
Instructions for Flow Cytometric Detection of ASC Specks as a Readout of Inflammasome Activation in Human Blood

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Blood Sample Processing
2.3. Freezing and Thawing Cells
2.4. Cell Culture
2.5. Flow Cytometry
2.6. Immunocytochemistry and Fluorescence Microscopy
2.7. Cytokine Determination
2.8. Statistical Analysis
3. Results
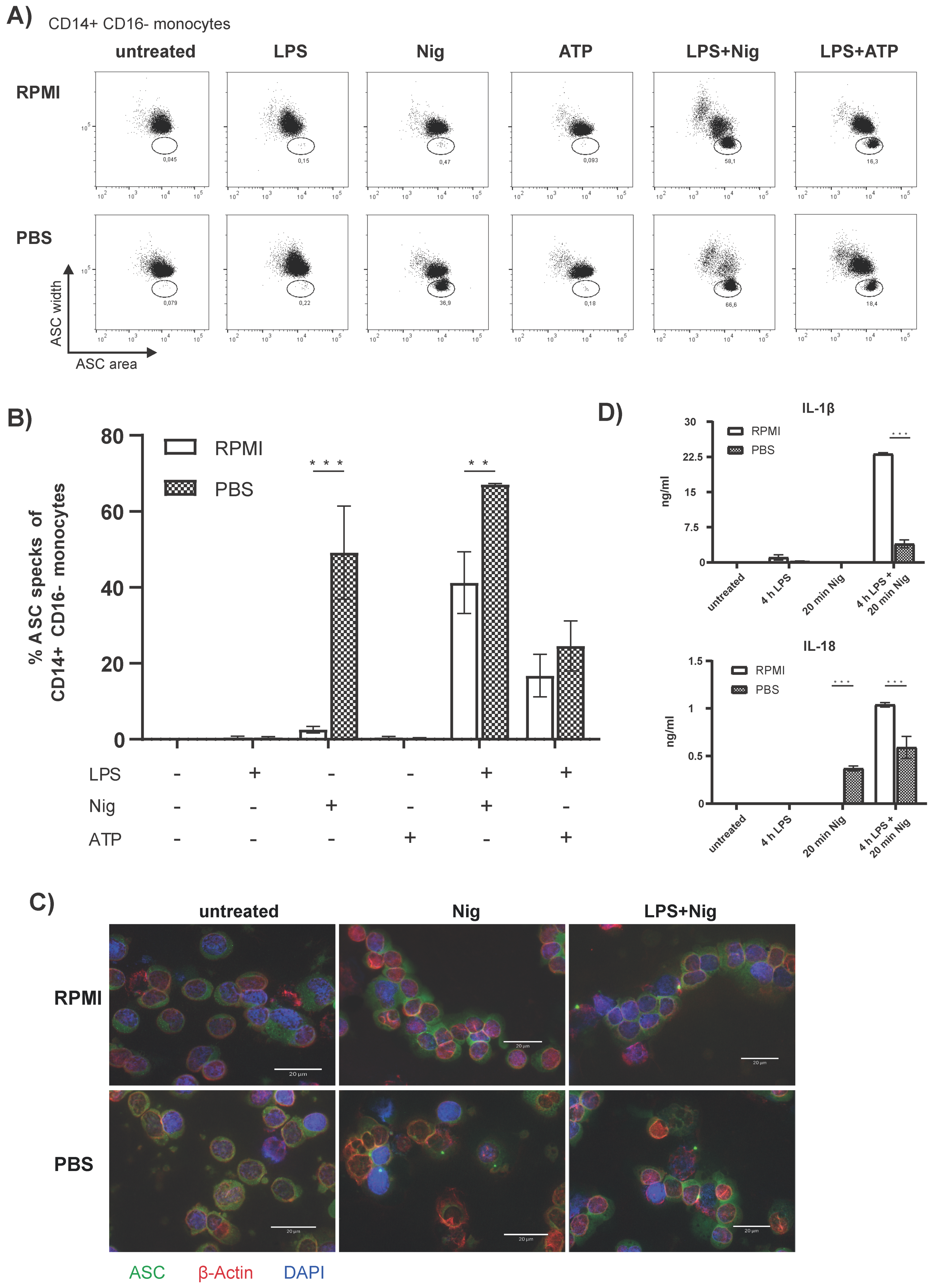
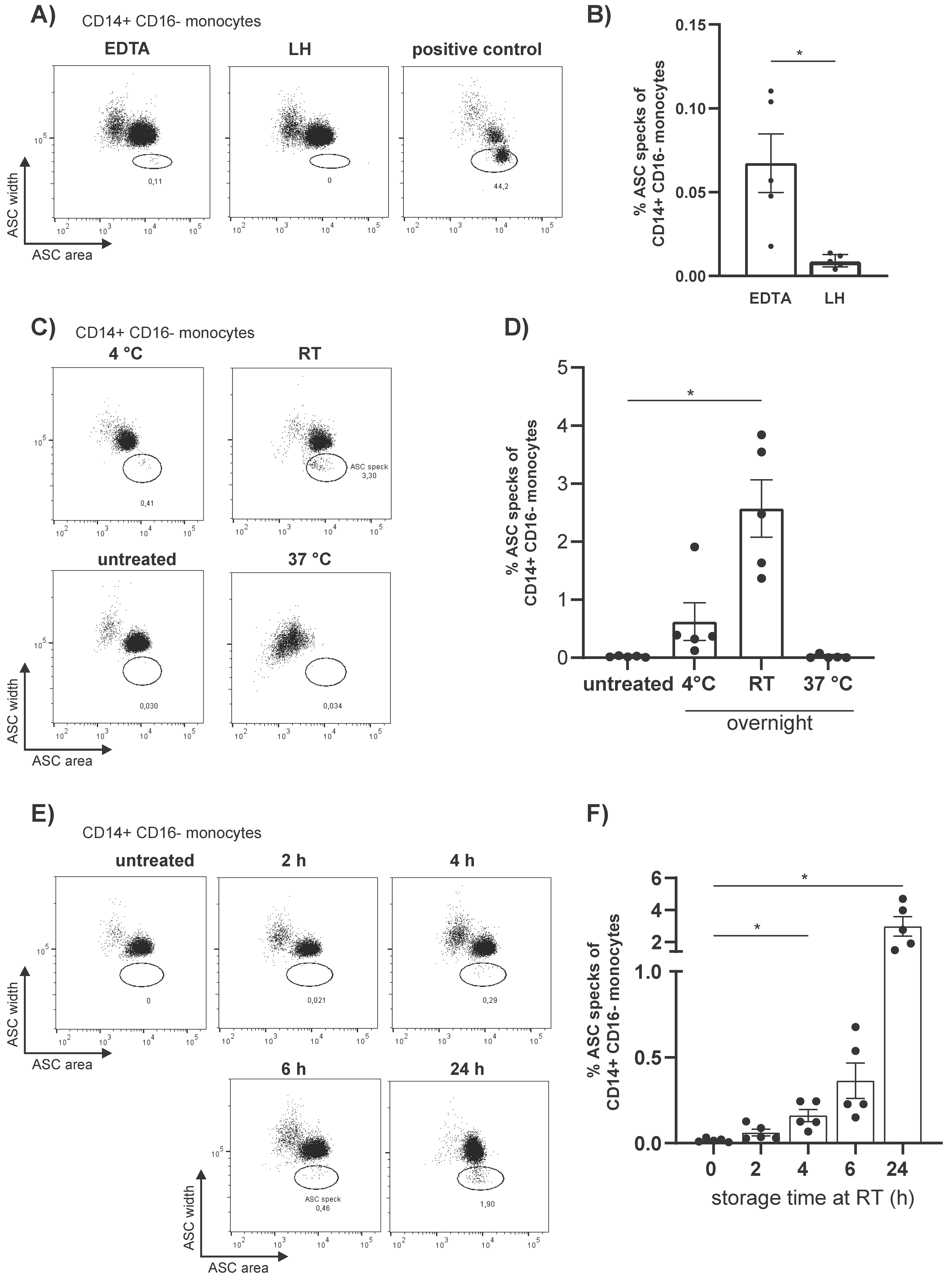
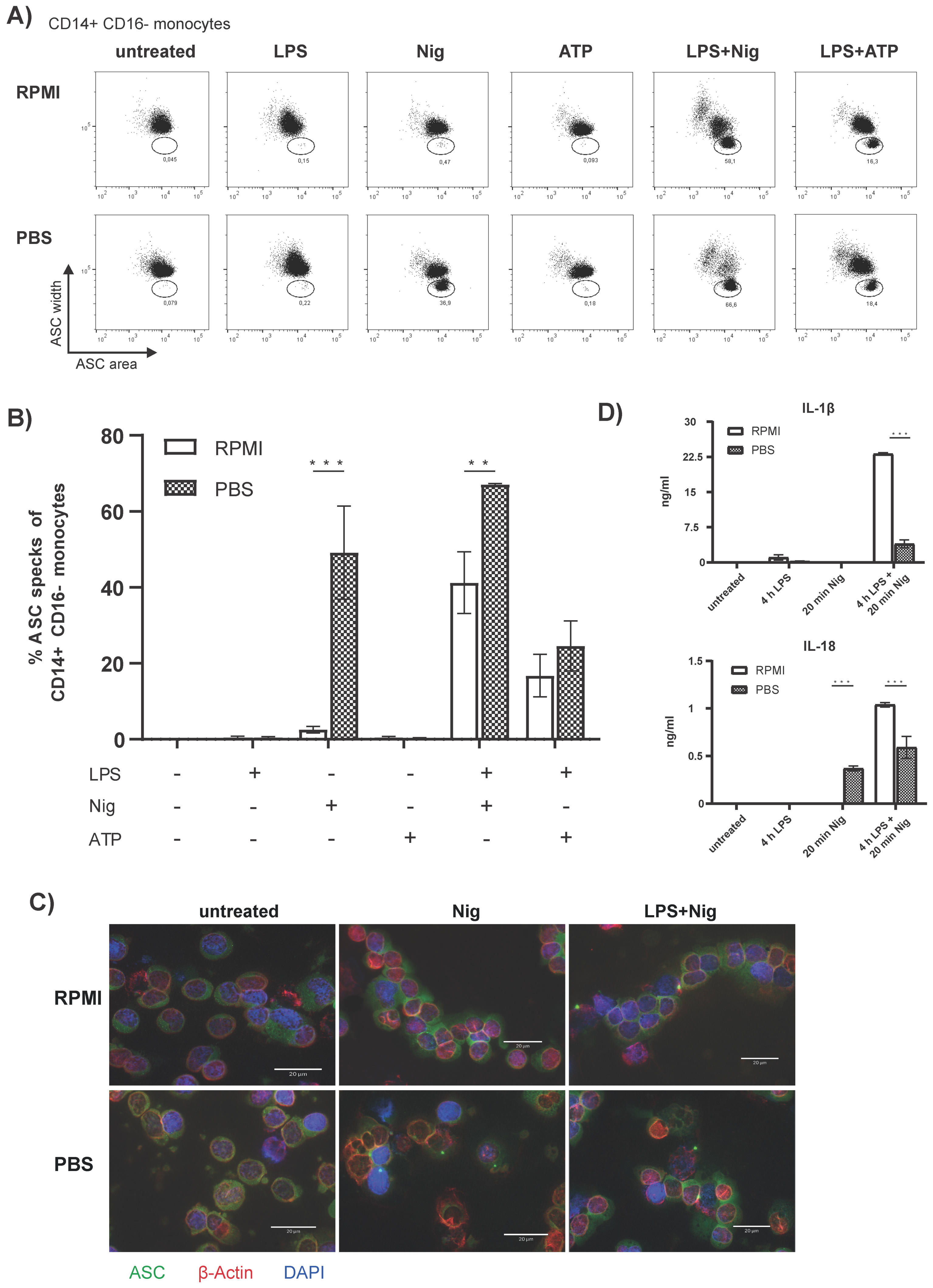
3.1. Flow Cytometric Detection of Spontaneous Intracellular ASC Specks Is Dependent on Anticoagulant, Storage Time, and Temperature
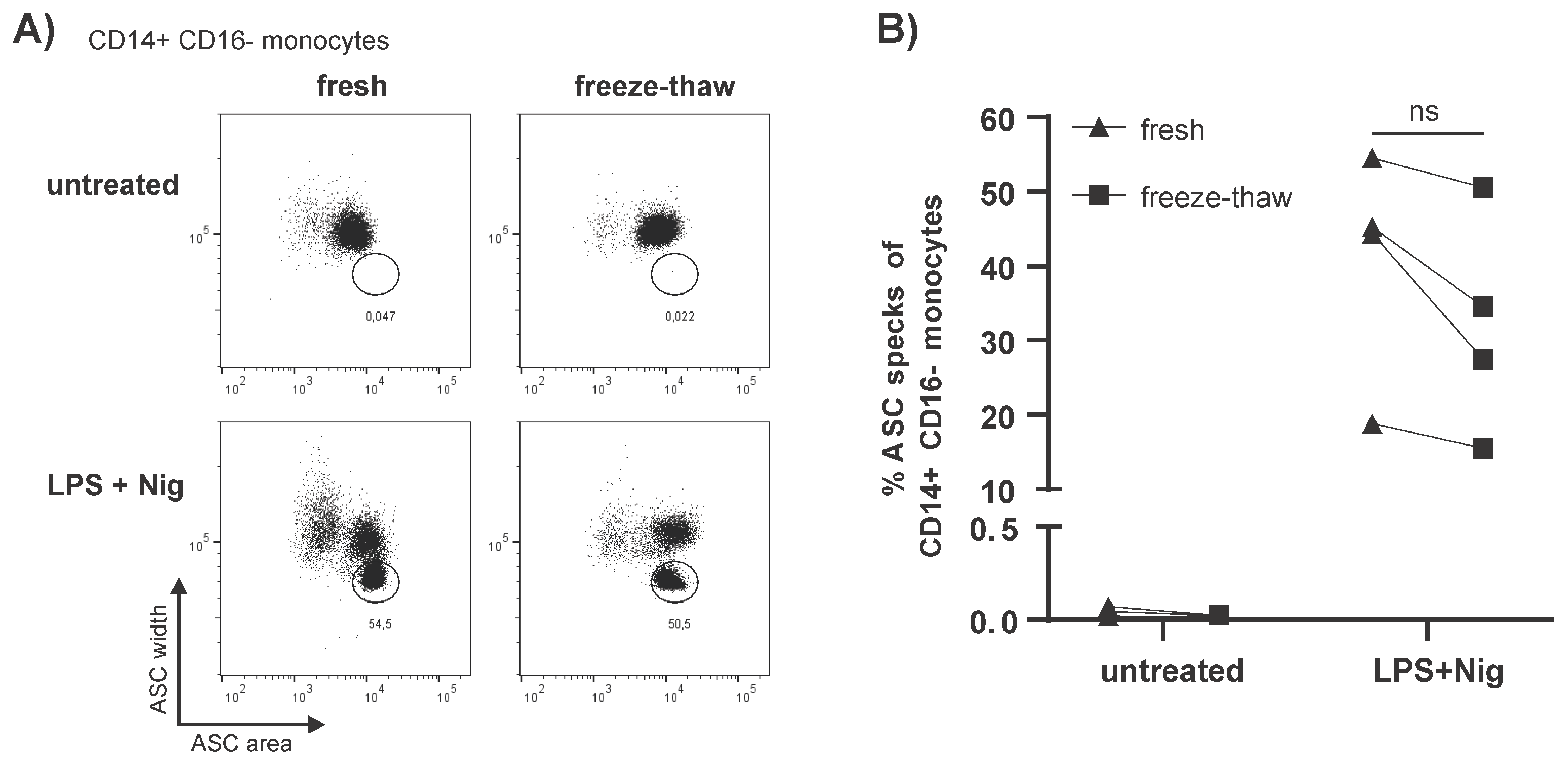
3.2. Storage of PBMCs in Liquid Nitrogen Does Not Impact Spontaneous ASC Speck Formation
3.3. Fast Generation of a Reliable ASC Speck Positive Control
3.4. The Monocytic THP1 Cell Line Represents a Valid ASC Speck Positive Control
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.-Y. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.K.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanneganti, T.-D.; Body-Malapel, M.; Amer, A.; Park, J.-H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded. RNA J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.-N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.G.; Dash, P.; Aldridge, J.R.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Amores-Iniesta, J.; Barberà-Cremades, M.; Martínez, C.M.; Pons, J.A.; Revilla-Nuin, B.; Martínez-Alarcón, L.; Di Virgilio, F.; Parrilla, P.; Baroja-Mazo, A.; Pelegrín, P. Extracellular ATP Activates the NLRP3 Inflammasome and Is an Early Danger Signal of Skin Allograft Rejection. Cell Rep. 2017, 21, 3414–3426. [Google Scholar] [CrossRef] [Green Version]
- Broderick, L.; De Nardo, D.; Franklin, B.S.; Hoffman, H.M.; Latz, E. The inflammasomes and autoinflammatory syndromes. Annu. Rev. Pathol. 2015, 10, 395–424. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Hopkins, L.J.; Nugent, E.; Cox, S.; Glück, I.M.; Tourlomousis, P.; Wright, J.A.; Cicuta, P.; Monie, T.P.; Bryant, C.E. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. USA 2014, 111, 7403–7408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sborgi, L.; Ravotti, F.; Dandey, V.P.; Dick, M.S.; Mazur, A.; Reckel, S.; Chami, M.; Scherer, S.; Huber, M.; Böckmann, A.; et al. Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 13237–13242. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Kanneganti, T.-D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2016, 16, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.J.; Wood, G.; Masters, S.L.; Richard, K.; Park, G.; Smith, B.J.; Kastner, D.L. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc. Natl. Acad. Sci. USA 2006, 103, 9982–9987. [Google Scholar] [CrossRef] [Green Version]
- Omenetti, A.; Carta, S.; Delfino, L.; Martini, A.; Gattorno, M.; Rubartelli, A. Increased NLRP3-dependent interleukin 1β secretion in patients with familial Mediterranean fever: Correlation with MEFV genotype. Ann. Rheum. Dis. 2014, 73, 462–469. [Google Scholar] [CrossRef]
- Masters, S.L.; Simon, A.; Aksentijevich, I.; Kastner, D.L. Horror autoinflammaticus: The molecular pathophysiology of autoinflammatory disease (*). Annu. Rev. Immunol. 2009, 27, 621–668. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.; Kanneganti, T.-D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 2019, 19, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, S.; Gagliani, N.; Zenewicz, L.A.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Vora., S.M.; Lieberman, J.; Wu, H. Inflammasome activation at the crux of severe COVID-19. Nat. Rev. Immunol. 2021, 21, 694–703. [Google Scholar] [CrossRef]
- Sepehri, Z.; Kiani, Z.; Afshari, M.; Kohan, F.; Dalvand, A.; Ghavami, S. Inflammasomes and type 2 diabetes: An updated systematic review. Immunol. Lett. 2017, 192, 97–103. [Google Scholar] [CrossRef]
- Kingsbury, S.R.; Conaghan, P.G.; McDermott, M.F. The role of the NLRP3 inflammasome in gout. J. Inflamm. Res. 2011, 4, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, F.; Mishra, N.; Ahrenstorf, G.; Franklin, B.S.; Latz, E.; Schmidt, R.E.; Bossaller, L. Evidence of inflammasome activation and formation of monocyte-derived ASC specks in HIV-1 positive patients. AIDS 2018, 32, 299–307. [Google Scholar] [CrossRef]
- Doitsh, G.; Galloway, N.L.K.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Oehrl, S.; Ahmad, F.; Brenner, T.; Uhle, F.; Nusshag, C.; Rupp, C.; Funck, F.; Meisel, S.; Weigand, M.A.; et al. Detection of In Vivo Inflammasome Activation for Predicting Sepsis Mortality. Front. Immunol. 2020, 11, 613745. [Google Scholar] [CrossRef] [PubMed]
- Barclay, W.; Shinohara, M.L. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol. 2017, 27, 213–219. [Google Scholar] [CrossRef]
- Freeman, L.C.; Ting, J.P.-Y. The pathogenic role of the inflammasome in neurodegenerative diseases. J. Neurochem. 2016, 136 (Suppl. 1), 29–38. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Sester, D.P.; Thygesen, S.J.; Sagulenko, V.; Vajjhala, P.R.; Cridland, J.A.; Vitak, N.; Chen, K.W.; Osborne, G.W.; Schroder, K.; Stacey, K.J. A novel flow cytometric method to assess inflammasome formation. J. Immunol. 2015, 194, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Lopez, N.; Romero, R.; Xu, Y.; Garcia-Flores, V.; Leng, Y.; Panaitescu, B.; Miller, D.; Abrahams, V.M.; Hassan, S.S. Inflammasome assembly in the chorioamniotic membranes during spontaneous labor at term. Am. J. Reprod. Immunol. 2017, 77, e12648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Nalda, A.; Fortuny, C.; Rey, L.; Bunney, T.D.; Alsina, L.; Esteve-Solé, A.; Bull, D.; Anton, M.C.; Basagaña, M.; Casals, F.; et al. Severe Autoinflammatory Manifestations and Antibody Deficiency Due to Novel Hypermorphic PLCG2 Mutations. J. Clin. Immunol. 2020, 40, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Mensa-Vilaro, A.; Teresa Bosque, M.; Magri, G.; Honda, Y.; Martínez-Banaclocha, H.; Casorran-Berges, M.; Sintes, J.; González-Roca, E.; Ruiz-Ortiz, E.; Heike, T.; et al. Brief Report: Late-Onset Cryopyrin-Associated Periodic Syndrome Due to Myeloid-Restricted Somatic NLRP3 Mosaicism. Arthritis Rheumatol. 2016, 68, 3035–3041. [Google Scholar] [CrossRef]
- Gritsenko, A.; Yu, S.; Martin-Sanchez, F.; Diaz-del-Olmo, I.; Nichols, E.-M.; Davis, D.M.; Brough, D.; Lopez-Castejon, G. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front. Immunol. 2020, 11, 565924. [Google Scholar] [CrossRef]
- Bossaller, L.; Chiang, P.-I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.K.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugawara, S.; Uehara, A.; Nochi, T.; Yamaguchi, T.; Ueda, H.; Sugiyama, A.; Hanzawa, K.; Kumagai, K.; Okamura, H.; Takada, H. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J. Immunol. 2001, 167, 6568–6575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.-C.; Goode, J.; Miething, C.; Göktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lage, S.L.; Dominical, V.M.; Wong, C.-S.; Sereti, I. Evaluation of Canonical Inflammasome Activation in Human Monocytes by Imaging Flow Cytometry. Front. Immunol. 2019, 10, 1284. [Google Scholar] [CrossRef]
- Apodaca, M.C.; Wright, A.E.; Riggins, A.M.; Harris, W.P.; Yeung, R.S.; Yu, L.; Morishima, C. Characterization of a whole blood assay for quantifying myeloid-derived suppressor cells. J. Immunother. Cancer 2019, 7, 230. [Google Scholar] [CrossRef]
- Tong, T.N.; Burke-Murphy, E.; Sakac, D.; Pendergrast, J.; Cserti-Gazdewich, C.; Laroche, V.; Branch, D.R. Optimal conditions for the performance of a monocyte monolayer assay. Transfusion 2016, 56, 2680–2690. [Google Scholar] [CrossRef]
- Diks, A.M.; Bonroy, C.; Teodosio, C.; Groenland, R.J.; de Mooij, B.; de Maertelaere, E.; Neirynck, J.; Philippé, J.; Orfao, A.; van Dongen, J.J.M.; et al. Impact of blood storage and sample handling on quality of high dimensional flow cytometric data in multicenter clinical research. J. Immunol. Methods 2019, 475, 112616. [Google Scholar] [CrossRef]
- Gray, E.; Hogwood, J.; Mulloy, B. The Anticoagulant and Antithrombotic Mechanisms of Heparin. In Heparin: A Century of Progress; Lever, R., Ed.; Springer: Heidelberg, Germany, 2012; pp. 43–61. [Google Scholar] [CrossRef]
- Jerram, A.; Guy, T.V.; Beutler, L.; Gunasegaran, B.; Sluyter, R.; de St Groth, B.F.; McGuire, H.M. Effects of storage time and temperature on highly multiparametric flow analysis of peripheral blood samples; implications for clinical trial samples. Biosci. Rep. 2021, 41, BSR20203827. [Google Scholar] [CrossRef] [PubMed]
- White, J.G.; Krivit, W. An Ultrastructural Basis for the Shape Changes Induced in Platelets by Chilling. Blood 1967, 30, 625–635. [Google Scholar] [CrossRef]
- Zucker, M.B.; Borrelli, J. Reversible Alterations in Platelet Morphology Produced by Anticoagulants and by Cold. Blood 1954, 9, 602–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perregaux, D.; Gabel, C.A. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Hentze, H.; Lin, X.Y.; Choi, M.S.K.; Porter, A.G. Critical role for cathepsin B in mediating caspase-1-dependent interleukin-18 maturation and caspase-1-independent necrosis triggered by the microbial toxin nigericin. Cell Death Differ. 2003, 10, 956–968. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, M.; Moehring, D.; Muñoz-Planillo, R.; Núñez, G.; Callaway, J.; Ting, J.; Scurria, M.; Ugo, T.; Bernad, L.; Cali, J.; et al. A bioluminescent caspase-1 activity assay rapidly monitors inflammasome activation in cells. J. Immunol. Methods 2017, 447, 1–13. [Google Scholar] [CrossRef]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Zhong, M.; Mittal, M.; Hong, Z.; Kanneganti, T.-D.; Rehman, J.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 2018, 49, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Franceschini, A.; Capece, M.; Chiozzi, P.; Falzoni, S.; Sanz, J.M.; Sarti, A.C.; Bonora, M.; Pinton, P.; Di Virgilio, F. The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J. 2015, 29, 2450–2461. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Puren, A.J.; Fantuzzi, G.; Dinarello, C.A. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc. Natl. Acad. Sci. USA 1999, 96, 2256–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wittmann, N.; Behrendt, A.-K.; Mishra, N.; Bossaller, L.; Meyer-Bahlburg, A. Instructions for Flow Cytometric Detection of ASC Specks as a Readout of Inflammasome Activation in Human Blood. Cells 2021, 10, 2880. https://doi.org/10.3390/cells10112880
Wittmann N, Behrendt A-K, Mishra N, Bossaller L, Meyer-Bahlburg A. Instructions for Flow Cytometric Detection of ASC Specks as a Readout of Inflammasome Activation in Human Blood. Cells. 2021; 10(11):2880. https://doi.org/10.3390/cells10112880
Chicago/Turabian StyleWittmann, Nico, Ann-Kathrin Behrendt, Neha Mishra, Lukas Bossaller, and Almut Meyer-Bahlburg. 2021. "Instructions for Flow Cytometric Detection of ASC Specks as a Readout of Inflammasome Activation in Human Blood" Cells 10, no. 11: 2880. https://doi.org/10.3390/cells10112880
APA StyleWittmann, N., Behrendt, A.-K., Mishra, N., Bossaller, L., & Meyer-Bahlburg, A. (2021). Instructions for Flow Cytometric Detection of ASC Specks as a Readout of Inflammasome Activation in Human Blood. Cells, 10(11), 2880. https://doi.org/10.3390/cells10112880