am. Coll. Cardiol. 1990, 15, 1594–1607. [Google Scholar] [CrossRef]
- Caldwell, B.J.; Trew, M.L.; Sands, G.B.; Hooks, D.A.; LeGrice, I.J.; Smaill, B.H. Three distinct directions of intramural activation reveal nonuniform side-to-side electrical coupling of ventricular myocytes. Circ. Arrhythmia Electrophysiol. 2009, 2, 433–440. [Google Scholar] [CrossRef]
- Gourdie, R.G. The cardiac gap junction has discrete functions in electrotonic and ephaptic coupling. Anat. Rec. 2019, 302, 93–100. [Google Scholar] [CrossRef]
- Page, E.; Shibata, Y. Permeable junctions between cardiac cells. Ann. Rev. Physiol. 1981, 43, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Dhein, S.; Seidel, T.; Salameh, A.; Jozwiak, J.; Hagen, A.; Kostelka, M.; Hindricks, G.; Mohr, F.-W. Remodeling of cardiac passive electrical properties and susceptibility to ventricular and atrial arrhythmias. Front. Physiol. 2014, 5, 424. [Google Scholar] [CrossRef]
- Vaidya, D.; Tamaddon, H.S.; Lo, C.W.; Taffet, S.M.; Delmar, M.; Morley, G.E.; Jalife, J. Null mutation of connexin43 causes slow propagation of ventricular activation in the late stages of mouse embryonic development. Circ. Res. 2001, 88, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- van Rijen, H.V.M.; Eckardt, D.; Degen, J.; Theis, M.; Ott, T.; Willecke, K.; Jongsma, H.J.; Opthof, T.; de Bakker, J.M.T. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 2004, 109, 1048–1055. [Google Scholar] [CrossRef]
- Kojodjojo, P.; Kanagaratnam, P.; Segal, O.R.; Hussain, W.; Peters, N.S. The effects of carbenoxolone on human myocardial conduction. a tool to investigate the role of gap junctional uncoupling in human arrhythmogenesis. J. Am. Coll. Cardiol. 2006, 48, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Eloff, B.C.; Gilat, E.; Wan, X.; Rosenbaum, D.S. Pharmacological modulation of cardiac gap junctions to enhance cardiac conduction: Evidence supporting a novel target for antiarrhythmic therapy. Circulation 2003, 108, 3157–3163. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, P.S.; Gray, R.; Kojodjojo, P.; Jabr, R.; Chowdhury, R.; Fry, C.H.; Peters, N.S. Relationship between gap-junctional conductance and conduction velocity in mammalian myocardium. Circ. Arrhythmia Electrophysiol. 2013, 6, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.M.; Rudy, Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997, 81, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Bukauskas, F.F.; Verselis, V.K. Gap junction channel gating. Biochim. Biophys. Acta 2004, 1662, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Sung, D.; Mills, R.W.; Schettler, J.; Narayan, S.M.; Omens, J.H.; McCulloch, A.D. Ventricular filling slows epicardial conduction and increases action potential duration in an optical mapping study of the isolated rabbit heart. J. Cardiovasc. Electrophysiol. 2003, 14, 739–749. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, B.L.; Pfeiffer, E.R.; Sundnes, J.; Wall, S.T.; McCulloch, A.D. Increased cell membrane capacitance is the dominant mechanism of stretch-dependent conduction slowing in the rabbit heart: A computational study. Cell Mol. Bioeng. 2015, 8, 237–246. [Google Scholar] [CrossRef][Green Version]
- Franz, M.R. Mechano-electrical feedback in ventricular myocardium. Cardiovasc. Res. 1996, 32, 15–24. [Google Scholar] [CrossRef]
- Mills, R.W.; Narayan, S.M.; McCulloch, A.D. Mechanisms of conduction slowing during myocardial stretch by ventricular volume loading in the rabbit. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1270–H1278. [Google Scholar] [CrossRef]
- Kohl, P.; Cooper, P.J.; Holloway, H. Effects of acute ventricular volume manipulation on in situ cardiomyocyte cell membrane configuration. Prog. Biophys. Mol. Biol. 2003, 82, 221–227. [Google Scholar] [CrossRef]
- Pfeiffer, E.R.; Wright, A.T.; Edwards, A.G.; Stowe, J.C.; McNall, K.; Tan, J.; Niesman, I.; Patel, H.H.; Roth, D.M.; Omens, J.H.; et al. Caveolae in ventricular myocytes are required for stretch-dependent conduction slowing. J. Mol. Cell. Cardiol. 2014, 76, 265–274. [Google Scholar] [CrossRef]
- Dhein, S.; Hammerath, S.B. Aspects of the intercellular communication in aged hearts: Effects of the gap junction uncoupler palmitoleic acid. Naunyn. Schmiedebergs. Arch. Pharmacol. 2001, 364, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Noorman, M.; van Rijen, H.V.M.; van Veen, T.A.B.; de Bakker, J.M.T.; Stein, M. Differences in distribution of fibrosis in the ventricles underlie dominant arrhythmia vulnerability of the right ventricle in senescent mice. Netherlands Hear. J. 2008, 16, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Koura, T.; Hara, M.; Takeuchi, S.; Ota, K.; Okada, Y.; Miyoshi, S.; Watanabe, A.; Shiraiwa, K.; Mitamura, H.; Kodama, I.; et al. Anisotropic conduction properties in canine atria analyzed by high-resolution optical mapping: Preferential direction of conduction block changes from longitudinal to transverse with increasing age. Circulation 2002, 105, 2092–2098. [Google Scholar] [CrossRef] [PubMed]
- Spach, M.S.; Dolber, P.C. Relating extracellular potentials and their derivatives to anisotropic propagation at a microscopic level in human cardiac muscle: Evidence for electrical uncoupling of side-to-side fiber connections with increasing age. Circ. Res. 1986, 58, 356–371. [Google Scholar] [CrossRef] [PubMed]
- van der Does, W.F.B.; Houck, C.A.; Heida, A.; van Schie, M.S.; van Schaagen, F.R.N.; Taverne, Y.J.H.J.; Bogers, A.J.J.C.; de Groot, N.M.S. Atrial electrophysiological characteristics of aging. J. Cardiovasc. Electrophysiol. 2021, 32, 903–912. [Google Scholar] [CrossRef]
- Signore, S.; Sorrentino, A.; Borghetti, G.; Cannata, A.; Meo, M.; Zhou, Y.; Kannappan, R.; Pasqualini, F.; O’Malley, H.; Sundman, M.; et al. Late Na + current and protracted electrical recovery are critical determinants of the aging myopathy. Nat. Commun. 2015, 6, 8803. [Google Scholar] [CrossRef]
- Macfarlane, P.W. The influence of age and sex on the electrocardiogram. Adv. Exp. Med. Biol. 2018, 1065, 93–106. [Google Scholar] [CrossRef]
- Rabkin, S.W. Impact of age and sex on QT prolongation in patients receiving psychotropics. Can. J. Psychiatry 2015, 60, 206–214. [Google Scholar] [CrossRef]
- Akar, F.G.; Spragg, D.D.; Tunin, R.S.; Kass, D.A.; Tomaselli, G.F. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ. Res. 2004, 95, 717–725. [Google Scholar] [CrossRef]
- Peters, N.S.; Green, C.R.; Poole-Wilson, P.A.; Severs, N.J. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation 1993, 88, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Pahor, M.; Bernabei, R.; Sgadari, A.; Gambassi, G., Jr.; Lo Giudice, P.; Pacifici, L.; Ramacci, M.T.; Lagrasta, C.; Olivetti, G.; Carbonin, P. Enalapril prevents cardiac fibrosis and arrhythmias in hypertensive rats. Hypertension 1991, 18, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Kozhevnikov, D.O.; Yamamoto, K.; Robotis, D.; Restivo, M.; El-Sherif, N. Electrophysiological mechanism of enhanced susceptibility of hypertrophied heart to acquired torsade de pointes arrhythmias: Tridimensional mapping of activation and recovery patterns. Circulation 2002, 105, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Kostin, S.; Klein, G.; Szalay, Z.; Hein, S.; Bauer, E.P.; Schaper, J. Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 2002, 54, 361–379. [Google Scholar] [CrossRef]
- Seidel, T.; Salameh, A.; Dhein, S. A simulation study of cellular hypertrophy and connexin lateralization in cardiac tissue. Biophys. J. 2010, 99, 2821–2830. [Google Scholar] [CrossRef]
- Kléber, A.G. Conduction of the impulse in the ischemic myocardium--implications for malignant ventricular arrhythmias. Experientia 1987, 43, 1056–1061. [Google Scholar] [CrossRef]
- Yao, J.A.; Hussain, W.; Patel, P.; Peters, N.S.; Boyden, P.A.; Wit, A.L. Remodeling of gap junctional channel function in epicardial border zone of healing canine infarcts. Circ. Res. 2003, 92, 437–443. [Google Scholar] [CrossRef]
- de Groot, J.R.; Coronel, R. Acute ischemia-induced gap junctional uncoupling and arrhythmogenesis. Cardiovasc. Res. 2004, 62, 323–334. [Google Scholar] [CrossRef]
- Gettes, L.S.; Reuter, H. Slow recovery from inactivation of inward currents in mammalian myocardial fibres. J. Physiol. 1974, 240, 703–724. [Google Scholar] [CrossRef]
- Kodama, I.; Wilde, A.; Janse, M.J.; Durrer, D.; Yamada, K. Combined effects of hypoxia, hyperkalemia and acidosis on membrane action potential and excitability of guinea-pig ventricular muscle. J. Mol. Cell. Cardiol. 1984, 16, 247–259. [Google Scholar] [CrossRef]
- Veenstra, R.D.; Joyner, R.W.; Wiedmann, R.T.; Young, M.L.; Tan, R.C. Effects of hypoxia, hyperkalemia, and metabolic acidosis on canine subendocardial action potential conduction. Circ. Res. 1987, 60, 93–101. [Google Scholar] [CrossRef]
- de Mello, W.C. Effect of intracellular injection of calcium and strontium on cell communication in the heart. J. Physiol. 1975, 250, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Kleber, G. The potential role of Ca2+ for electrical cell-to-cell uncoupling and conduction block in myocardial tissue. Basic Res. Cardiol. 1992, 87, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; McHowat, J.; Saffitz, J.E.; Yamada, K.A.; Corr, P.B. Inhibition of gap junctional conductance by long-chain acylcarnitines and their preferential accumulation in junctional sarcolemma during hypoxia. Circ. Res. 1993, 72, 879–889. [Google Scholar] [CrossRef]
- Connolly, A.; Trew, M.L.; Smaill, B.H.; Plank, G.; Bishop, M.J. Local gradients in electrotonic loading modulate the local effective refractory period: Implications for arrhythmogenesis in the infarct border zone. IEEE Trans. Biomed. Eng. 2015, 62, 2251–2259. [Google Scholar] [CrossRef]
- Thompson, S.A.; Copeland, C.R.; Reich, D.H.; Tung, L. Mechanical coupling between myofibroblasts and cardiomyocytes slows electric conduction in fibrotic cell monolayers. Circulation 2011, 123, 2083–2093. [Google Scholar] [CrossRef] [PubMed]
- Manjarrez-Marmolejo, J.; Franco-Pérez, J. Gap junction blockers: An overview of their effects on induced seizures in animal models. Curr. Neuropharmacol. 2016, 14, 759–771. [Google Scholar] [CrossRef]
- Davidson, J.S.; Baumgarten, I.M.; Harley, E.H. Reversible inhibition of intercellular junctional communication by glycyrrhetinic acid. Biochem. Biophys. Res. Commun. 1986, 134, 29–36. [Google Scholar] [CrossRef]
- Weingart, R.; Bukauskas, F.F. Long-chain n-alkanols and arachidonic acid interfere with the V(m)-sensitive gating mechanism of gap junction channels. Pflugers Arch. 1998, 435, 310–319. [Google Scholar] [CrossRef]
- Tse, G.; Yeo, J.M.; Tse, V.; Kwan, J.; Sun, B. Gap junction inhibition by heptanol increases ventricular arrhythmogenicity by reducing conduction velocity without affecting repolarization properties or myocardial refractoriness in Langendorff-perfused mouse hearts. Mol. Med. Rep. 2016, 14, 4069–4074. [Google Scholar] [CrossRef]
- Bastide, B.; Hervé, J.C.; Cronier, L.; Délèze, J. Rapid onset and calcium independence of the gap junction uncoupling induced by heptanol in cultured heart cells. Pflügers Arch. 1995, 429, 386–393. [Google Scholar] [CrossRef]
- Cotter, M.L.; Boitano, S.; Vagner, J.; Burt, J.M. Lipidated connexin mimetic peptides potently inhibit gap junction-mediated Ca2+-wave propagation. Am. J. Physiol. Cell Physiol. 2018, 315, C141–C154. [Google Scholar] [CrossRef]
- Kim, Y.; Griffin, J.M.; Harris, P.W.R.; Chan, S.H.C.; Nicholson, L.F.B.; Brimble, M.A.; O’Carroll, S.J.; Green, C.R. Characterizing the mode of action of extracellular Connexin43 channel blocking mimetic peptides in an in vitro ischemia injury model. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 68–78. [Google Scholar] [CrossRef]
- Burt, J.M.; Massey, K.D.; Minnich, B.N. Uncoupling of cardiac cells by fatty acids: Structure-activity relationships. Am. J. Physiol. 1991, 260, 439–448. [Google Scholar] [CrossRef]
- Dhein, S.; Krüsemann, K.; Schaefer, T. Effects of the gap junction uncoupler palmitoleic acid on the activation and repolarization wavefronts in isolated rabbit hearts. Br. J. Pharmacol. 1999, 128, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.P.; Walker, R.; Lux, R.L.; Ershler, P.R.; Menlove, R.; Williams, M.R.; Krall, R.; Moddrelle, D. Conduction velocity depression and drug-Induced ventricular tachyarrhythmias. effects of lidocaine in the intact canine heart. Circulation 1990, 81, 1024–1038. [Google Scholar] [CrossRef]
- Clarkson, C.W.; Matsubara, T.; Hondeghem, L.M. Slow inactivation of Vmax in guinea pig ventricular myocardium. Am. J. Physiol. 1984, 16, H645–654. [Google Scholar] [CrossRef]
- Matthews, G.D.K.; Guzadhur, L.; Sabir, I.N.; Grace, A.A.; Huang, C.L.-H. Action potential wavelength restitution predicts alternans and arrhythmia in murine Scn5a+/− hearts. J. Physiol. 2013, 591, 4167–4188. [Google Scholar] [CrossRef]
- Gallagher, J.D. Effects of halothane and quinidine on intracardiac conduction and QTc interval in pentobarbital-anesthetized dogs. Anesth. Analg. 1992, 75, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, D.; Delfaut, P.; Adamantidis, M.; Cardinal, R.; Klug, D.; Kacet, S.; Dupuis, B. Differential effects of quinidine, flecainide, and cibenzoline on anisotropic conduction in the isolated porcine heart. J. Cardiovasc. Electrophysiol. 1998, 9, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.J.; Rogers, J.; Perdichizzi, A.P.; Colvin, A.A.; Catterall, W.A. cAMP-dependent phosphorylation of two sites in the α subunit of the cardiac sodium channel. J. Biol. Chem. 1996, 271, 28837–28843. [Google Scholar] [CrossRef]
- Lu, T.; Lee, H.C.; Kabat, J.A.; Shibata, E.F. Modulation of rat cardiac sodium channel by the stimulatory G protein α subunit. J. Physiol. 1999, 518, 371–384. [Google Scholar] [CrossRef]
- Ozaki, S.; Nakaya, H.; Gotoh, Y.; Azuma, M.; Kemmostu, O.; Kanno, M. Effects of halothane and enflurane on conduction velocity and maximum rate of rise of action potential upstroke in guinea pig papillary muscles. Anesth. Analg. 1989, 68, 219–225. [Google Scholar] [CrossRef]
- Weigt, H.U.; Rehmert, G.C.; Bosnjak, Z.J.; Kwok, W.M. Conformational state-dependent effects of halothane on cardiac Na+ current. Anesthesiology 1997, 87, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Schmeling, W.T.; Warltier, D.C.; McDonald, D.J.; Madsen, K.E.; Atlee, J.L.; Kampine, J.P. Prolongation of the QT interval by enflurane, isoflurane, and halothane in humans. Anesth. Analg. 1991, 72, 137–144. [Google Scholar] [CrossRef]
- Saito, H.; Kambayashi, R.; Goto, A.; Hagiwara-Nagasawa, M.; Hoshiai, K.; Nunoi, Y.; Izumi-Nakaseko, H.; Akie, Y.; Takei, Y.; Matsumoto, A.; et al. In vivo analysis of concentration-dependent effects of halothane or isoflurane inhalation on the electrocardiographic and hemodynamic variables in dogs. J. Pharmacol. Sci. 2021, 145, 268–272. [Google Scholar] [CrossRef]
- Posnack, N.G.; Jaimes, R., 3rd; Asfour, H.; Swift, L.M.; Wengrowski, A.M.; Sarvazyan, N.; Kay, M.W. Bisphenol A exposure and cardiac electrical conduction in excised rat hearts. Environ. Health Perspect. 2014, 122, 384–390. [Google Scholar] [CrossRef]
- Cobellis, L.; Colacurci, N.; Trabucco, E.; Carpentiero, C.; Grumetto, L. Measurement of bisphenol A and bisphenol B levels in human blood sera from healthy and endometriotic women. Biomed. Chromatogr. 2009, 23, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Guida, G.; Sorbo, A.R.; Fenici, R.; Brisinda, D. Predictive value of unshielded magnetocardiographic mapping to differentiate atrial fibrillation patients from healthy subjects. Ann. Noninvasive Electrocardiol. 2018, 23, e12569. [Google Scholar] [CrossRef]
- Sorbo, A.R.; Lombardi, G.; Brocca, L.L.; Guida, G.; Fenici, R.; Brisinda, D. Unshielded magnetocardiography: Repeatability and reproducibility of automatically estimated ventricular repolarization parameters in 204 healthy subjects. Ann. Noninvasive Electrocardiol. 2018, 23, e12526. [Google Scholar] [CrossRef]
- Aita, S.; Ogata, K.; Yoshida, K.; Inaba, T.; Kosuge, H.; Machino, T.; Tsumagari, Y.; Hattori, A.; Ito, Y.; Komatsu, Y.; et al. Noninvasive mapping of premature ventricular contractions by merging magnetocardiography and computed tomography. JACC Clin. Electrophysiol. 2019, 5, 1144–1157. [Google Scholar] [CrossRef]
- Mäntynen, V.; Konttila, T.; Stenroos, M. Investigations of sensitivity and resolution of ECG and MCG in a realistically shaped thorax model. Phys. Med. Biol. 2014, 59, 7141–7158. [Google Scholar] [CrossRef]
- Christoph, J.; Chebbok, M.; Richter, C.; Schröder-Schetelig, J.; Bittihn, P.; Stein, S.; Uzelac, I.; Fenton, F.H.; Hasenfuß, G.; Gilmour, R.F., Jr.; et al. Electromechanical vortex filaments during cardiac fibrillation. Nature 2018, 555, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.K.; Yan, P.; Kang, J.; Abou, D.S.; Le, H.N.D.; Jha, A.K.; Thorek, D.L.J.; Kang, J.U.; Rahmim, A.; Wong, D.F.; et al. Listening to membrane potential: Photoacoustic voltage-sensitive dye recording. J. Biomed. Opt. 2017, 22, 45006. [Google Scholar] [CrossRef] [PubMed]
- Trew, M.; LeGrice, I.; Smaill, B.; Pullan, A. A finite volume method for modeling dis-continuous electrical activation in cardiac tissue. Ann. Biomed. Eng. 2005, 33, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Austin, T.A.; Trew, M.L.; Pullan, A.J. Solving the cardiac bidomain equations for dis-continuous conductivities. IEEE Trans. Biomed. Eng. 2006, 53, 1265–1272. [Google Scholar] [CrossRef]
- Faber, G.M.; Rudy, Y. Action potential and contractility changes in [Na+]i overloaded cardiac myocytes: A simulation study. Biophys. J. 2000, 78, 2392–2404. [Google Scholar] [CrossRef]
- Rush, S.; Larsen, H. A practical algorithm for solving dynamic membrane equations. IEEE Trans. Biomed. Eng. 1978, 25, 389–392. [Google Scholar] [CrossRef]
- Hooks, D.A.; Trew, M.L.; Caldwell, B.A.; Sands, G.B.; LeGrice, I.J.; Smaill, B.H. Lami-nar arrangement of ventricular myocytes influences electrical behavior of the heart. Circ. Res. 2007, 101, e103–e112. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, B.; Trew, M.L.; Zgierski-Johnston, C.M. Cardiac Conduction Velocity, Remodeling and Arrhythmogenesis. Cells 2021, 10, 2923. https://doi.org/10.3390/cells10112923
Han B, Trew ML, Zgierski-Johnston CM. Cardiac Conduction Velocity, Remodeling and Arrhythmogenesis. Cells. 2021; 10(11):2923. https://doi.org/10.3390/cells10112923
Chicago/Turabian StyleHan, Bo, Mark L. Trew, and Callum M. Zgierski-Johnston. 2021. "Cardiac Conduction Velocity, Remodeling and Arrhythmogenesis" Cells 10, no. 11: 2923. https://doi.org/10.3390/cells10112923
APA StyleHan, B., Trew, M. L., & Zgierski-Johnston, C. M. (2021). Cardiac Conduction Velocity, Remodeling and Arrhythmogenesis. Cells, 10(11), 2923. https://doi.org/10.3390/cells10112923