
Adenosine Receptor Antagonists to Combat Cancer and to Boost Anti-Cancer Chemotherapy and Immunotherapy

 ,
,  , and
, and
Abstract
:1. Introduction
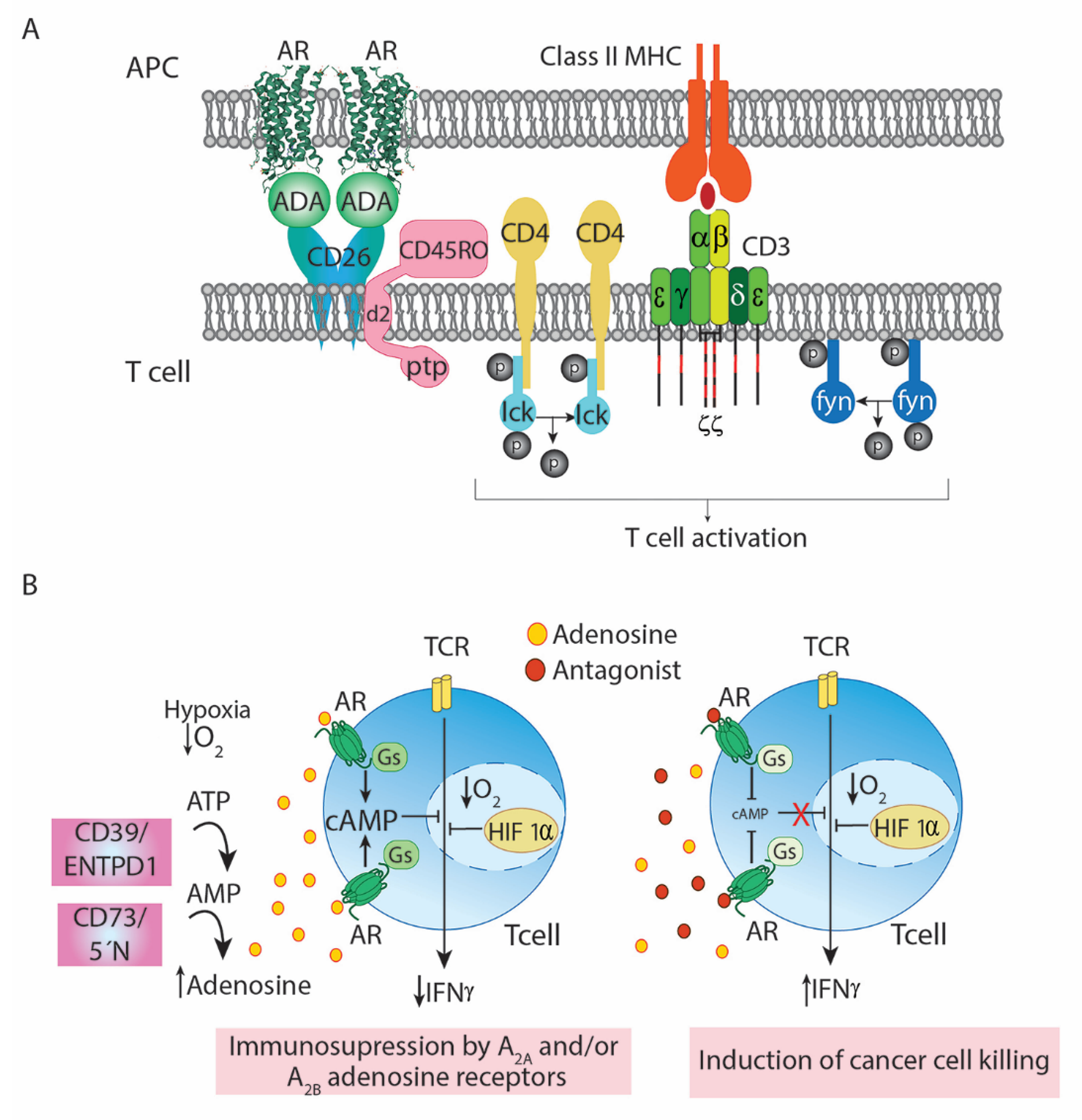
2. Adenosine and Adenosine Receptors
3. Therapeutic Drugs Acting on ARs
4. Adenosine and Adenosine Deaminase in the Cells of the Immune System
5. A2AR Antagonists to Boost Anti-Cancer Immunotherapy
6. AR Ligands in Chemotherapeutic Approaches
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bar-Yehuda, S.; Barer, F.; Volfsson, L.; Fishman, P. Resistance of Muscle to Tumor Metastases: A Role for A3 Adenosine Receptor Agonists. Neoplasia 2001, 3, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. The concise guide to pharmacology 2021/22: G protein-coupled receptors. Br. J. Pharmacol. 2021, 178, S27–S156. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does it Exert its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Spychala, J. Tumor-promoting functions of adenosine. Pharmacol. Ther. 2000, 87, 161–173. [Google Scholar] [CrossRef]
- Stemmer, S.M.; Manojlovic, N.S.; Marinca, M.V.; Petrov, P.; Cherciu, N.; Ganea, D.; Ciuleanu, T.E.; Pusca, I.A.; Beg, M.S.; Purcell, W.T.; et al. Namodenoson in advanced hepatocellular carcinoma and child–pugh B cirrhosis: Randomized placebo-controlled clinical trial. Cancers 2021, 13, 187. [Google Scholar] [CrossRef] [PubMed]
- Boison, D.; Yegutkin, G.G. Adenosine Metabolism: Emerging Concepts for Cancer Therapy. Cancer Cell 2019, 36, 582–596. [Google Scholar] [CrossRef]
- Fishman, P.; Bar-Yehuda, S.; Synowitz, M.; Powell, J.D.; Klotz, K.N.; Gessi, S.; Borea, P.A. Adenosine Receptors and Cancer. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2009; pp. 399–441. [Google Scholar]
- Merighi, S.; Battistello, E.; Giacomelli, L.; Varani, K.; Vincenzi, F.; Borea, P.A.; Gessi, S. Targeting A3 and A2A adenosine receptors in the fight against cancer. Expert Opin. Ther. Targets 2019, 23, 669–678. [Google Scholar] [CrossRef]
- Drury, A.N.; Szent-Györgyi, A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J. Physiol. 1929, 68, 213–237. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.M.; Berne, R.M. Coronary Vasodilator Properties of Purine and Pyrimidine Derivatives. Circ. Res. 1956, 4, 343–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berne, R.M. The Role of Adenosine in the Regulation of Coronary Blood Flow. Circ. Res. 1980, 47, 807–813. [Google Scholar] [CrossRef] [Green Version]
- Jenner, P.; Mori, A.; Hauser, R.; Morelli, M.; Fredholm, B.B.; Chen, J.F. Adenosine, adenosine A 2A antagonists, and Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 406–413. [Google Scholar] [CrossRef]
- Jenner, P. An Overview of adenosine A2A receptor antagonists in Parkinson’s disease. Int. Rev. Neurobiol. 2014, 119, 71–86. [Google Scholar] [CrossRef]
- Mizuno, Y.; Kondo, T. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov. Disord. 2013, 28, 1138–1141. [Google Scholar] [CrossRef] [Green Version]
- Saki, M.; Yamada, K.; Koshimura, E.; Sasaki, K.; Kanda, T. In vitro pharmacological profile of the A2A receptor antagonist istradefylline. Naunyn Schmiedeberg’s Arch. Pharmacol. 2013, 386, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Mizuno, Y.; Japanese Istradefylline Study Group. A long-term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin. Neuropharmacol. 2015, 38, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, M.D. The future of pharmacologic stress: Selective a2a adenosine receptor agonists. Am. J. Cardiol. 2004, 94, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P.; Curran, M.P. Regadenoson. Am. J. Cardiovasc. Drugs 2012, 10, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Hendel, R.C.; Bateman, T.M.; Cerqueira, M.D.; Iskandrian, A.E.; Leppo, J.A.; Blackburn, B.; Mahmarian, J.J. Initial clinical experience with regadenoson, a novel selective A 2A agonist for pharmacologic stress single-photon emission computed tomography myocardial perfusion imaging. J. Am. Coll. Cardiol. 2005, 46, 2069–2075. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.; Weingart, J.; Nduom, E.K.; Harfi, T.T.; George, R.T.; McAreavey, D.; Ye, X.; Anders, N.M.; Peer, C.; Figg, W.D.; et al. The effect of an adenosine A2A agonist on intra-tumoral concentrations of temozolomide in patients with recurrent glioblastoma. Fluids Barriers CNS 2018, 15, 2. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.; George, R.T.; Lodge, M.A.; Piotrowski, A.; Wahl, R.L.; Gujar, S.K.; Grossman, S.A. The effect of regadenoson on the integrity of the human blood–brain barrier, a pilot study. J. Neurooncol. 2017, 132, 513–519. [Google Scholar] [CrossRef]
- Byrne, E.M.; Johnson, J.; McRae, A.F.; Nyholt, D.R.; Medland, S.E.; Gehrman, P.R.; Heath, A.C.; Madden, P.A.F.; Montgomery, G.W.; Chenevix-Trench, G.; et al. A Genome-Wide Association Study of Caffeine-Related Sleep Disturbance: Confirmation of a Role for a Common Variant in the Adenosine Receptor. Sleep 2012, 35, 967–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, I.; Albasanz, J.L.; Martín, M. Effect of Caffeine Chronically Consumed During Pregnancy on Adenosine A 1 and A 2A Receptors Signaling in Both Maternal and Fetal Heart from Wistar Rats. J. Caffeine Res. 2014, 4, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Osswald, H. Adenosine Receptors and the Kidney. In Adenosine Receptors in Health and Disease; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 193, pp. 443–470. [Google Scholar] [CrossRef]
- Spielman, W.S.; Arend, L.J. Adenosine receptors and signaling in the kidney. Hypertension 1991, 17, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Pawelczyk, T.; Grden, M.; Rzepko, R.; Sakowicz, M.; Szutowicz, A. Region-Specific Alterations of Adenosine Receptors Expression Level in Kidney of Diabetic Rat. Am. J. Pathol. 2005, 167, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Vitzthum, H.; Weiss, B.; Bachleitner, W.; Krämer, B.K.; Kurtz, A. Gene expression of adenosine receptors along the nephron. Kidney Int. 2004, 65, 1180–1190. [Google Scholar] [CrossRef] [Green Version]
- Blanco, J.; Canela, E.I.; Sayós, J.; Mallol, J.; Lluis, C.; Franco, R. Adenine nucleotides and adenosine metabolism in pig kidney proximal tubule membranes. J. Cell. Physiol. 1993, 157, 77–83. [Google Scholar] [CrossRef]
- Blanco, J.; Canela, E.I.; Mallol, J.; Lluís, C.; Franco, R. Characterization of adenosine receptors in brush-border membranes from pig kidney. Br. J. Pharmacol. 1992, 107, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Kazemi, M.H.; Raoofi Mohseni, S.; Hojjat-Farsangi, M.; Anvari, E.; Ghalamfarsa, G.; Mohammadi, H.; Jadidi-Niaragh, F. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J. Cell. Physiol. 2018, 233, 2032–2057. [Google Scholar] [CrossRef] [Green Version]
- De Araújo, J.B.; Kerkhoff, V.V.; de Oliveira Maciel, S.F.V.; de Resende e Silva, D.T. Targeting the purinergic pathway in breast cancer and its therapeutic applications. Purinergic Signal. 2021, 17, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Seitz, L.; Jin, L.; Leleti, M.; Ashok, D.; Jeffrey, J.; Rieger, A.; Tiessen, R.G.; Arold, G.; Tan, J.B.L.; Powers, J.P.; et al. Safety, tolerability, and pharmacology of AB928, a novel dual adenosine receptor antagonist, in a randomized, phase 1 study in healthy volunteers. Investig. New Drugs 2019, 37, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.; Williams, C.C.; Creelan, B.C.; Tanvetyanon, T.; Gray, J.E.; Haura, E.B.; Thapa, R.; Chen, D.-T.; Beg, A.A.; Boyle, T.A.; et al. Phase I/II study of the A2AR antagonist NIR178 (PBF-509), an oral immunotherapy, in patients (pts) with advanced NSCLC. J. Clin. Oncol. 2018, 36, 9089. [Google Scholar] [CrossRef]
- Knudsen, B.B.; Dissing, J. Adenosine deaminase deficiency in a child with severe combined immunodeficiency. Clin. Genet. 1973, 4, 344–347. [Google Scholar] [CrossRef]
- Booth, C.; Gaspar, H.B. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biologics 2009, 3, 349–358. [Google Scholar] [CrossRef]
- Hershfield, M.S.; Kurtzberg, J.; Aiyar, V.N.; Suh, E.J.; Schiff, R. Abnormalities in S-Adenosylhomocysteine Hydrolysis, ATP Catabolism, and Lymphoid Differentiation in Adenosine Deaminase Deficiency. Ann. N. Y. Acad. Sci. 1985, 451, 78–86. [Google Scholar] [CrossRef]
- Blaese, R.M.; Culver, K.W.; Miller, A.D.; Carter, C.S.; Fleisher, T.; Clerici, M.; Shearer, G.; Chang, L.; Chiang, Y.; Tolstoshev, P.; et al. T lymphocyte-directed gene therapy for ADA-SCID: Initial trial results after 4 years. Science 1995, 270, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Hirschhorn, R. Adenosine deaminase deficiency and immunodeficiencies. Fed. Proc. 1977, 36, 2166–2170. [Google Scholar]
- Rieger, C.H.L.; Lustig, J.V.; Hirschhorn, R.; Rothberg, R.M. Reconstitution of T-cell function in severe combined immunodeficiency disease following transplantation of early embryonic liver cells. J. Pediatr. 1977, 90, 707–712. [Google Scholar] [CrossRef]
- Polmar, S.H.; Stern, R.C.; Schwartz, A.L.; Wetzler, E.M.; Chase, P.A.; Hirschhorn, R. Enzyme Replacement Therapy for Adenosine Deaminase Deficiency and Severe Combined Immunodeficiency. N. Engl. J. Med. 1976, 295, 1337–1343. [Google Scholar] [CrossRef]
- Hirschhorn, R.; Beratis, N.; Rosen, F.S. Characterization of residual enzyme activity in fibroblasts from patients with adenosine deaminase deficiency and combined immunodeficiency: Evidence for a mutant enzyme. Proc. Natl. Acad. Sci. USA 1976, 73, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Polmar, S.H.; Wetzler, E.M.; Stern, R.C.; Hirschhorn, R. Restoration of in-vitro lymphocyte responses with exogenous adenosine deaminase in a patient with severe combined immunodeficiency. Lancet 1975, 306, 743–746. [Google Scholar] [CrossRef]
- Hirschhorn, R. Therapy of Genetic Disorders. N. Engl. J. Med. 1987, 316, 623–624. [Google Scholar] [CrossRef]
- Hirschhorn, K.; Hirschhorn, R.; Hirschhorn, J.N. A Conversation with Kurt and Rochelle Hirschhorn. Annu. Rev. Genom. Hum. Genet. 2017, 18, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Hershfield, M.S.; Buckley, R.H.; Greenberg, M.L.; Melton, A.L.; Schiff, R.; Hatem, C.; Kurtzberg, J.; Markert, M.L.; Kobayashi, R.H.; Kobayashi, A.L.; et al. Treatment of Adenosine Deaminase Deficiency with Polyethylene Glycol–Modified Adenosine Deaminase. N. Engl. J. Med. 1987, 316, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Grunebaum, E.; Mazzolari, E.; Porta, F.; Dallera, D.; Atkinson, A.; Reid, B.; Notarangelo, L.D.; Roifman, C.M. Bone marrow transplantation for severe combined immune deficiency. J. Am. Med. Assoc. 2006, 295, 508–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Shepherd, R.K.; Duling, B.R.; Linden, J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J. Clin. Investig. 1997, 100, 2849–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Pacheco, R.; Gatell, J.M.; Gallart, T.; Lluis, C. Enzymatic and extraenzymatic role of adenosine deaminase 1 in T-cell-dendritic cell contacts and in alterations of the immune function. Crit. Rev. Immunol. 2007, 27, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Climent, N.; Martinez-Navio, J.M.; Gil, C.; Garcia, F.; Rovira, C.; Hurtado, C.; Miralles, L.; Gatell, J.M.; Gallart, T.; Mallol, J.; et al. Adenosine deaminase enhances T-cell response elicited by dendritic cells loaded with inactivated HIV. Immunol. Cell Biol. 2009, 87, 634–639. [Google Scholar] [CrossRef]
- Martinez-Navio, J.M.J.M.; Climent, N.; Pacheco, R.; Garcia, F.; Plana, M.; Nomdedeu, M.; Oliva, H.; Rovira, C.; Miralles, L.; Gatell, J.M.J.M.; et al. Immunological dysfunction in HIV-1-infected individuals caused by impairment of adenosine deaminase-induced costimulation of T-cell activation. Immunology 2009, 128, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Casanova, V.; Naval-Macabuhay, I.; Massanella, M.; Rodríguez-García, M.; Blanco, J.; Gatell, J.M.; García, F.; Gallart, T.; Lluis, C.; Mallol, J.; et al. Adenosine deaminase enhances the immunogenicity of human dendritic cells from healthy and HIV-infected individuals. PLoS ONE 2012, 7, e51287. [Google Scholar] [CrossRef] [Green Version]
- Naval-Macabuhay, I.; Casanova, V.; Navarro, G.; Garcia, F.; Leon, A.; Miralles, L.; Rovira, C.; Martinez-Navio, J.M.; Gallart, T.; Mallol, J.; et al. Adenosine deaminase regulates Treg expression in autologous T cell-dendritic cell cocultures from patients infected with HIV-1. J. Leukoc. Biol. 2016, 99, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Navio, J.M.; Casanova, V.; Pacheco, R.; Naval-Macabuhay, I.; Climent, N.; Garcia, F.; Gatell, J.M.; Mallol, J.; Gallart, T.; Lluis, C.; et al. Adenosine deaminase potentiates the generation of effector, memory, and regulatory CD4+ T cells. J. Leukoc. Biol. 2011, 89, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Valenzuela, A.; Lluis, C.; Blanco, J. Enzymatic and extraenzymatic role of ecto-adenosine deaminase in lymphocytes. Immunol. Rev. 1998, 161, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Centelles, J.J.; Huguet, J.; Echevarne, F.; Colomer, D.; Vives-Corrons, J.L.; Franco, R. Surface expression of adenosine deaminase in mitogen-stimulated lymphocytes. Clin. Exp. Immunol. 1993, 93, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Ciruela, F.; Saura, C.; Canela, E.I.E.I.; Mallol, J.; Lluis, C.; Franco, R. Adenosine deaminase affects ligand-induced signalling by interacting with cell surface adenosine receptors. FEBS Lett. 1996, 380, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, R.; Martinez-Navio, J.M.M.; Lejeune, M.; Climent, N.; Oliva, H.; Gatell, J.M.M.; Gallart, T.; Mallol, J.; Lluis, C.; Franco, R. CD26, adenosine deaminase, and adenosine receptors mediate costimulatory signals in the immunological synapse. Proc. Natl. Acad. Sci. USA 2005, 102, 9583–9588. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Navio, J.M.; Climent, N.; Gallart, T.; Lluis, C.; Franco, R. An old enzyme for current needs: Adenosine deaminase and a dendritic cell vaccine for HIV. Immunol. Cell Biol. 2012, 90, 594–600. [Google Scholar] [CrossRef]
- Shimoyama, M.; Tobinai, K.; Yamaguchi, K.; Hirashima, K.; Itoh, S.; Konishi, H.; Mikuni, C.; Togawa, A.; Hotta, T.; Toyoda, N.; et al. Treatment of Hairy Cell Leukemia with Deoxycoformycin (YK-176). Jpn. J. Clin. Oncol. 1992, 22, 406–410. [Google Scholar] [CrossRef]
- Gracia, E.; Farré, D.; Cortés, A.; Ferrer-Costa, C.; Orozco, M.; Mallol, J.; Lluís, C.; Canela, E.I.; McCormick, P.J.; Franco, R.; et al. The catalytic site structural gate of adenosine deaminase allosterically modulates ligand binding to adenosine receptors. FASEB J. 2013, 27, 1048–1061. [Google Scholar] [CrossRef]
- Gracia, E.; Cortés, A.; Meana, J.J.; García-Sevilla, J.; Herhsfield, M.S.; Canel, E.I.; Mallol, J.; Lluís, C.; Franco, R.; Casadó, V. Human adenosine deaminase as an allosteric modulator of human A1 adenosine receptor: Abolishment of negative cooperativity for [3H](R)-pia binding to the caudate nucleus. J. Neurochem. 2008, 107, 161–170. [Google Scholar] [CrossRef]
- Franco, R.; Casadó, V.; Ciruela, F.; Saura, C.; Mallol, J.; Canela, E.I.; Lluis, C. Cell surface adenosine deaminase: Much more than an ectoenzyme. Prog. Neurobiol. 1997, 52, 283–294. [Google Scholar] [CrossRef]
- Kjaergaard, J.; Hatfield, S.; Jones, G.; Ohta, A.; Sitkovsky, M. A 2A Adenosine Receptor Gene Deletion or Synthetic A 2A Antagonist Liberate Tumor-Reactive CD8 + T Cells from Tumor-Induced Immunosuppression. J. Immunol. 2018, 201, 782–791. [Google Scholar] [CrossRef] [Green Version]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [Green Version]
- Willingham, S.B.; Hotson, A.N.; Miller, R.A. Targeting the A2AR in cancer; early lessons from the clinic. Curr. Opin. Pharmacol. 2020, 53, 126–133. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Chern, Y.; Franco, R.; Sitkovsky, M. Aspects of the general biology of adenosine A2A signaling. Prog. Neurobiol. 2007, 83, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.V. Lessons from the A2A adenosine receptor antagonist– enabled tumor regression and survival in patients with treatment-refractory renal cell cancer. Cancer Discov. 2020, 10, 16–19. [Google Scholar] [CrossRef]
- Sitkovsky, M.V.; Hatfield, S.; Abbott, R.; Belikoff, B.; Lukashev, D.; Ohta, A. Hostile, Hypoxia-A2-Adenosinergic Tumor Biology as the Next Barrier to Overcome for Tumor Immunologists. Cancer Immunol. Res. 2014, 2, 598–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, S.M.; Sitkovsky, M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1α driven immunosuppression and improve immunotherapies of cancer. Curr. Opin. Pharmacol. 2016, 29, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.M.; Hellmann, M.D.; Shepard, D.R.; et al. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov. 2020, 10, 40–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mediavilla-Varela, M.; Castro, J.; Chiappori, A.; Noyes, D.; Hernandez, D.C.; Allard, B.; Stagg, J.; Antonia, S.J. A Novel Antagonist of the Immune Checkpoint Protein Adenosine A2a Receptor Restores Tumor-Infiltrating Lymphocyte Activity in the Context of the Tumor Microenvironment. Neoplasia 2017, 19, 530–536. [Google Scholar] [CrossRef]
- Naing, A.; Gainor, J.F.; Gelderblom, H.; Forde, P.M.; Butler, M.O.; Lin, C.C.; Sharma, S.; Ochoa De Olza, M.; Varga, A.; Taylor, M.; et al. A first-in-human phase 1 dose escalation study of spartalizumab (PDR001), an anti-PD-1 antibody, in patients with advanced solid tumors. J. Immunother. Cancer 2020, 8, e000530. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, K.; Sitkovsky, M.V. Adenosine A2A receptor is involved in cell surface expression of A2B receptor. J. Biol. Chem. 2010, 285, 39271–39288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, S.; Navarro, G.; Borroto-Escuela, D.; Seibt, B.F.; Ammon, C.; de Filippo, E.; Danish, A.; Lacher, S.K.; Červinková, B.; Rafehi, M.; et al. Adenosine A2A receptor ligand recognition and signaling is blocked by A2B receptors. Oncotarget 2018, 9, 13593–13611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnad, T.; Navarro, G.; Lahesmaa, M.; Reverte-Salisa, L.; Copperi, F.; Cordomi, A.; Naumann, J.; Hochhäuser, A.; Haufs-Brusberg, S.; Wenzel, D.; et al. Adenosine/A2B Receptor Signaling Ameliorates the Effects of Aging and Counteracts Obesity. Cell Metab. 2020, 32, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Peppelenbosch, M.P.; Sprengers, D.; Kwekkeboom, J. TIGIT, the Next Step Towards Successful Combination Immune Checkpoint Therapy in Cancer. Front. Immunol. 2021, 12, 699895. [Google Scholar] [CrossRef]

{kind=link}
| Target * | +/− Combination Therapy | Company /Institution | Cancer Type | Clinicaltrials.Gov Identifier | Study Phase |
|---|---|---|---|---|---|
| A2AR | Inupadenant + EOS-448 (anti-TIGIT mAB) | iTeos Therapeutics | AST | NCT05060432 | 1, 2 (NR) |
| A2AR | Ciforadenant +/− atezolizumab (mAb against PD-1) | Corvus Pharmaceuticals, Inc. | Prostate (advanced/incurable) | NCT02655822 | 1, 1b (R, OL) |
| A2AR | PBF-509 +/− antibody against PD-1 | Palobiofarma SL | NSLC (advanced) | NCT02403193 | 1, 2 (NR, OL) |
| A2AR | Taminadenant + Spartalizumab (mAb against PD-1) + DFF332 (Hif2α inhibitor) | Novartis Pharmaceuticals | Renal (advanced) | NCT04895748 | 1, 1b (NR, OL) |
| A2AR | NIR178 +/− Spartalizumab (mAb against PD-1 | Novartis Pharmaceuticals | Solid tumor, NHL | NCT03207867 | 2 (NR, OL) |
| A2AR | PBF -509 | Palobiofarma SL | NA (safety assessment) | NCT01691924 | 1 (R) |
| A2AR | PBF -509 | Fundació Institut de Recerca de l’Hospital de la Santa Creu i Sant Pau | Cancer (general) | NCT02111330 | 1 (R, DB, PC) |
| A2AR | Ciforadenant + daratumumab (mAb against CD38) | Corvus Pharmaceuticals, Inc. | Multiple Myeloma | NCT04280328 | 1 (OL) |
| A2AR | TT-10 | Tarus Therapeutics | Renal, prostate, NSCLC (AST) | NCT04969315 | 1, 2 (OL) |
| A2BR | TT-4 | Tarus Therapeutics | Gastrointestinal, hepatocellular, prostate | NCT04976660 | 1, 2 (OL) |
| A2AR/A2BR | Arm 1: AB928/Etrumadenant + zimberelimab (mAb against PD-1) + enzalutamide Arm 2: AB928/Etrumadenant + zimberelimab (mAb against PD-1) + docetaxel | Arcus Biosciences, Inc. | Prostate (advanced/incurable) | NCT04381832 | 1, 2 (R, OL) |
| A2AR/A2BR | AB928/Etrumadenant (dual antagonist) + zimberelimab (mAb against PD-1) | Arcus Biosciences, Inc. | Advanced malignancies | NCT03629756 | 1 (NR, OL) |
| A2AR/A2BR | AB928/Etrumadenant (dual antagonist) + Cisplatin/Radiation Therapy + Zimberelimab (mAb against PD-1) | Jennifer Choe in collaboration with Arcus Biosciences Inc | Head and neck | NCT04892875 | 1 (NR, OL) |
| A2AR/A2BR | Arm 1. AB928/Etrumadenant (dual antagonist) + zimberelimab (mAb against PD-1) + standard chemotherapeutic regime (mFOLFOX-6 + bevacizumab) Arm 2: AB928/Etrumadenant zimberelimab (mAb against PD-1) + AB680 (CD73 inhibitor) | Arcus Biosciences Inc. | Colon (metastatic) | NCT04660812 | 1, 2 (R, OL) |
| A2AR/A2BR | AB928/Etrumadenant (dual antagonist) + Carboplatin and Pemetrexed +/− Zimberelimab (mAb against PD-1) | Arcus Biosciences Inc. | Lung | NCT03846310 | 1 (NR, OL) |
| A2AR/A2BR | Arm A: AB928/Etrumadenant (dual antagonist) + Pegylated liposomal doxorubicin (PLD) Arm B: Etrumadenant + nanoparticle albumin-bound paclitaxel (NP) Arm C: Etrumadenant +PLD + IPI-549 (phosphoinositide-3-kinase-gamma inhibitor) | Arcus Biosciences Inc. | Breast (TN) or gynecologic | NCT03719326 | 1 (NR, OL) |
| A2AR/A2BR | AB928/Etrumadenant (dual antagonist) + mFOLFOX | Arcus Biosciences Inc. | Gastrointestinal | NCT03720678 | 1 (NR, OL) |
| A2BR | PBF-1129 | Palobiofarma SL | NSCLC | NCT03274479 | 1 (NR, OL) |
| A2AR | Arm 1: AZD4635 + Durvalumab (mAb against PD-L1) Arm 2: AZD4635 + Oleclumab (mAb against CD73) Arm 3: AZD4635 + Durvalumab + Oleclumab | AstraZeneca | Prostate | NCT04089553 | 1, 2 (NR, OL) |
| A2BR | TT-4 | Tarus Therapeutics | Gastrointestinal, hepatocellular, prostate | NCT04976660 | 1, 2 (OL) |
| A2BR | TT-4 | Tarus Therapeutics | Gastrointestinal, hepatocellular, prostate | NCT04976660 | 1, 2 (OL) |
| A2BR | TT-4 | Tarus Therapeutics | Gastrointestinal, hepatocellular, prostate | NCT04976660 | 1, 2 (OL) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, R.; Rivas-Santisteban, R.; Navarro, G.; Reyes-Resina, I. Adenosine Receptor Antagonists to Combat Cancer and to Boost Anti-Cancer Chemotherapy and Immunotherapy. Cells 2021, 10, 2831. https://doi.org/10.3390/cells10112831
Franco R, Rivas-Santisteban R, Navarro G, Reyes-Resina I. Adenosine Receptor Antagonists to Combat Cancer and to Boost Anti-Cancer Chemotherapy and Immunotherapy. Cells. 2021; 10(11):2831. https://doi.org/10.3390/cells10112831
Chicago/Turabian StyleFranco, Rafael, Rafael Rivas-Santisteban, Gemma Navarro, and Irene Reyes-Resina. 2021. "Adenosine Receptor Antagonists to Combat Cancer and to Boost Anti-Cancer Chemotherapy and Immunotherapy" Cells 10, no. 11: 2831. https://doi.org/10.3390/cells10112831
APA StyleFranco, R., Rivas-Santisteban, R., Navarro, G., & Reyes-Resina, I. (2021). Adenosine Receptor Antagonists to Combat Cancer and to Boost Anti-Cancer Chemotherapy and Immunotherapy. Cells, 10(11), 2831. https://doi.org/10.3390/cells10112831