
A-Kinase Anchoring Protein 2 Promotes Protection against Myocardial Infarction

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Animal Procedures
2.3. Echocardiography
2.4. Immunohistochemistry
2.5. Infarct Size Determination
2.6. TUNEL Assay
2.7. Lectin Staining and Vessel Density Determination
2.8. RNA Preparation and Quantitative Real-Time PCR
2.9. Rat neonatal Ventricular Myocytes Preparation
2.10. AKAP2 Immunoprecipitation and Shotgun Proteomic Analysis
2.11. SDS–PAGE and Western Blotting
2.12. Plasmids and Constructs
2.13. Cell Culture
2.14. Production of Lentiviruses
2.15. Lentiviral Infection
2.16. Luciferase Activity Assay
2.17. PKA-Anchoring Inhibitory Peptides
2.18. ELISA Assays
2.19. Statistical Analysis
3. Results
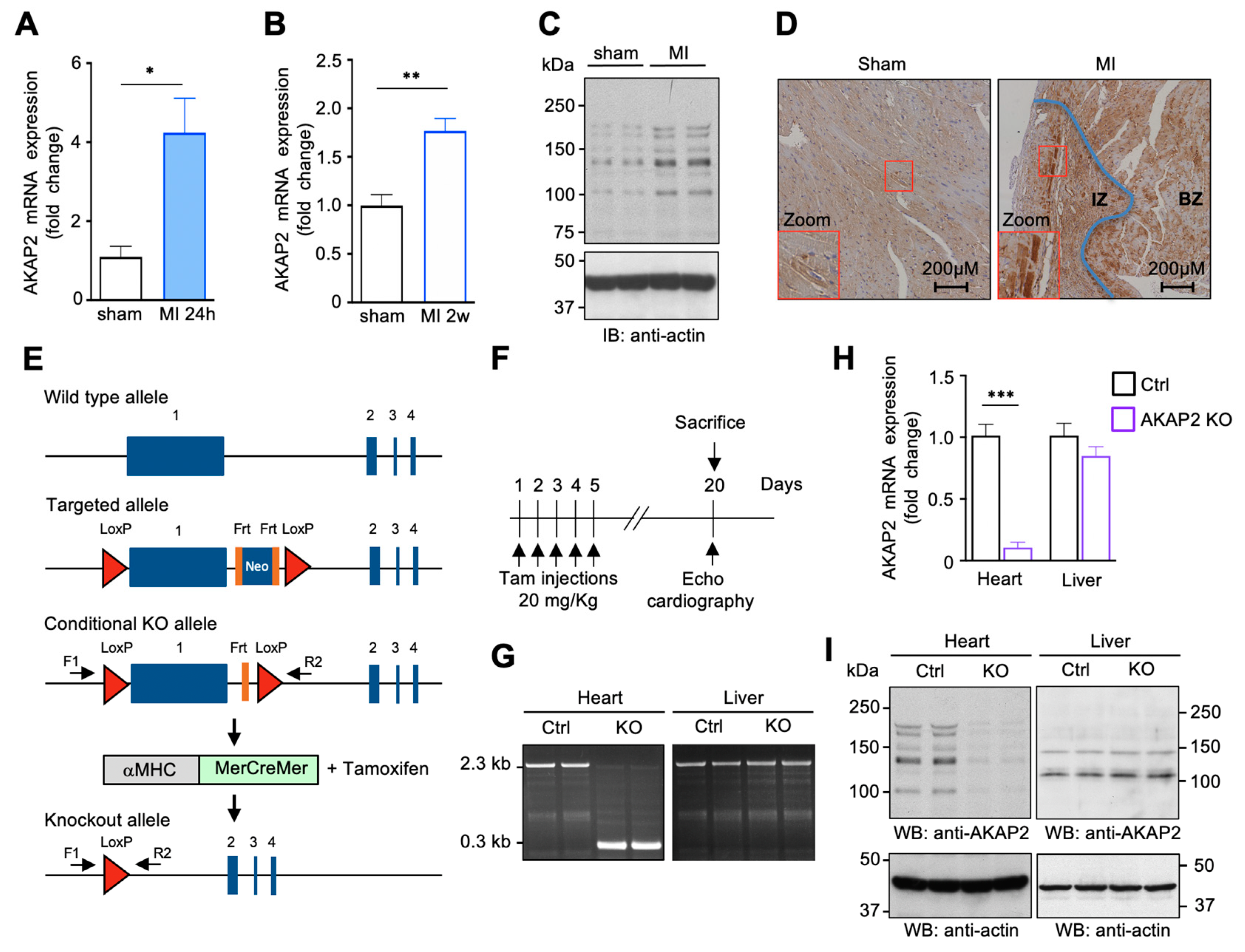
3.1. AKAP2 Is Upregulated in Cardiomyocytes after Myocardial Infarction
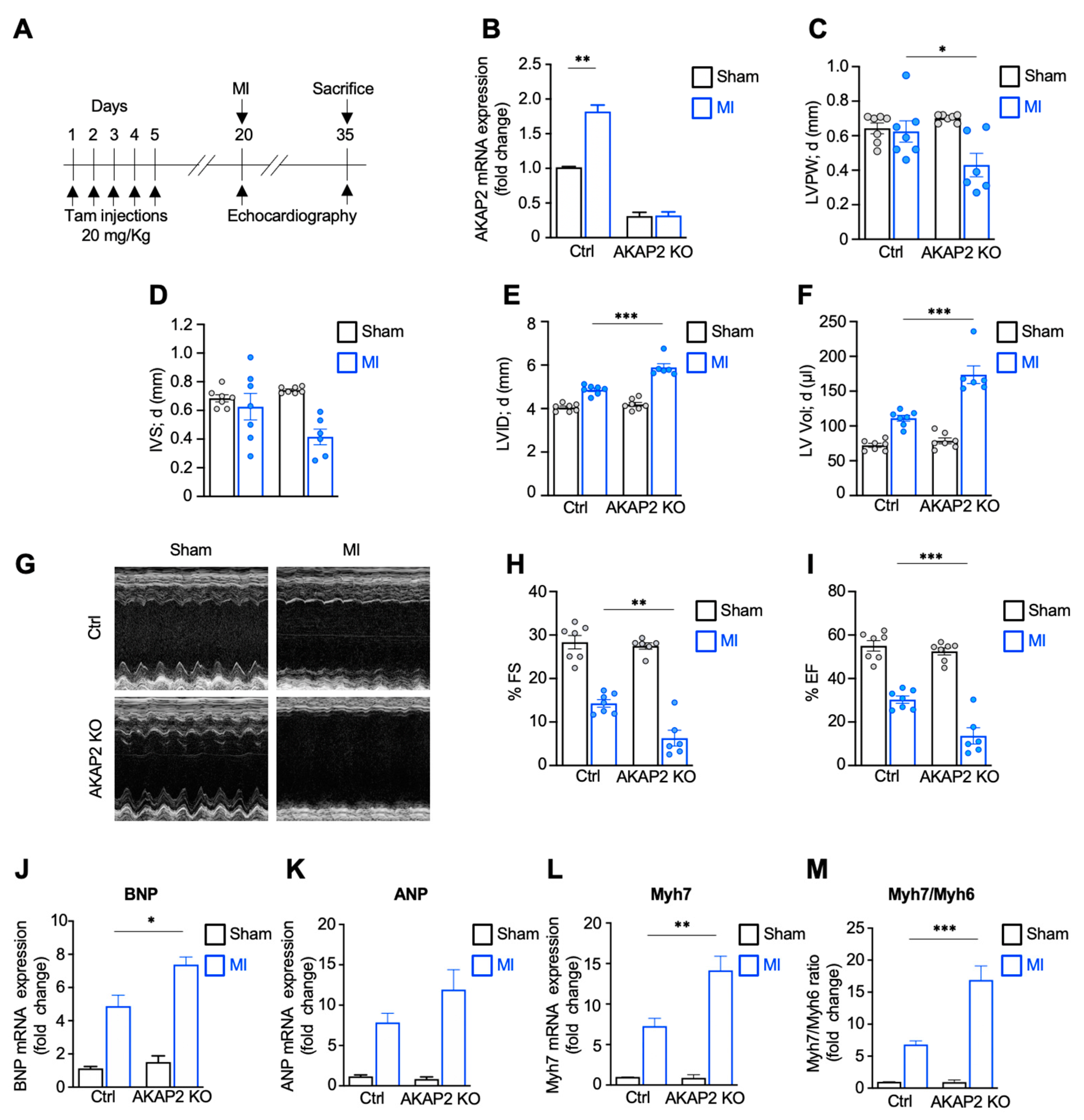
3.2. Targeted Deletion of AKAP2 in Adult Cardiomyocytes Exacerbates Cardiac Dysfunction after MI
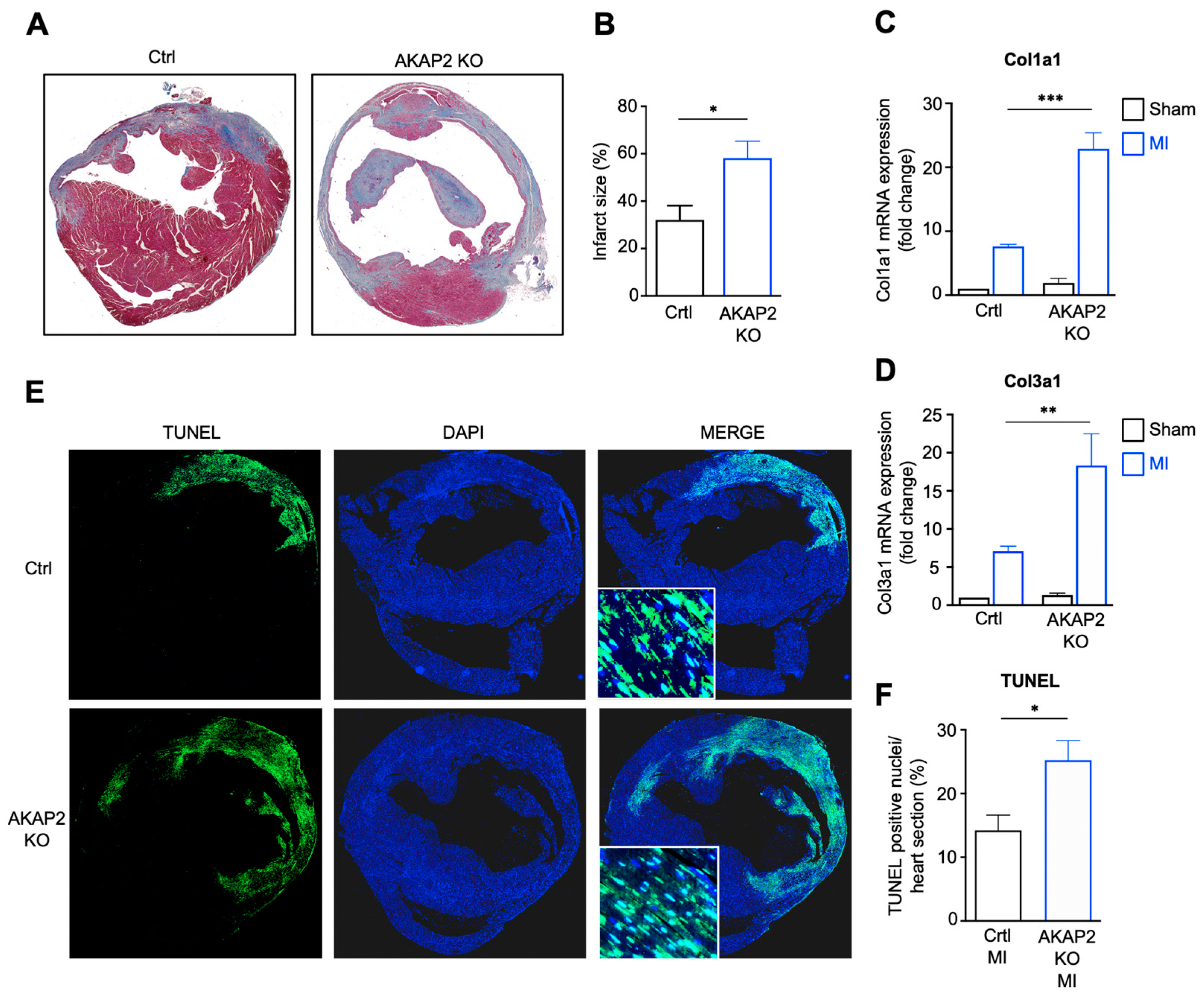
3.3. AKAP2 Knockout in Cardiomyocytes Enhances Myocardial Apoptosis, Fibrosis and Infarct Size after LAD Ligation
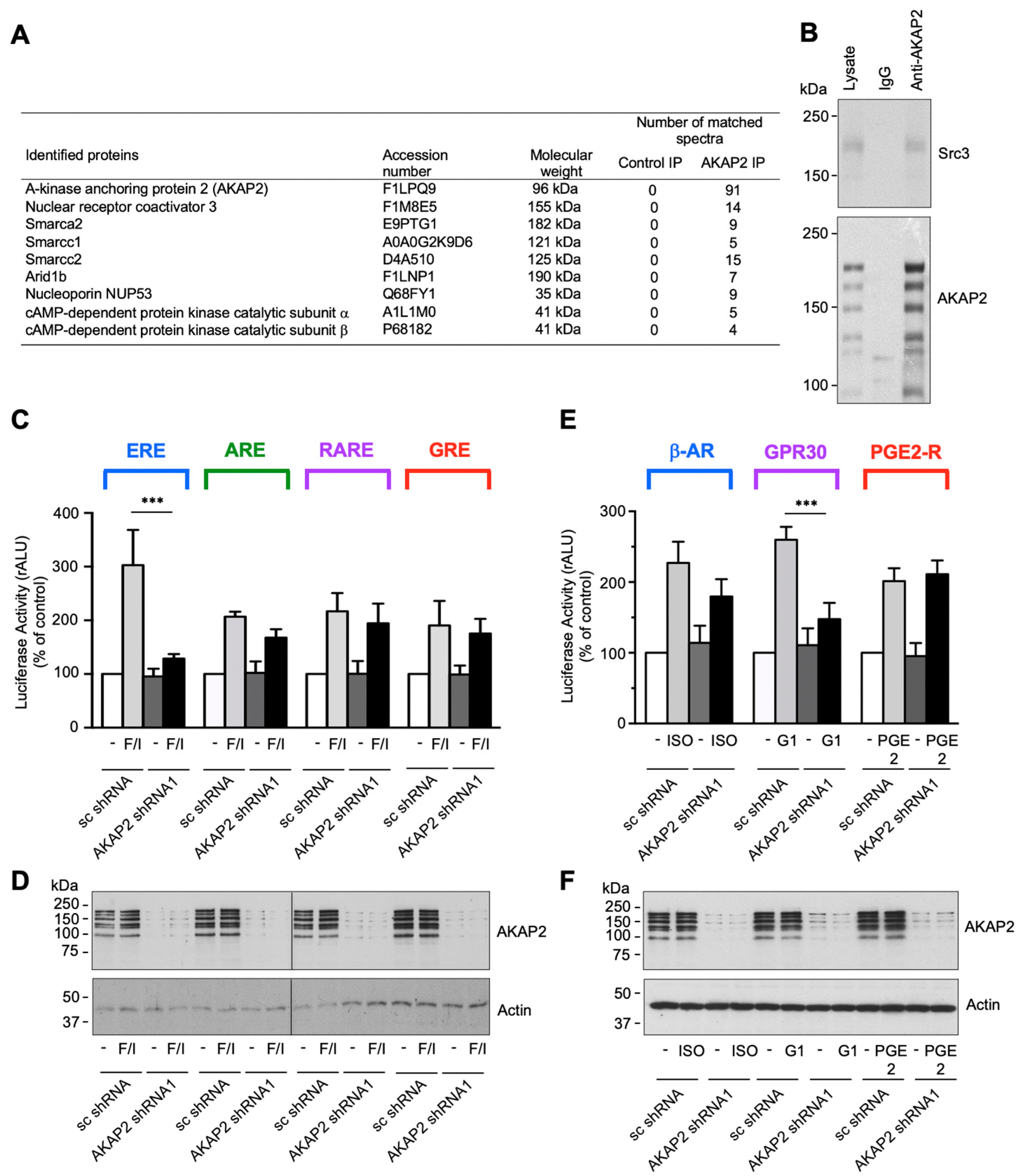
3.4. AKAP2 Assembles a Steroid-Receptor Coactivator-3-Based Signaling Complex Involved in Estrogen Receptor Activation
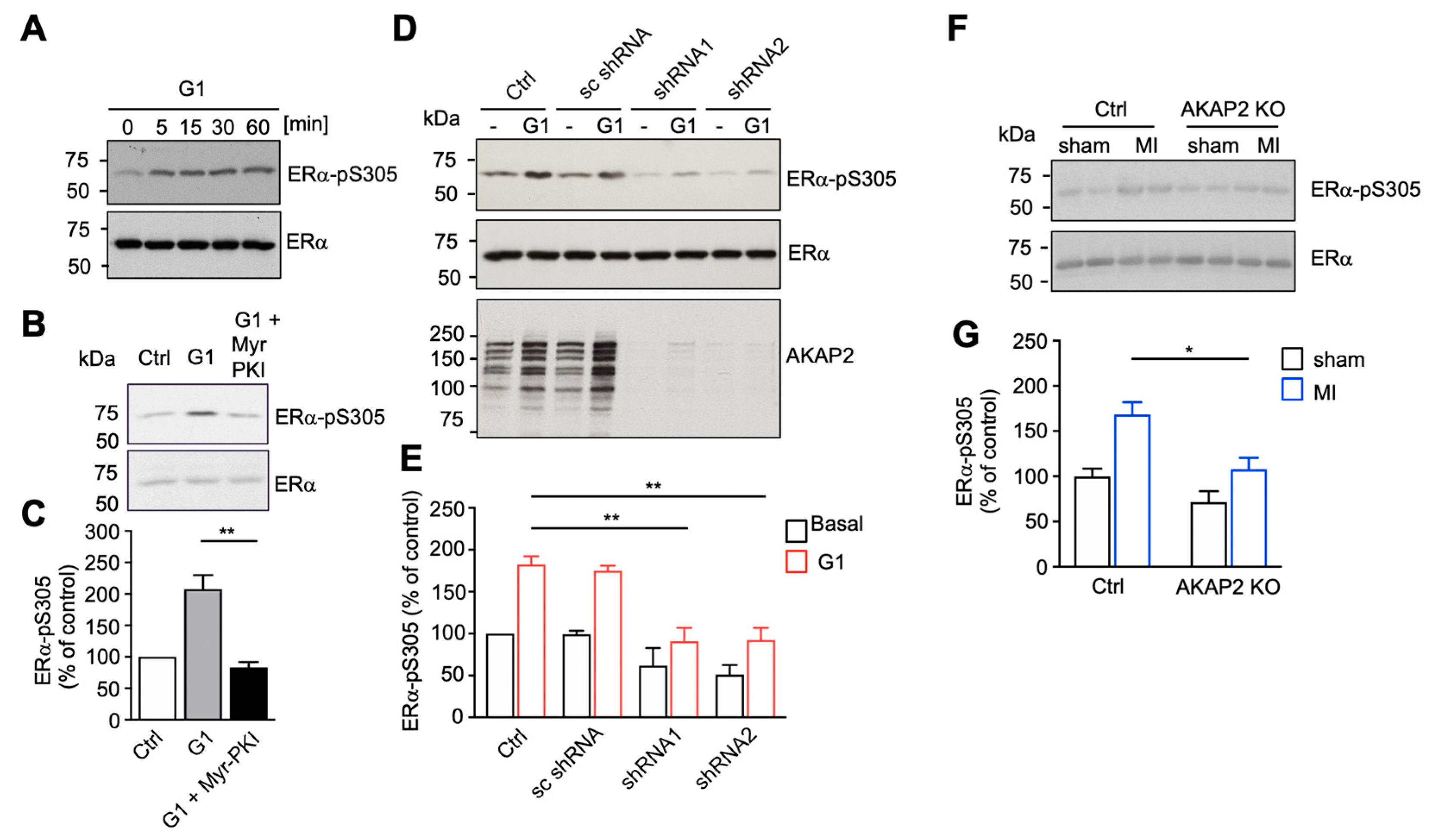
3.5. AKAP2 Favors PKA-Mediated Phosphorylation of ERα
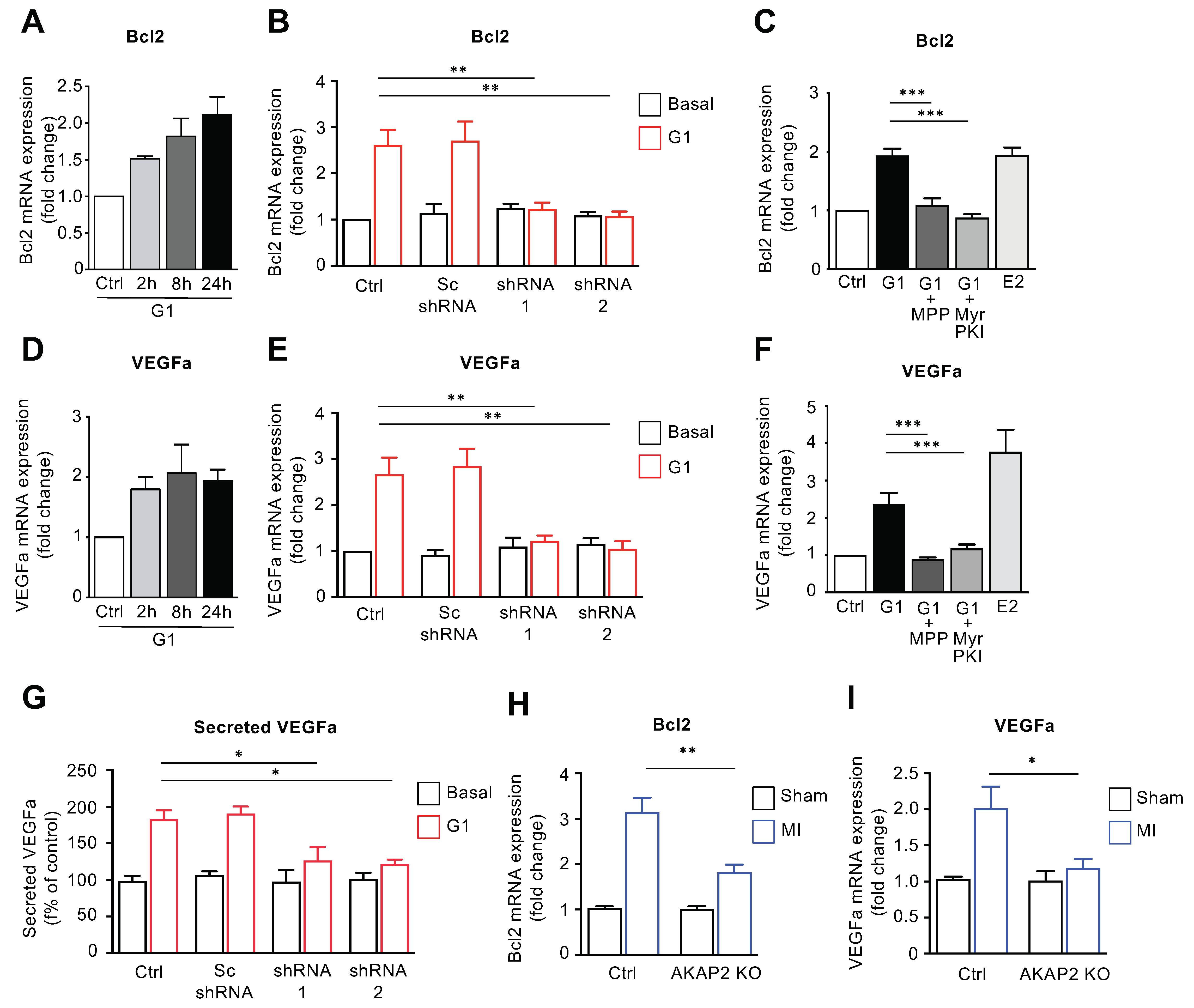
3.6. AKAP2 Promotes ERα-Mediated Regulation of Bcl2 and VEGFa in Cardiomyocytes
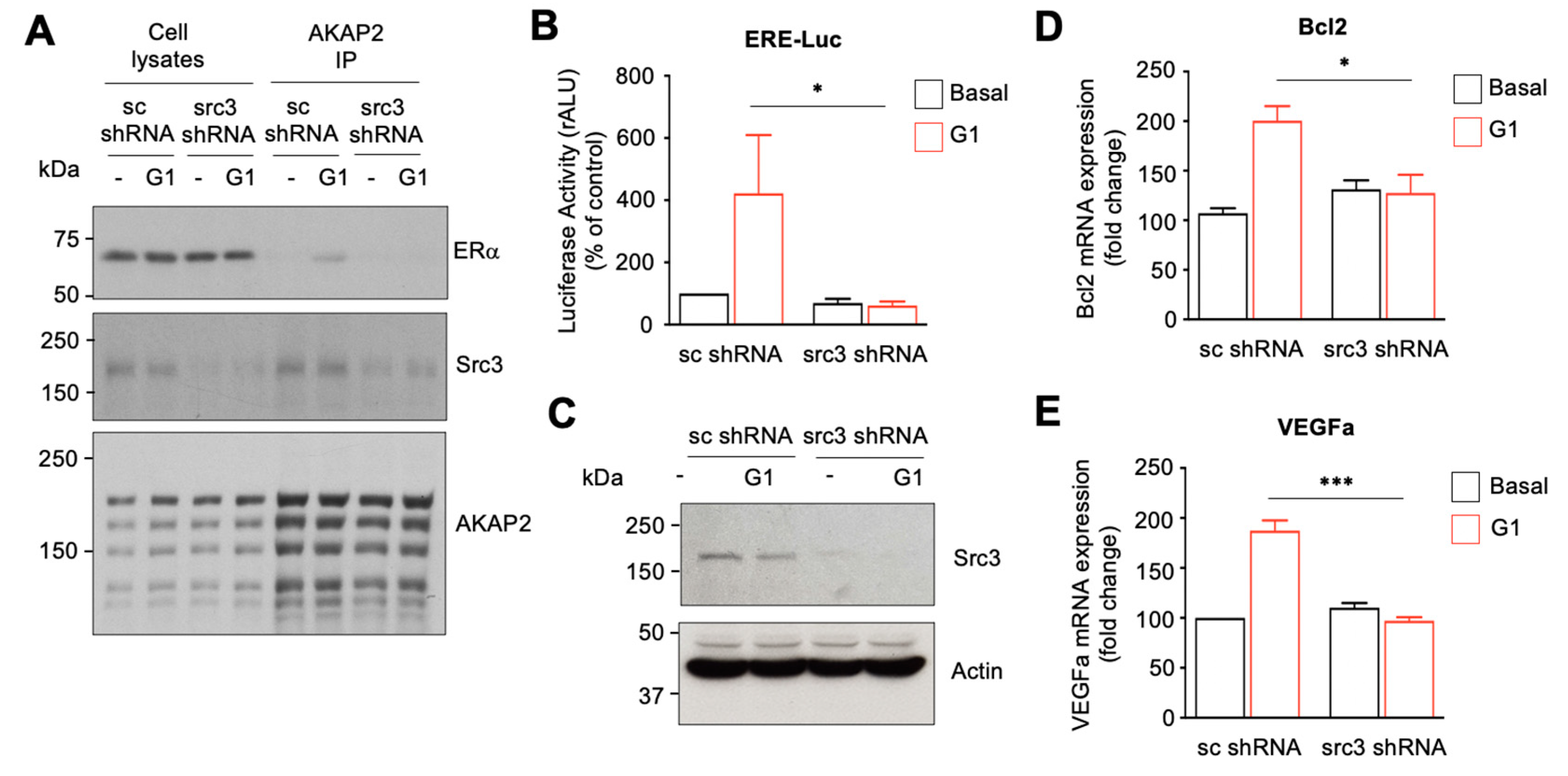
3.7. The AKAP2–Src3 Complex Mediates GPR30-Induced ER Activation in Cardiomyocytes
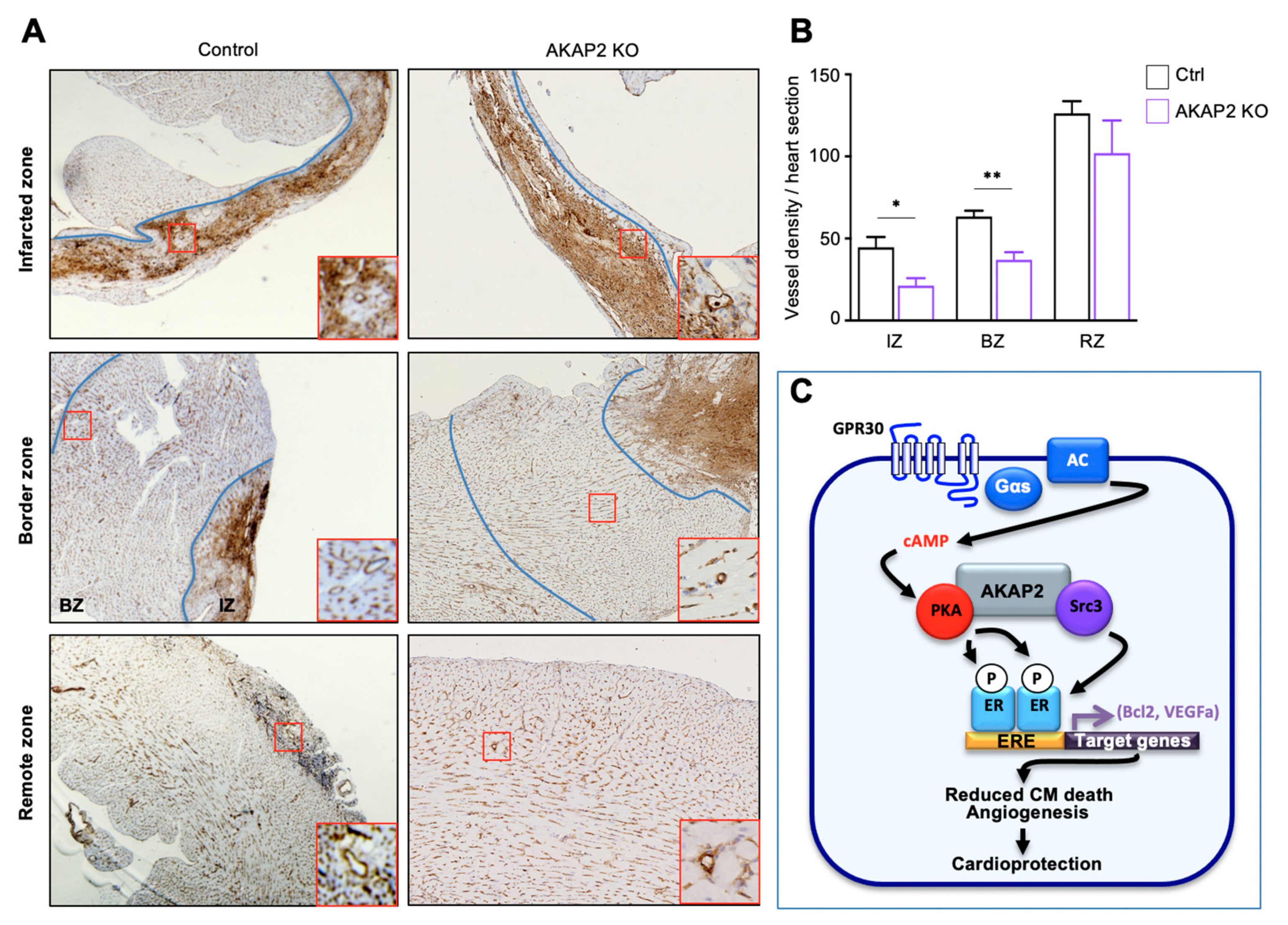
3.8. AKAP2 Knockout in Adult Cardiomyocytes Reduces Vessel Number in Infarcted Hearts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Burchfield, J.S.; Xie, M.; Hill, J.A. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef]
- Morissette, M.R.; Rosenzweig, A. Targeting survival signaling in heart failure. Curr. Opin. Pharmacol. 2005, 5, 165–170. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardio.l 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Scott, J.D.; Dessauer, C.W.; Tasken, K. Creating order from chaos: Cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Scott, J.D.; Hirsch, E. Anchoring proteins as regulators of signaling pathways. Circ. Res. 2012, 111, 482–492. [Google Scholar] [CrossRef]
- Bucko, P.J.; Scott, J.D. Drugs That Regulate Local Cell Signaling: AKAP Targeting as a Therapeutic Option. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Omar, M.H.; Scott, J.D. AKAP Signaling Islands: Venues for Precision Pharmacology. Trends Pharmacol. Sci. 2020, 41, 933–946. [Google Scholar] [CrossRef]
- Chen, L.; Marquardt, M.L.; Tester, D.J.; Sampson, K.J.; Ackerman, M.J.; Kass, R.S. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 20990–20995. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Scholten, A.; Vos, M.A.; van Veen, T.A. Anchored protein kinase A signalling in cardiac cellular electrophysiology. J. Cell Mol. Med. 2014, 18, 2135–2146. [Google Scholar] [CrossRef]
- Lygren, B.; Carlson, C.R.; Santamaria, K.; Lissandron, V.; McSorley, T.; Lorenz, D.; Wiesner, B.; Rosenthal, W.; Zaccolo, M.; Tasken, K.; et al. AKAP-complex regulates the Ca2+ reuptake into heart sarcoplasmic reticulum. EMBO Rep. 2007, 8, 1061–1067. [Google Scholar] [CrossRef]
- Jones, B.W.; Brunet, S.; Gilbert, M.L.; Nichols, C.B.; Su, T.; Westenbroek, R.E.; Scott, J.D.; Catterall, W.A.; McKnight, G.S. Cardiomyocytes from AKAP7 knockout mice respond normally to adrenergic stimulation. Proc. Natl. Acad. Sci. USA 2012, 109, 17099–17104. [Google Scholar] [CrossRef]
- Diviani, D.; Osman, H.; Delaunay, M.; Kaiser, S. The role of A-kinase anchoring proteins in cardiac oxidative stress. Biochem. Soc. Trans. 2019, 47, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Passariello, C.L.; Li, J.; Dodge-Kafka, K.; Kapiloff, M.S. mAKAP-a master scaffold for cardiac remodeling. J. Cardiovasc. Pharmacol. 2015, 65, 218–225. [Google Scholar] [CrossRef]
- Appert-Collin, A.; Cotecchia, S.; Nenniger-Tosato, M.; Pedrazzini, T.; Diviani, D. The A-kinase anchoring protein (AKAP)-Lbc-signaling complex mediates alpha1 adrenergic receptor-induced cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 10140–10145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Malik, S.; Pang, J.; Wang, H.; Park, K.M.; Yule, D.I.; Blaxall, B.C.; Smrcka, A.V. Phospholipase Cepsilon hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 2013, 153, 216–227. [Google Scholar] [CrossRef]
- Dodge-Kafka, K.L.; Soughayer, J.; Pare, G.C.; Carlisle Michel, J.J.; Langeberg, L.K.; Kapiloff, M.S.; Scott, J.D. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 2005, 437, 574–578. [Google Scholar] [CrossRef]
- Diviani, D.; Reggi, E.; Arambasic, M.; Caso, S.; Maric, D. Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochim. Biophys. Acta 2016, 1863, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, M.; Osman, H.; Kaiser, S.; Diviani, D. The Role of Cyclic AMP Signaling in Cardiac Fibrosis. Cells 2019, 9, 69. [Google Scholar] [CrossRef]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, M.D.; Li, J.; Passariello, C.L.; Gayanilo, M.; Thakur, H.; Dayan, J.; Dodge-Kafka, K.; Kapiloff, M.S. The scaffold protein muscle A-kinase anchoring protein beta orchestrates cardiac myocyte hypertrophic signaling required for the development of heart failure. Circ. Heart Fail. 2014, 7, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tan, Y.; Passariello, C.L.; Martinez, E.C.; Kritzer, M.D.; Li, X.; Li, X.; Li, Y.; Yu, Q.; Ohgi, K.; et al. Signalosome-Regulated Serum Response Factor Phosphorylation Determining Myocyte Growth in Width Versus Length as a Therapeutic Target for Heart Failure. Circulation 2020, 142, 2138–2154. [Google Scholar] [CrossRef]
- Ibarrola, J.; Sadaba, R.; Martinez-Martinez, E.; Garcia-Pena, A.; Arrieta, V.; Alvarez, V.; Fernandez-Celis, A.; Gainza, A.; Cachofeiro, V.; Santamaria, E.; et al. Aldosterone Impairs Mitochondrial Function in Human Cardiac Fibroblasts via A-Kinase Anchor Protein 12. Sci. Rep. 2018, 8, 6801. [Google Scholar] [CrossRef]
- Perrino, C.; Feliciello, A.; Schiattarella, G.G.; Esposito, G.; Guerriero, R.; Zaccaro, L.; Del Gatto, A.; Saviano, M.; Garbi, C.; Carangi, R.; et al. AKAP121 downregulation impairs protective cAMP signals, promotes mitochondrial dysfunction, and increases oxidative stress. Cardiovasc. Res. 2010, 88, 101–110. [Google Scholar] [CrossRef]
- Kim, H.; Scimia, M.C.; Wilkinson, D.; Trelles, R.D.; Wood, M.R.; Bowtell, D.; Dillin, A.; Mercola, M.; Ronai, Z.A. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 2011, 44, 532–544. [Google Scholar] [CrossRef]
- Li, L.; Li, J.; Drum, B.M.; Chen, Y.; Yin, H.; Guo, X.; Luckey, S.W.; Gilbert, M.L.; McKnight, G.S.; Scott, J.D.; et al. Loss of AKAP150 promotes pathological remodelling and heart failure propensity by disrupting calcium cycling and contractile reserve. Cardiovasc. Res. 2017, 113, 147–159. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Boccella, N.; Paolillo, R.; Cattaneo, F.; Trimarco, V.; Franzone, A.; D’Apice, S.; Giugliano, G.; Rinaldi, L.; Borzacchiello, D.; et al. Loss of Akap1 Exacerbates Pressure Overload-Induced Cardiac Hypertrophy and Heart Failure. Front. Physiol. 2018, 9, 558. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Cattaneo, F.; Pironti, G.; Magliulo, F.; Carotenuto, G.; Pirozzi, M.; Polishchuk, R.; Borzacchiello, D.; Paolillo, R.; Oliveti, M.; et al. Akap1 Deficiency Promotes Mitochondrial Aberrations and Exacerbates Cardiac Injury Following Permanent Coronary Ligation via Enhanced Mitophagy and Apoptosis. PLoS ONE 2016, 11, e0154076. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Felsmesser, M.; Casadevall, A.; Rubin, C.S. Molecular characterization of a cDNA that encodes six isoforms of a novel murine A kinase anchor protein. J. Biol. Chem. 1998, 273, 6533–6541. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G.; Reichow, S.L.; O’Neill, S.E.; Weisbrod, C.R.; Langeberg, L.K.; Bruce, J.E.; Gonen, T.; Scott, J.D. AKAP2 anchors PKA with aquaporin-0 to support ocular lens transparency. EMBO Mol. Med. 2012, 4, 15–26. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, B.; Li, Y.; Dai, Y.; Li, P.; Li, L.; Xu, J.; Li, L.; Wu, P. AKAP2 overexpression modulates growth plate chondrocyte functions through ERK1/2 signaling. Bone 2021, 146, 115875. [Google Scholar] [CrossRef]
- Thakkar, A.; Aljameeli, A.; Thomas, S.; Shah, G.V. A-kinase anchoring protein 2 is required for calcitonin-mediated invasion of cancer cells. Endocr. Relat. Cancer 2016, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Takagawa, J.; Zhang, Y.; Wong, M.L.; Sievers, R.E.; Kapasi, N.K.; Wang, Y.; Yeghiazarians, Y.; Lee, R.J.; Grossman, W.; Springer, M.L. Myocardial infarct size measurement in the mouse chronic infarction model: Comparison of area- and length-based approaches. J. Appl. Physiol. 2007, 102, 2104–2111. [Google Scholar] [CrossRef]
- Ren, G.; Michael, L.H.; Entman, M.L.; Frangogiannis, N.G. Morphological characteristics of the microvasculature in healing myocardial infarcts. J. Histochem. Cytochem. 2002, 50, 71–79. [Google Scholar] [CrossRef] [PubMed]
- del Vescovo, C.D.; Cotecchia, S.; Diviani, D. A-kinase-anchoring protein-Lbc anchors IkappaB kinase beta to support interleukin-6-mediated cardiomyocyte hypertrophy. Mol. Cell. Biol. 2013, 33, 14–27. [Google Scholar] [CrossRef]
- Crippa, S.; Nemir, M.; Ounzain, S.; Ibberson, M.; Berthonneche, C.; Sarre, A.; Boisset, G.; Maison, D.; Harshman, K.; Xenarios, I.; et al. Comparative transcriptome profiling of the injured zebrafish and mouse hearts identifies miRNA-dependent repair pathways. Cardiovasc. Res. 2016, 110, 73–84. [Google Scholar] [CrossRef]
- Mullany, L.K.; Rohira, A.D.; Leach, J.P.; Kim, J.H.; Monroe, T.O.; Ortiz, A.R.; Stork, B.; Gaber, M.W.; Sarkar, P.; Sikora, A.G.; et al. A steroid receptor coactivator stimulator (MCB-613) attenuates adverse remodeling after myocardial infarction. Proc. Natl. Acad. Sci. USA 2020, 117, 31353–31364. [Google Scholar] [CrossRef] [PubMed]
- Lonard, D.M.; O’Malley, B.W. The expanding cosmos of nuclear receptor coactivators. Cell 2006, 125, 411–414. [Google Scholar] [CrossRef]
- Gaillard, E.; Bruck, N.; Brelivet, Y.; Bour, G.; Lalevee, S.; Bauer, A.; Poch, O.; Moras, D.; Rochette-Egly, C. Phosphorylation by PKA potentiates retinoic acid receptor alpha activity by means of increasing interaction with and phosphorylation by cyclin H/cdk7. Proc. Natl. Acad. Sci. USA 2006, 103, 9548–9553. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, G.; Wu, J.; French, J.; Kim, J.; Moniri, N.H.; Daaka, Y. Androgens transduce the G alphas-mediated activation of protein kinase A in prostate cells. Cancer Res. 2008, 68, 3225–3231. [Google Scholar] [CrossRef]
- Rangarajan, P.N.; Umesono, K.; Evans, R.M. Modulation of glucocorticoid receptor function by protein kinase A. Mol. Endocrinol. 1992, 6, 1451–1457. [Google Scholar] [PubMed]
- de Leeuw, R.; Flach, K.; Bentin Toaldo, C.; Alexi, X.; Canisius, S.; Neefjes, J.; Michalides, R.; Zwart, W. PKA phosphorylation redirects ERalpha to promoters of a unique gene set to induce tamoxifen resistance. Oncogene 2013, 32, 3543–3551. [Google Scholar] [CrossRef]
- Deschamps, A.M.; Murphy, E.; Sun, J. Estrogen receptor activation and cardioprotection in ischemia reperfusion injury. Trends Cardiovasc. Med. 2010, 20, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.; Fajardo, G.; Zhao, M.; Urashima, T.; Powers, J.; Berry, G.; Kobilka, B.K. Differential cardioprotective/cardiotoxic effects mediated by beta-adrenergic receptor subtypes. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2441–H2449. [Google Scholar] [CrossRef]
- Xiao, C.Y.; Yuhki, K.; Hara, A.; Fujino, T.; Kuriyama, S.; Yamada, T.; Takayama, K.; Takahata, O.; Karibe, H.; Taniguchi, T.; et al. Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4. Circulation 2004, 109, 2462–2468. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Arterburn, J.B.; Smith, H.O.; Oprea, T.I.; Sklar, L.A.; Hathaway, H.J. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu. Rev. Physiol. 2008, 70, 165–190. [Google Scholar] [CrossRef]
- Chen, D.; Pace, P.E.; Coombes, R.C.; Ali, S. Phosphorylation of human estrogen receptor alpha by protein kinase A regulates dimerization. Mol. Cell. Biol. 1999, 19, 1002–1015. [Google Scholar] [CrossRef]
- van Rooij, E.; Fielitz, J.; Sutherland, L.B.; Thijssen, V.L.; Crijns, H.J.; Dimaio, M.J.; Shelton, J.; De Windt, L.J.; Hill, J.A.; Olson, E.N. Myocyte enhancer factor 2 and class II histone deacetylases control a gender-specific pathway of cardioprotection mediated by the estrogen receptor. Circ. Res. 2010, 106, 155–165. [Google Scholar] [CrossRef]
- Lee, S.H.; Wolf, P.L.; Escudero, R.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N. Engl. J. Med. 2000, 342, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, I.Y.; Mahtta, D.; Pepine, C.J. Medical Therapy for Heart Failure Caused by Ischemic Heart Disease. Circ. Res. 2019, 124, 1520–1535. [Google Scholar] [CrossRef]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the cAMP Signaling Pathway. Pharmacol. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, R.S.; Gregorian, C.; Drenan, R.M.; Xiang, Y.; Regan, J.W.; Insel, P.A. Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase. J. Biol. Chem. 2001, 276, 42063–42069. [Google Scholar] [CrossRef]
- Coleman, K.M.; Dutertre, M.; El-Gharbawy, A.; Rowan, B.G.; Weigel, N.L.; Smith, C.L. Mechanistic differences in the activation of estrogen receptor-alpha (ER alpha)- and ER beta-dependent gene expression by cAMP signaling pathway(s). J. Biol. Chem. 2003, 278, 12834–12845. [Google Scholar] [CrossRef]
- Houtman, R.; de Leeuw, R.; Rondaij, M.; Melchers, D.; Verwoerd, D.; Ruijtenbeek, R.; Martens, J.W.; Neefjes, J.; Michalides, R. Serine-305 phosphorylation modulates estrogen receptor alpha binding to a coregulator peptide array, with potential application in predicting responses to tamoxifen. Mol. Cancer Ther. 2012, 11, 805–816. [Google Scholar] [CrossRef]
- Bentin Toaldo, C.; Alexi, X.; Beelen, K.; Kok, M.; Hauptmann, M.; Jansen, M.; Berns, E.; Neefjes, J.; Linn, S.; Michalides, R.; et al. Protein Kinase A-induced tamoxifen resistance is mediated by anchoring protein AKAP13. BMC Cancer 2015, 15, 588. [Google Scholar] [CrossRef]
- Albanito, L.; Madeo, A.; Lappano, R.; Vivacqua, A.; Rago, V.; Carpino, A.; Oprea, T.I.; Prossnitz, E.R.; Musti, A.M.; Ando, S.; et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007, 67, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Sirianni, R.; Chimento, A.; Ruggiero, C.; De Luca, A.; Lappano, R.; Ando, S.; Maggiolini, M.; Pezzi, V. The novel estrogen receptor, G protein-coupled receptor 30, mediates the proliferative effects induced by 17beta-estradiol on mouse spermatogonial GC-1 cell line. Endocrinology 2008, 149, 5043–5051. [Google Scholar] [CrossRef]
- Notas, G.; Kampa, M.; Pelekanou, V.; Castanas, E. Interplay of estrogen receptors and GPR30 for the regulation of early membrane initiated transcriptional effects: A pharmacological approach. Steroids 2012, 77, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Liu, Y.; Sun, D.; Zhou, C.; Liu, A.; Xu, C.; Hao, Y.; Li, D.; Yan, C.; Sun, H. Chronic activation of the G protein-coupled receptor 30 with agonist G-1 attenuates heart failure. PLoS ONE 2012, 7, e48185. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.E.; Singh, H.; Lu, R.; Olde, B.; Leeb-Lundberg, L.M.; Bopassa, J.C. G Protein-Coupled Estrogen Receptor 1 Mediates Acute Estrogen-Induced Cardioprotection via MEK/ERK/GSK-3beta Pathway after Ischemia/Reperfusion. PLoS ONE 2015, 10, e0135988. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, X.; Chou, J.; Lin, M.; Ferrario, C.M.; Zapata-Sudo, G.; Groban, L. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: A sex-specific gene profiling analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1870–1882. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Males | Females | |||
|---|---|---|---|---|
| Control | AKAP2 KO | Control | AKAP2 KO | |
| n | 27 | 32 | 29 | 20 |
| Heart Rate (beads/min) | 470 ± 8 | 499 ± 11 | 477 ± 9 | 490 ± 14 |
| Body Weight (g) | 29.43 ± 1.00 | 28.41 ± 0.69 | 22.46 ± 0.31 | 22.71 ± 0.45 |
| LVPW; d (mm) | 0.66 ± 0.01 | 0.69 ± 0.01 | 0.66 ± 0.01 | 0.69 ± 0.02 |
| LVPW; s (mm) | 0.88 ± 0.02 | 0.95 ± 0.03 | 0.92 ± 0.02 | 0.94 ± 0.03 |
| IVS; d (mm) | 0.68 ± 0.01 | 0.71 ± 0.01 | 0.69 ± 0.01 | 0.72 ± 0.03 |
| IVS; s (mm) | 1 ± 0.02 | 1.03 ± 0.02 | 1.01 ± 0.02 | 1.02 ± 0.03 |
| LVID; d (mm) | 4.28 ± 0.05 | 4.15 ± 0.12 | 3.87 ± 0.06 | 3.95 ± 0.08 |
| LVID; s (mm) | 3.29 ± 0.07 | 3.16 ± 0.11 | 2.86 ± 0.08 | 2.94 ± 0.09 |
| LV Vol.; d (µL) | 82.66 ± 2.3 | 77.4 ± 4.84 | 65.35 ± 2.43 | 68.95 ± 3.09 |
| LV Vol.; s (µL) | 44.65 ± 2.02 | 41.27 ± 3.41 | 32.25 ± 2.07 | 34.37 ± 2.61 |
| FS (%) | 23.19 ± 0.99 | 24.05 ± 0.93 | 26.45 ± 1.23 | 25.82 ± 1.34 |
| EF (%) | 46.39 ± 1.62 | 47.91 ± 1.6 | 51.87 ± 1.87 | 50.86 ± 2.12 |
| Control | AKAP2 KO | |||
|---|---|---|---|---|
| Sham | 2 Weeks Post-MI | Sham | 2 Weeks Post-MI | |
| Male mice | ||||
| n | 7 | 7 | 7 | 6 |
| Heart Rate (beats/min) | 515 ± 18 | 509 ± 37 | 563 ± 32 | 537 ± 38 ns |
| Body Weight (g) | 29.23 ± 1.12 | 27.82 ± 1.13 | 29.57 ± 1.11 | 28.37 ± 0.83 ns |
| LVPWd (mm) | 0.64 ± 0.03 | 0.62 ± 0.06 | 0.70 ± 0.01 | 0.43 ± 0.07 * |
| IVSd (mm) | 0.68 ± 0.03 | 0.63 ± 0.09 | 0.74 ± 0.01 | 0.42 ± 0.05 ns |
| LVIDd (mm) | 4.05 ± 0.07 | 4.86 ± 0.08 | 4.19 ± 0.09 | 5.89 ± 0.18 *** |
| LV Volume d (µl) | 72.19 ± 2.82 | 111.10 ± 4.14 | 78.66 ± 4.16 | 173.60 ± 12.67 *** |
| %FS | 28.35 ± 1.54 | 14.30 ± 0.87 | 26.73 ± 0.98 | 6.30 ± 1.81 ** |
| %EF | 55.03 ± 2.42 | 30.30 ± 1.65 | 52.48 ± 1.62 | 13.65 ± 3.73 *** |
| Female mice | ||||
| n | 8 | 6 | 6 | 8 |
| Heart Rate (beats/min) | 509 ± 20 | 505 ± 37 | 576 ± 12 | 519 ± 24 ns |
| Body Weight (g) | 22.91 ± 0.62 | 22.23 ± 0.58 | 21.62 ± 0.91 | 23.18 ± 0.44 ns |
| LVPWd (mm) | 0.63 ± 0.02 | 0.65 ± 0.08 | 0.68 ± 0.03 | 0.68 ± 0.14 ns |
| IVSd (mm) | 0.67 ± 0.01 | 0.61 ± 0.02 | 0.75 ± 0.03 | 0.37 ± 0.03 *** |
| LVIDd (mm) | 3.81 ± 0.09 | 4.12 ± 0.17 | 3.76 ± 0.05 | 5.04 ± 0.23 ** |
| LV Volume (µl) | 62.88 ± 3.55 | 79.93 ± 8.60 | 60.47 ± 2.04 | 122.90 ± 13.19 * |
| %FS | 27.74 ± 1.21 | 16.89 ± 3.83 | 32.55 ± 2.76 | 7.30 ± 1.26 * |
| %EF | 54.37 ± 1.87 | 34.89 ± 7.16 | 61.11 ± 4.26 | 16.07 ± 2.68 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maric, D.; Paterek, A.; Delaunay, M.; López, I.P.; Arambasic, M.; Diviani, D. A-Kinase Anchoring Protein 2 Promotes Protection against Myocardial Infarction. Cells 2021, 10, 2861. https://doi.org/10.3390/cells10112861
Maric D, Paterek A, Delaunay M, López IP, Arambasic M, Diviani D. A-Kinase Anchoring Protein 2 Promotes Protection against Myocardial Infarction. Cells. 2021; 10(11):2861. https://doi.org/10.3390/cells10112861
Chicago/Turabian StyleMaric, Darko, Aleksandra Paterek, Marion Delaunay, Irene Pérez López, Miroslav Arambasic, and Dario Diviani. 2021. "A-Kinase Anchoring Protein 2 Promotes Protection against Myocardial Infarction" Cells 10, no. 11: 2861. https://doi.org/10.3390/cells10112861
APA StyleMaric, D., Paterek, A., Delaunay, M., López, I. P., Arambasic, M., & Diviani, D. (2021). A-Kinase Anchoring Protein 2 Promotes Protection against Myocardial Infarction. Cells, 10(11), 2861. https://doi.org/10.3390/cells10112861