
Molecular Mechanisms of mtDNA-Mediated Inflammation

 , and
, and 
Abstract
:1. Introduction
2. mtDNA as a Danger Signal
2.1. The Evolution of the Mitochondrion and Its Inflammatogenic Role
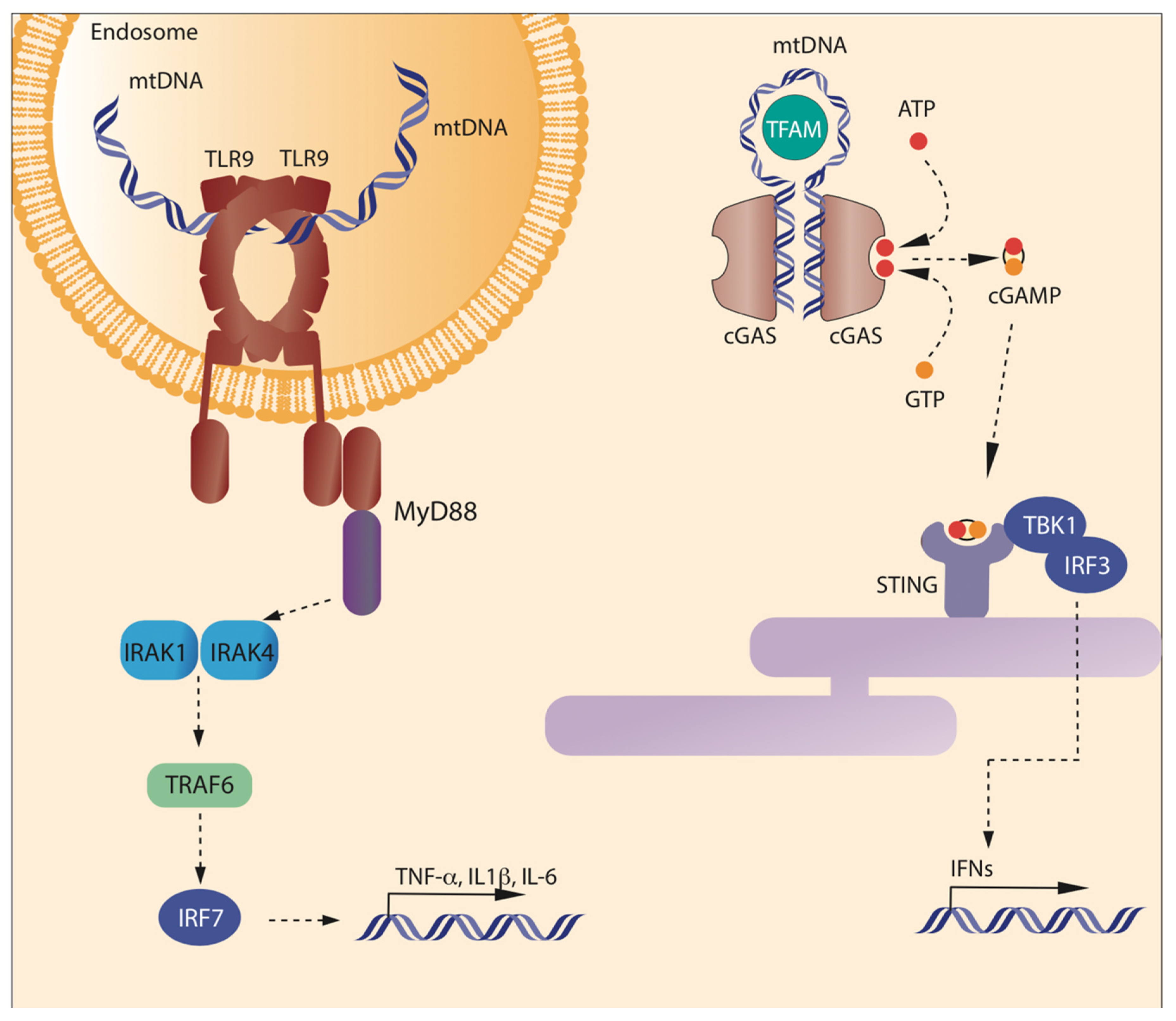
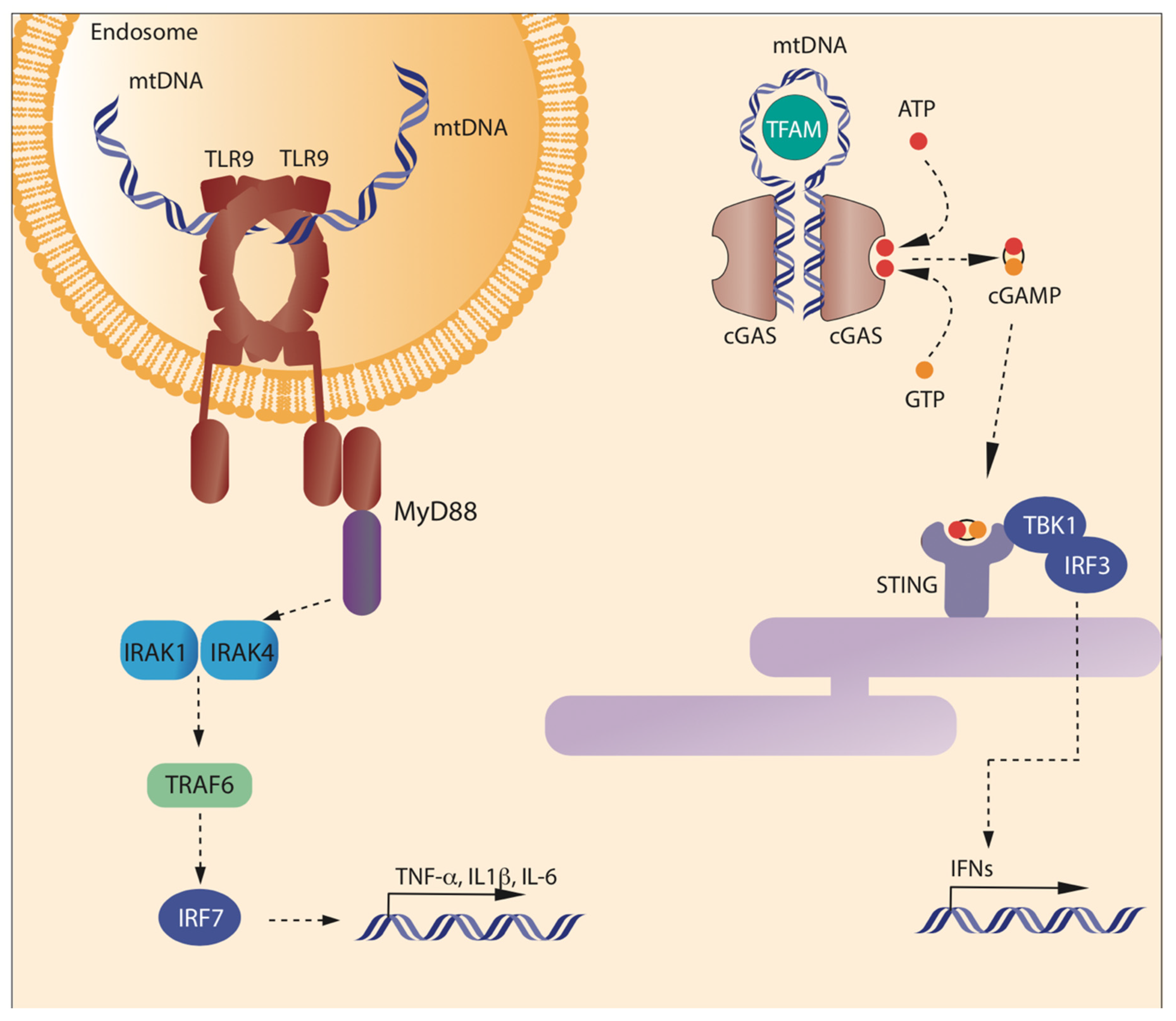
2.2. Receptors and Pathways Activated by mtDNA
2.2.1. mtDNA and Stimulation of TLR9
2.2.2. mtDNA and Activation of NLRP3 Inflammasome
2.2.3. mtDNA and cGAS-STING
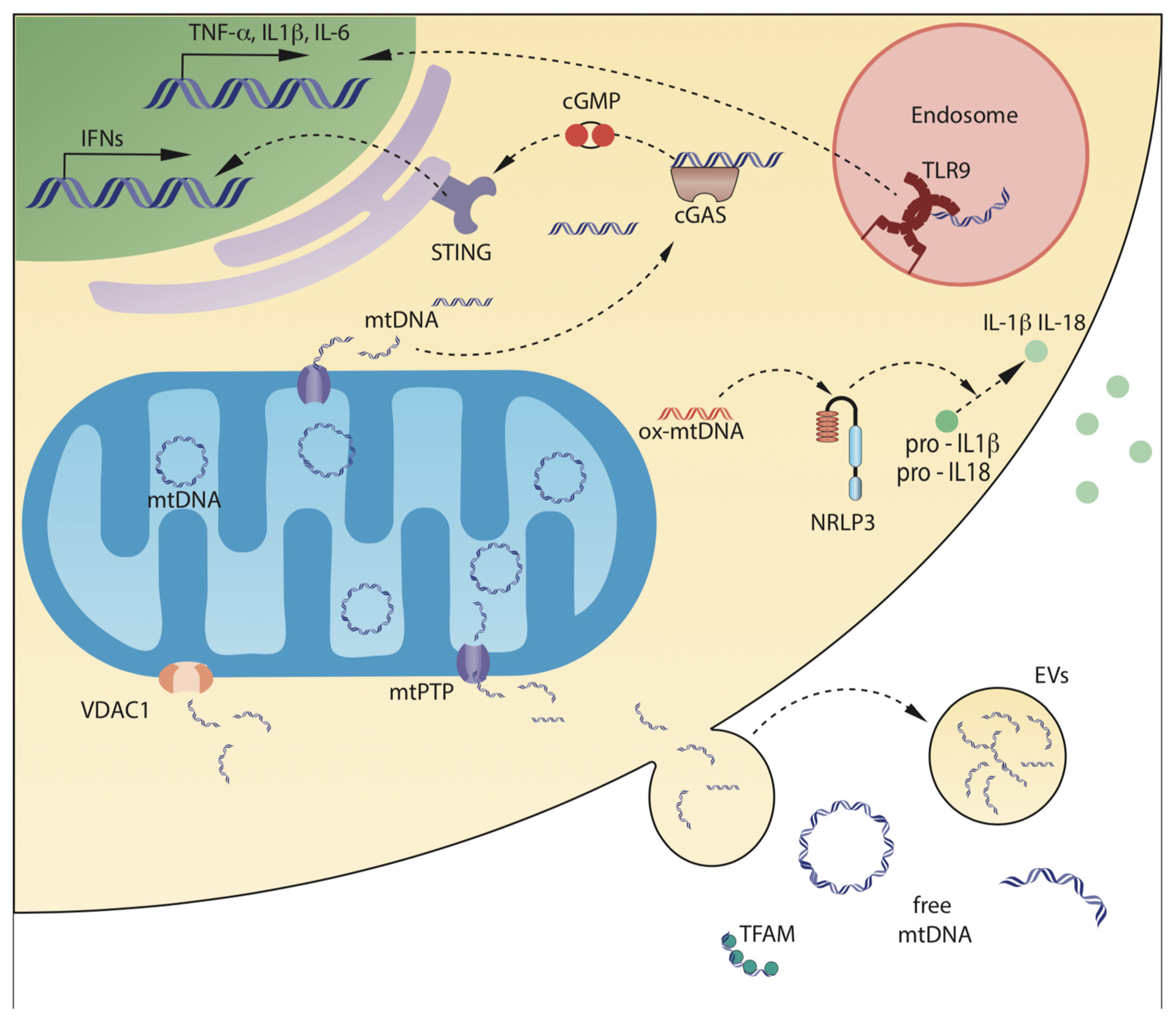
3. Mechanisms of Release of mtDNA in Biological Fluids
3.1. Release of mtDNA from Mitochondria to the Cytosol
3.1.1. mtDNA Release Mediated by Mitochondrial Outer Membrane Permeabilization (MOMP)
3.1.2. mtDNA Release Mediated by Mitochondrial Permeability Transition Pore (mPTP)
3.2. Mechanisms of Release of mtDNA outside the Cells
3.2.1. Active Release of mtDNA: Extracellular Traps (ETs)
3.2.2. Active Release of mtDNA Mediated by Extracellular Vesicles (EVs)
3.2.3. Passive Release of mtDNA
3.3. Anatomical Source of mtDNA
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic Instruction of Immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef]
- Dela Cruz, C.S.; Kang, M.J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Roger, A.J.; Munoz-Gomez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [Green Version]
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 255–274. [Google Scholar] [CrossRef]
- Bonen, L.; Cunningham, R.S.; Gray, M.W.; Doolittle, W.F. Wheat embryo mitochondrial 18S ribosomal RNA: Evidence for its prokaryotic nature. Nucleic Acids Res. 1977, 4, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, R.M.; Dayhoff, M.O. Origins of prokaryotes, eukaryotes, mitochondria, and chloroplasts. Science 1978, 199, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Oyaizu, Y.; Oyaizu, H.; Olsen, G.J.; Woese, C.R. Mitochondrial origins. Proc. Natl. Acad. Sci. USA 1985, 82, 4443–4447. [Google Scholar] [CrossRef] [Green Version]
- Spang, A.; Saw, J.H.; Jorgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaremba-Niedzwiedzka, K.; Caceres, E.F.; Saw, J.H.; Backstrom, D.; Juzokaite, L.; Vancaester, E.; Seitz, K.W.; Anantharaman, K.; Starnawski, P.; Kjeldsen, K.U.; et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 2017, 541, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wu, M. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci. Rep. 2015, 5, 7949. [Google Scholar] [CrossRef] [Green Version]
- Kausar, S.; Yang, L.; Abbas, M.N.; Hu, X.; Zhao, Y.; Zhu, Y.; Cui, H. Mitochondrial DNA: A Key Regulator of Anti-Microbial Innate Immunity. Genes 2020, 11, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Wei, X.; Wei, Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 2016, 7, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C.A., Jr. Innate immunity: Impact on the adaptive immune response. Curr. Opin. Immunol. 1997, 9, 4–9. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Godfrey, V.; Zaki, M.H. Cytosolic Nucleic Acid Sensors in Inflammatory and Autoimmune Disorders. Int. Rev. Cell Mol. Biol. 2019, 344, 215–253. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef]
- Bauer, S.; Kirschning, C.J.; Hacker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.K.; Mullen, G.E.; Leifer, C.A.; Mazzoni, A.; Davies, D.R.; Segal, D.M. Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol. 2003, 24, 528–533. [Google Scholar] [CrossRef]
- Bella, J.; Hindle, K.L.; McEwan, P.A.; Lovell, S.C. The leucine-rich repeat structure. Cell Mol. Life Sci. 2008, 65, 2307–2333. [Google Scholar] [CrossRef] [PubMed]
- Pohar, J.; Kuznik Krajnik, A.; Jerala, R.; Bencina, M. Minimal sequence requirements for oligodeoxyribonucleotides activating human TLR9. J. Immunol. 2015, 194, 3901–3908. [Google Scholar] [CrossRef] [Green Version]
- Ohto, U.; Shibata, T.; Tanji, H.; Ishida, H.; Krayukhina, E.; Uchiyama, S.; Miyake, K.; Shimizu, T. Structural basis of CpG and inhibitory DNA recognition by Toll-like receptor 9. Nature 2015, 520, 702–705. [Google Scholar] [CrossRef]
- Pohar, J.; Lainscek, D.; Fukui, R.; Yamamoto, C.; Miyake, K.; Jerala, R.; Bencina, M. Species-Specific Minimal Sequence Motif for Oligodeoxyribonucleotides Activating Mouse TLR9. J. Immunol. 2015, 195, 4396–4405. [Google Scholar] [CrossRef] [Green Version]
- Pohar, J.; Lainscek, D.; Ivicak-Kocjan, K.; Cajnko, M.M.; Jerala, R.; Bencina, M. Short single-stranded DNA degradation products augment the activation of Toll-like receptor 9. Nat. Commun. 2017, 8, 15363. [Google Scholar] [CrossRef]
- Ohto, U.; Ishida, H.; Shibata, T.; Sato, R.; Miyake, K.; Shimizu, T. Toll-like Receptor 9 Contains Two DNA Binding Sites that Function Cooperatively to Promote Receptor Dimerization and Activation. Immunity 2018, 48, 649–658.e644. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Itagaki, K.; Hauser, C.J. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 2010, 34, 55–59. [Google Scholar] [CrossRef]
- Julian, M.W.; Shao, G.; Bao, S.; Knoell, D.L.; Papenfuss, T.L.; VanGundy, Z.C.; Crouser, E.D. Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J. Immunol. 2012, 189, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Chen, X.; Sun, T.; Li, Q.; Zhang, R.; Zhang, J.; Zhong, J. Significance of Serum mtDNA Concentration in Lung Injury Induced by Hip Fracture. Shock 2015, 44, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Jia, Z.; Gong, W. Circulating Mitochondrial DNA Stimulates Innate Immune Signaling Pathways to Mediate Acute Kidney Injury. Front. Immunol. 2021, 12, 680648. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin. Exp. Rheumatol. 2016, 34, 12–16. [Google Scholar] [PubMed]
- Ward, G.A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; Meyer, B.S.; McLemore, A.F.; Vincelette, N.D.; Lam, N.B.; Aldrich, A.L.; Al Ali, N.H.; Padron, E.; et al. Oxidized mitochondrial DNA released after inflammasome activation is a disease biomarker for myelodysplastic syndromes. Blood Adv. 2021, 5, 2216–2228. [Google Scholar] [CrossRef] [PubMed]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Kranzusch, P.J.; Lee, A.S.; Berger, J.M.; Doudna, J.A. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013, 3, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lassig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e618. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-kappaB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Sprenger, H.G.; MacVicar, T.; Bahat, A.; Fiedler, K.U.; Hermans, S.; Ehrentraut, D.; Ried, K.; Milenkovic, D.; Bonekamp, N.; Larsson, N.G.; et al. Cellular pyrimidine imbalance triggers mitochondrial DNA-dependent innate immunity. Nat. Metab. 2021, 3, 636–650. [Google Scholar] [CrossRef]
- Kerur, N.; Fukuda, S.; Banerjee, D.; Kim, Y.; Fu, D.; Apicella, I.; Varshney, A.; Yasuma, R.; Fowler, B.J.; Baghdasaryan, E.; et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 2018, 24, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhou, M.; Bao, X.; Pan, D.; Jiao, M.; Liu, X.; Li, F.; Li, C.Y. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Lepelley, A.; Della Mina, E.; Van Nieuwenhove, E.; Waumans, L.; Fraitag, S.; Rice, G.I.; Dhir, A.; Fremond, M.L.; Rodero, M.P.; Seabra, L.; et al. Enhanced cGAS-STING-dependent interferon signaling associated with mutations in ATAD3A. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Thorsness, P.E.; Fox, T.D. Nuclear mutations in Saccharomyces cerevisiae that affect the escape of DNA from mitochondria to the nucleus. Genetics 1993, 134, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Ponce, L. Preprint Highlight: ER-lysosome lipid transfer protein VPS13C/PARK23 prevents aberrant mtDNA- dependent STING signaling. Mol. Biol. Cell 2021, 32, 1110. [Google Scholar] [CrossRef]
- Gkirtzimanaki, K.; Kabrani, E.; Nikoleri, D.; Polyzos, A.; Blanas, A.; Sidiropoulos, P.; Makrigiannakis, A.; Bertsias, G.; Boumpas, D.T.; Verginis, P. IFNalpha Impairs Autophagic Degradation of mtDNA Promoting Autoreactivity of SLE Monocytes in a STING-Dependent Fashion. Cell Rep. 2018, 25, 921–933.e925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fermaintt, C.S.; Takahashi-Ruiz, L.; Liang, H.; Mooberry, S.L.; Risinger, A.L. Eribulin activates the cGAS-STING pathway via the cytoplasmic accumulation of mtDNA. Mol. Pharmacol. 2021, 100, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Farge, G.; Falkenberg, M. Organization of DNA in Mammalian Mitochondria. Int. J. Mol. Sci. 2019, 20, 2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef]
- Kim, E.K.; Cho, Y.M. Response to Comment on Kim et al. The Effect of a Smartphone-Based, Patient-Centered Diabetes Care System in Patients With Type 2 Diabetes: A Randomized, Controlled Trial for 24 Weeks. Diabetes Care 2019;42:3-9. Diabetes Care 2019, 42, e126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Israelson, A.; Brdiczka, D.; Sheu, S.S. The voltage-dependent anion channel (VDAC): Function in intracellular signalling, cell life and cell death. Curr. Pharm. Des. 2006, 12, 2249–2270. [Google Scholar] [CrossRef] [Green Version]
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef] [Green Version]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.R.; Cao, L.B.; Yang, Y.L.; Hua, D.; Hu, M.M.; Shu, H.B. VRK2 is involved in the innate antiviral response by promoting mitostress-induced mtDNA release. Cell. Mol. Immunol. 2021, 18, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta 2009, 1787, 1402–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef]
- Rasheed, A.; Wen, W.; Gao, F.; Zhai, S.; Jin, H.; Liu, J.; Guo, Q.; Zhang, Y.; Dreisigacker, S.; Xia, X.; et al. Development and validation of KASP assays for genes underpinning key economic traits in bread wheat. Theor. Appl. Genet. 2016, 129, 1843–1860. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.; Garcia, J.J.; Correa, F.; Chavez, E. The permeability transition pore as a pathway for the release of mitochondrial DNA. Life Sci. 2005, 76, 2873–2880. [Google Scholar] [CrossRef] [PubMed]
- Nasi, M.; Bianchini, E.; De Biasi, S.; Gibellini, L.; Neroni, A.; Mattioli, M.; Pinti, M.; Iannone, A.; Mattioli, A.V.; Simone, A.M.; et al. Increased plasma levels of mitochondrial DNA and pro-inflammatory cytokines in patients with progressive multiple sclerosis. J. Neuroimmunol. 2020, 338, 577107. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Hajizadeh, S.; DeGroot, J.; TeKoppele, J.M.; Tarkowski, A.; Collins, L.V. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res. Ther. 2003, 5, R234–R240. [Google Scholar] [CrossRef] [Green Version]
- Al Amir Dache, Z.; Otandault, A.; Tanos, R.; Pastor, B.; Meddeb, R.; Sanchez, C.; Arena, G.; Lasorsa, L.; Bennett, A.; Grange, T.; et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 2020, 34, 3616–3630. [Google Scholar] [CrossRef] [Green Version]
- Stephens, O.R.; Grant, D.; Frimel, M.; Wanner, N.; Yin, M.; Willard, B.; Erzurum, S.C.; Asosingh, K. Characterization and origins of cell-free mitochondria in healthy murine and human blood. Mitochondrion 2020, 54, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Hu, W.; Yu, H.; Wang, H.; Zhao, Y.; Korngold, R.; Zhao, Y. Existence of Circulating Mitochondria in Human and Animal Peripheral Blood. Int. J. Mol. Sci. 2020, 21, 2122. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- McIlroy, D.J.; Jarnicki, A.G.; Au, G.G.; Lott, N.; Smith, D.W.; Hansbro, P.M.; Balogh, Z.J. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J. Crit. Care 2014, 29, 1133.e1–1133.e5. [Google Scholar] [CrossRef] [Green Version]
- Itagaki, K.; Kaczmarek, E.; Lee, Y.T.; Tang, I.T.; Isal, B.; Adibnia, Y.; Sandler, N.; Grimm, M.J.; Segal, B.H.; Otterbein, L.E.; et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PLoS ONE 2015, 10, e0120549. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [Green Version]
- Van der Windt, D.J.; Sud, V.; Zhang, H.; Varley, P.R.; Goswami, J.; Yazdani, H.O.; Tohme, S.; Loughran, P.; O’Doherty, R.M.; Minervini, M.I.; et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018, 68, 1347–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, V.; Zychlinsky, A. Beneficial suicide: Why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumburu, L.; Ghosh-Choudhary, S.; Seifuddin, F.T.; Barbu, E.A.; Yang, S.; Ahmad, M.M.; Wilkins, L.H.W.; Tunc, I.; Sivakumar, I.; Nichols, J.S.; et al. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood 2021, 137, 3116–3126. [Google Scholar] [CrossRef]
- Conceicao-Silva, F.; Reis, C.S.M.; De Luca, P.M.; Leite-Silva, J.; Santiago, M.A.; Morrot, A.; Morgado, F.N. The Immune System Throws Its Traps: Cells and Their Extracellular Traps in Disease and Protection. Cells 2021, 10, 1891. [Google Scholar] [CrossRef]
- Caielli, S.; Athale, S.; Domic, B.; Murat, E.; Chandra, M.; Banchereau, R.; Baisch, J.; Phelps, K.; Clayton, S.; Gong, M.; et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016, 213, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Gold, J.A.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef]
- Costanza, M.; Poliani, P.L.; Portararo, P.; Cappetti, B.; Musio, S.; Pagani, F.; Steinman, L.; Colombo, M.P.; Pedotti, R.; Sangaletti, S. DNA threads released by activated CD4(+) T lymphocytes provide autocrine costimulation. Proc. Natl. Acad. Sci. USA 2019, 116, 8985–8994. [Google Scholar] [CrossRef] [Green Version]
- Ueki, S.; Melo, R.C.; Ghiran, I.; Spencer, L.A.; Dvorak, A.M.; Weller, P.F. Eosinophil extracellular DNA trap cell death mediates lytic release of free secretion-competent eosinophil granules in humans. Blood 2013, 121, 2074–2083. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Martinez, E.; Hernandez-Gonzalez, L.; Ramos-Martinez, I.; Perez-Campos Mayoral, L.; Lopez-Cortes, G.I.; Perez-Campos, E.; Mayoral Andrade, G.; Hernandez-Huerta, M.T.; Jose, M.V. Multiple Origins of Extracellular DNA Traps. Front. Immunol. 2021, 12, 621311. [Google Scholar] [CrossRef] [PubMed]
- Peachman, K.K.; Lyles, D.S.; Bass, D.A. Mitochondria in eosinophils: Functional role in apoptosis but not respiration. Proc. Natl. Acad. Sci. USA 2001, 98, 1717–1722. [Google Scholar] [CrossRef] [Green Version]
- Cossarizza, A.; Pinti, M.; Moretti, L.; Bricalli, D.; Bianchi, R.; Troiano, L.; Fernandez, M.G.; Balli, F.; Brambilla, P.; Mussini, C.; et al. Mitochondrial functionality and mitochondrial DNA content in lymphocytes of vertically infected human immunodeficiency virus-positive children with highly active antiretroviral therapy-related lipodystrophy. J. Infect. Dis. 2002, 185, 299–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, C.C.; Wardini, A.B.; Vieira, M.; Passos, L.S.A.; Martinelli, P.M.; Neves, E.G.A.; Antonelli, L.; Barbosa, D.F.; Velikkakam, T.; Gutseit, E.; et al. Human CD8+ T Cells Release Extracellular Traps Co-Localized With Cytotoxic Vesicles That Are Associated With Lesion Progression and Severity in Human Leishmaniasis. Front. Immunol. 2020, 11, 594581. [Google Scholar] [CrossRef]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Cantaluppi, V.; Biancone, L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010, 78, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Tkach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Muhsin-Sharafaldine, M.R.; McLellan, A.D. Tumor-Derived Apoptotic Vesicles: With Death They Do Part. Front. Immunol. 2018, 9, 957. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [Green Version]
- Marostica, G.; Gelibter, S.; Gironi, M.; Nigro, A.; Furlan, R. Extracellular Vesicles in Neuroinflammation. Front. Cell Dev. Biol. 2020, 8, 623039. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Gronborg, M.; Mobius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Rider, M.A.; Bundy, J.L.; Liu, X.; Singh, R.K.; Meckes, D.G., Jr. Proteomic profiling of NCI-60 extracellular vesicles uncovers common protein cargo and cancer type-specific biomarkers. Oncotarget 2016, 7, 86999–87015. [Google Scholar] [CrossRef]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017, 6, e22187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. 2010, 117, 1–4. [Google Scholar] [CrossRef]
- Guescini, M.; Guidolin, D.; Vallorani, L.; Casadei, L.; Gioacchini, A.M.; Tibollo, P.; Battistelli, M.; Falcieri, E.; Battistin, L.; Agnati, L.F.; et al. C2C12 myoblasts release micro-vesicles containing mtDNA and proteins involved in signal transduction. Exp. Cell Res. 2010, 316, 1977–1984. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Johansson, C.; Xiao, F.; Stone, P.R.; James, J.L.; Chen, Q.; Cree, L.M.; Chamley, L.W. Antiphospholipid antibodies increase the levels of mitochondrial DNA in placental extracellular vesicles: Alarmin-g for preeclampsia. Sci. Rep. 2017, 7, 16556. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Xu, M.J.; Koritzinsky, E.H.; Zhou, Z.; Wang, W.; Cao, H.; Yuen, P.S.; Ross, R.A.; Star, R.A.; Liangpunsakul, S.; et al. Mitochondrial DNA-enriched microparticles promote acute-on-chronic alcoholic neutrophilia and hepatotoxicity. JCI Insight 2017, 2, e92634. [Google Scholar] [CrossRef]
- Ye, W.; Tang, X.; Yang, Z.; Liu, C.; Zhang, X.; Jin, J.; Lyu, J. Plasma-derived exosomes contribute to inflammation via the TLR9-NF-kappaB pathway in chronic heart failure patients. Mol. Immunol. 2017, 87, 114–121. [Google Scholar] [CrossRef]
- Zhu, M.; Barbas, A.S.; Lin, L.; Scheuermann, U.; Bishawi, M.; Brennan, T.V. Mitochondria Released by Apoptotic Cell Death Initiate Innate Immune Responses. Immunohorizons 2018, 2, 384–397. [Google Scholar] [CrossRef]
- Emre, Y.; Hurtaud, C.; Nubel, T.; Criscuolo, F.; Ricquier, D.; Cassard-Doulcier, A.M. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem. J. 2007, 402, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Puhm, F.; Afonyushkin, T.; Resch, U.; Obermayer, G.; Rohde, M.; Penz, T.; Schuster, M.; Wagner, G.; Rendeiro, A.F.; Melki, I.; et al. Mitochondria Are a Subset of Extracellular Vesicles Released by Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial Cells. Circ. Res. 2019, 125, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Bernimoulin, M.; Waters, E.K.; Foy, M.; Steele, B.M.; Sullivan, M.; Falet, H.; Walsh, M.T.; Barteneva, N.; Geng, J.G.; Hartwig, J.H.; et al. Differential stimulation of monocytic cells results in distinct populations of microparticles. J. Thromb. Haemost. 2009, 7, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Pare, A.; Rousseau, M.; Naika, G.S.; Levesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef]
- Lei, L.; Spradling, A.C. Single-Cell Lineage Analysis of Oogenesis in Mice. Methods Mol. Biol. 2017, 1463, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todkar, K.; Chikhi, L.; Desjardins, V.; El-Mortada, F.; Pepin, G.; Germain, M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 2021, 12, 1971. [Google Scholar] [CrossRef]
- Tixeira, R.; Caruso, S.; Paone, S.; Baxter, A.A.; Atkin-Smith, G.K.; Hulett, M.D.; Poon, I.K. Defining the morphologic features and products of cell disassembly during apoptosis. Apoptosis 2017, 22, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Koppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal. 2009, 2, ra81. [Google Scholar] [CrossRef] [PubMed]
- Berda-Haddad, Y.; Robert, S.; Salers, P.; Zekraoui, L.; Farnarier, C.; Dinarello, C.A.; Dignat-George, F.; Kaplanski, G. Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1alpha. Proc. Natl. Acad. Sci. USA 2011, 108, 20684–20689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miliotis, S.; Nicolalde, B.; Ortega, M.; Yepez, J.; Caicedo, A. Forms of extracellular mitochondria and their impact in health. Mitochondrion 2019, 48, 16–30. [Google Scholar] [CrossRef]
- Jiang, L.; Paone, S.; Caruso, S.; Atkin-Smith, G.K.; Phan, T.K.; Hulett, M.D.; Poon, I.K.H. Determining the contents and cell origins of apoptotic bodies by flow cytometry. Sci. Rep. 2017, 7, 14444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Yao, Y.; Wu, G.; Lv, T.; Luo, L.; Song, Y. The plasma mitochondrial DNA is an independent predictor for post-traumatic systemic inflammatory response syndrome. PLoS ONE 2013, 8, e72834. [Google Scholar] [CrossRef]
- Simmons, J.D.; Lee, Y.L.; Mulekar, S.; Kuck, J.L.; Brevard, S.B.; Gonzalez, R.P.; Gillespie, M.N.; Richards, W.O. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann. Surg. 2013, 258, 591–596, discussion 596–598. [Google Scholar] [CrossRef] [Green Version]
- Aswani, A.; Manson, J.; Itagaki, K.; Chiazza, F.; Collino, M.; Wupeng, W.L.; Chan, T.K.; Wong, W.S.F.; Hauser, C.J.; Thiemermann, C.; et al. Scavenging Circulating Mitochondrial DNA as a Potential Therapeutic Option for Multiple Organ Dysfunction in Trauma Hemorrhage. Front. Immunol. 2018, 9, 891. [Google Scholar] [CrossRef]
- Yamanouchi, S.; Kudo, D.; Yamada, M.; Miyagawa, N.; Furukawa, H.; Kushimoto, S. Plasma mitochondrial DNA levels in patients with trauma and severe sepsis: Time course and the association with clinical status. J. Crit. Care 2013, 28, 1027–1031. [Google Scholar] [CrossRef]
- Qin, C.Y.; Zhang, H.W.; Gu, J.; Xu, F.; Liang, H.M.; Fan, K.J.; Shen, J.Y.; Xiao, Z.H.; Zhang, E.Y.; Hu, J. Mitochondrial DNAinduced inflammatory damage contributes to myocardial ischemia reperfusion injury in rats: Cardioprotective role of epigallocatechin. Mol. Med. Rep. 2017, 16, 7569–7576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.M.; Cui, L.; White, J.; Kuck, J.; Ruchko, M.V.; Wilson, G.L.; Alexeyev, M.; Gillespie, M.N.; Downey, J.M.; Cohen, M.V. Mitochondrially targeted Endonuclease III has a powerful anti-infarct effect in an in vivo rat model of myocardial ischemia/reperfusion. Basic Res. Cardiol. 2015, 110, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Hann, H.W.; Wan, S.; Hann, R.S.; Wang, C.; Lai, Y.; Ye, X.; Evans, A.; Myers, R.E.; Ye, Z.; et al. Cell-free circulating mitochondrial DNA content and risk of hepatocellular carcinoma in patients with chronic HBV infection. Sci. Rep. 2016, 6, 23992. [Google Scholar] [CrossRef] [PubMed]
- Singel, K.L.; Grzankowski, K.S.; Khan, A.; Grimm, M.J.; D’Auria, A.C.; Morrell, K.; Eng, K.H.; Hylander, B.; Mayor, P.C.; Emmons, T.R.; et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br. J. Cancer 2019, 120, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef]
- McIlroy, D.J.; Bigland, M.; White, A.E.; Hardy, B.M.; Lott, N.; Smith, D.W.; Balogh, Z.J. Cell necrosis-independent sustained mitochondrial and nuclear DNA release following trauma surgery. J. Trauma Acute Care Surg. 2015, 78, 282–288. [Google Scholar] [CrossRef]
- Hauser, C.J.; Sursal, T.; Rodriguez, E.K.; Appleton, P.T.; Zhang, Q.; Itagaki, K. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and P44/42 MAP kinase. J. Orthop. Trauma 2010, 24, 534–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurairajah, K.; Briggs, G.D.; Balogh, Z.J. The source of cell-free mitochondrial DNA in trauma and potential therapeutic strategies. Eur. J. Trauma Emerg. Surg. 2018, 44, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Chan, S.J.; Mandeville, E.T.; Park, J.H.; Bruzzese, M.; Montaner, J.; Arai, K.; Rosell, A.; Lo, E.H. Protective Effects of Endothelial Progenitor Cell-Derived Extracellular Mitochondria in Brain Endothelium. Stem Cells 2018, 36, 1404–1410. [Google Scholar] [CrossRef] [Green Version]
- Munakata, Y.; Shirasuna, K.; Kuwayama, T.; Iwata, H. Cell-free DNA in medium is associated with the maturation ability of in vitro cultured oocytes. J. Reprod. Dev. 2019, 65, 171–175. [Google Scholar] [CrossRef] [Green Version]
- Stigliani, S.; Orlando, G.; Massarotti, C.; Casciano, I.; Bovis, F.; Anserini, P.; Ubaldi, F.M.; Remorgida, V.; Rienzi, L.; Scaruffi, P. Non-invasive mitochondrial DNA quantification on Day 3 predicts blastocyst development: A prospective, blinded, multi-centric study. Mol. Hum. Reprod. 2019, 25, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Liou, C.W.; Chen, S.D.; Hsu, T.Y.; Chuang, J.H.; Wang, P.W.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; Lin, T.K.; et al. Mitochondrial transfer from Wharton’s jelly-derived mesenchymal stem cells to mitochondria-defective cells recaptures impaired mitochondrial function. Mitochondrion 2015, 22, 31–44. [Google Scholar] [CrossRef]
- Kansaku, K.; Munakata, Y.; Shirasuna, K.; Kuwayama, T.; Iwata, H. Mitochondrial cell-free DNA secreted from porcine granulosa cells. Zygote 2019, 27, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Varhaug, K.N.; Barro, C.; Bjornevik, K.; Myhr, K.M.; Torkildsen, O.; Wergeland, S.; Bindoff, L.A.; Kuhle, J.; Vedeler, C. Neurofilament light chain predicts disease activity in relapsing-remitting MS. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e422. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Pathology | Observation/Mechanism | Type of Study/Sample | Reference | |
|---|---|---|---|---|
| Trauma | Systemic inflammatory response syndrome (SIRS) | mtDNA plasma levels were significantly higher in trauma patients, and a correlation between mtDNA levels and clinical severity was observed | Blood from patients with SIRS | [148,149] |
| Multiple organic dysfunction syndrome (MODS) | mtDNA concentrations were predictive for the development of MODS (in patients) or organ dysfunction (in an animal model) | Plasma from severely injured patients | [150] [149] | |
| Traumatic injury and shock | Release of mtDNA triggers the development of severe tissue injury | Plasma from an animal model of trauma | [150] | |
| Trauma | TLR9-mediated NETs formation triggered by mtDNA | Plasma from trauma patients | [94] | |
| Trauma and sepsis | An increase in mtDNA plasma levels was observed, although differences in disease course and prognostic were observed, suggesting that the mechanism of release of mtDNA is different between the two groups | Blood from patients presenting trauma or severe sepsis | [151] | |
| Trauma and haemorrhagic shock | mtDNA release triggers the activation of neutrophils | Rat model of trauma and haemorrhagic shock | [34] | |
| Autoimmune origin | Multiple sclerosis (MS) | mtDNA level, together with other pro-inflammatory cytokines, was observed to be higher in patients with progressive forms of MS, which probably contributes to the systemic inflammation present in the pathology | Plasma from MS patients | [84] |
| Increased levels of cf-mtDNA in patients with relapsing-remitting form of MS An inverse correlation was observed between cf-mtDNA and disease duration | CSF from MS patients | [165] | ||
| Lupus-like disease | VDAC-mediated mtDNA release | In vitro and in vivo animal model of SLE | [74] | |
| Neutrophil-mediated ox-mtDNA release | Blood from SLE patients | [101] | ||
| NETosis inductors triggered the release of ox-mtDNA, leading to STING activation | Blood from patients with CGD or SLE In vitro models Iv vivo animal model of CGD or SLE | [91] | ||
| Cancer | Advanced epithelial ovarian cancer (EOC) | The levels of mtDNA ascites were correlated with worse outcome in EOC patients | Blood from EOC patients | [155] |
| Hormonal therapy-resistant breast cancer | mtDNA release via exosomes | Blood from patients with breast cancer In vitro and in vivo animal cancer models | [128] | |
| Myelodysplastic syndromes (MDS) | Ox-mtDNA release after inflammasome activation | Blood from patients with MDS In vitro cancer model | [49] | |
| Other | Sickle cell disease | Increased circulating levels of cf-mtDNA Activation of NETs formation and cGAS-STING pathway triggered by elevated levels of cf-mtDNA The results showed a mitochondrial retention by circulating SCD red blood cells, therefore the authors suggested that this could be the source of the elevated levels of cf-mtDNA | Blood from SCD patients | [99] |
| Non-alcoholic steatohepatitis | mtDNA activation of TLR9 | Blood from patients with non-alcohol steatohepatitis In vivo models of NASH | [38] | |
| Macular degeneration | cGAS activation by mtDNA, released into cytosol by Alu-RNA accumulation | In vivo animal model of RPE degeneration | [61] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells 2021, 10, 2898. https://doi.org/10.3390/cells10112898
De Gaetano A, Solodka K, Zanini G, Selleri V, Mattioli AV, Nasi M, Pinti M. Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells. 2021; 10(11):2898. https://doi.org/10.3390/cells10112898
Chicago/Turabian StyleDe Gaetano, Anna, Kateryna Solodka, Giada Zanini, Valentina Selleri, Anna Vittoria Mattioli, Milena Nasi, and Marcello Pinti. 2021. "Molecular Mechanisms of mtDNA-Mediated Inflammation" Cells 10, no. 11: 2898. https://doi.org/10.3390/cells10112898
APA StyleDe Gaetano, A., Solodka, K., Zanini, G., Selleri, V., Mattioli, A. V., Nasi, M., & Pinti, M. (2021). Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells, 10(11), 2898. https://doi.org/10.3390/cells10112898