
Progesterone Induced Blocking Factor Reduces Hypertension and Placental Mitochondrial Dysfunction in Response to sFlt-1 during Pregnancy

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Infusion of sFlt-1/PIBF into Pregnant Sprague Dawley Rats
2.3. Isolation of Mitochondria
2.4. Mitochondrial Respiration
2.5. Mitochondrial ROS
2.6. Endothelial Mitochondrial ROS
2.7. Statistical Analysis
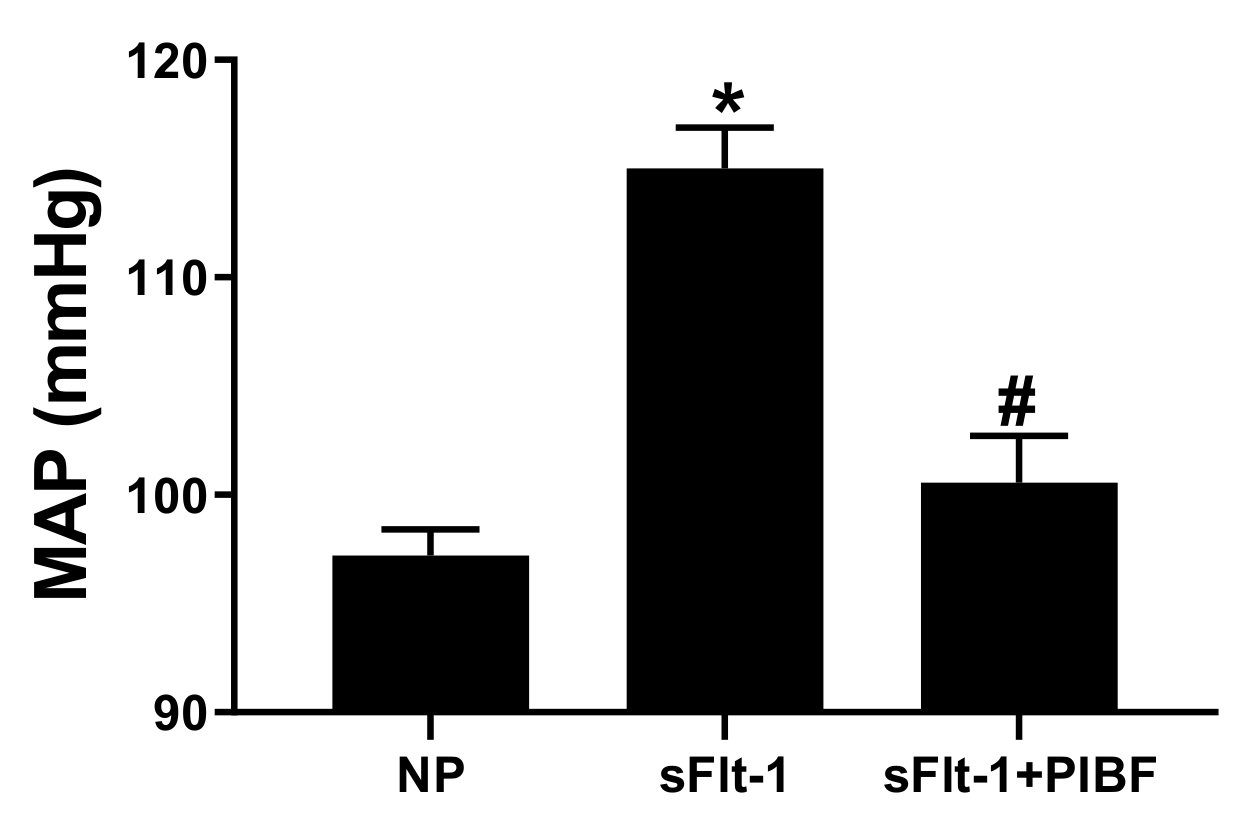
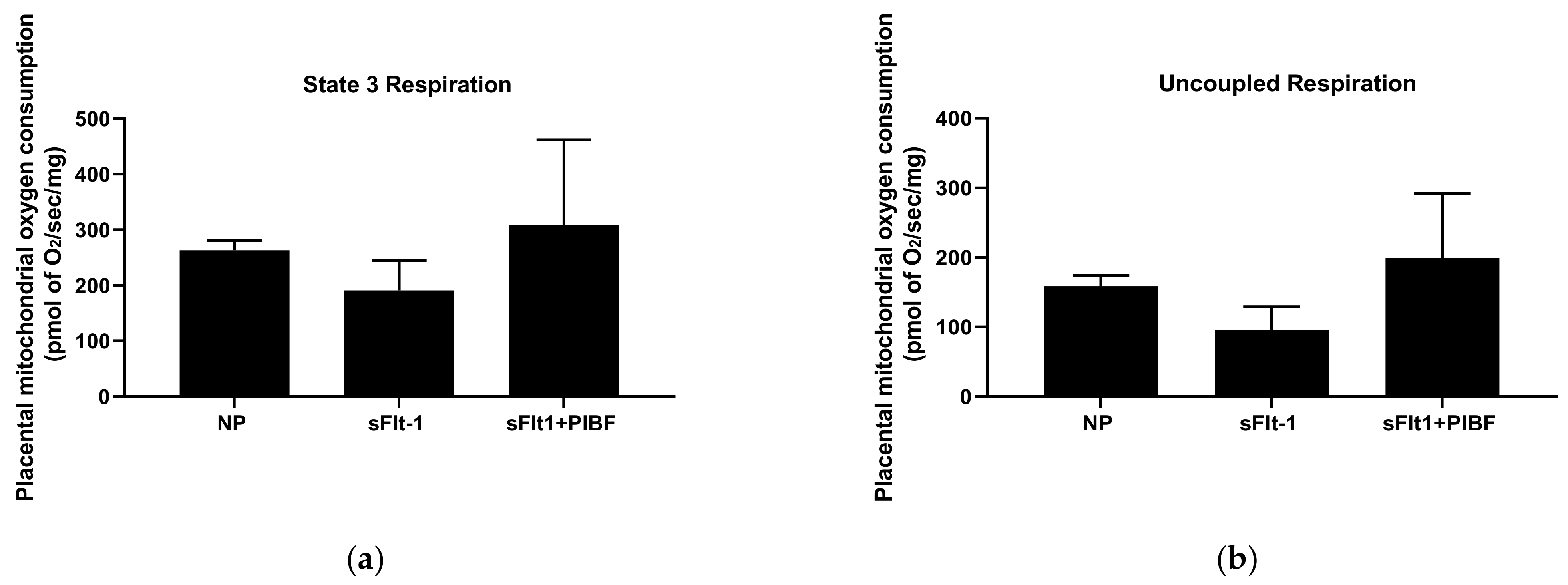
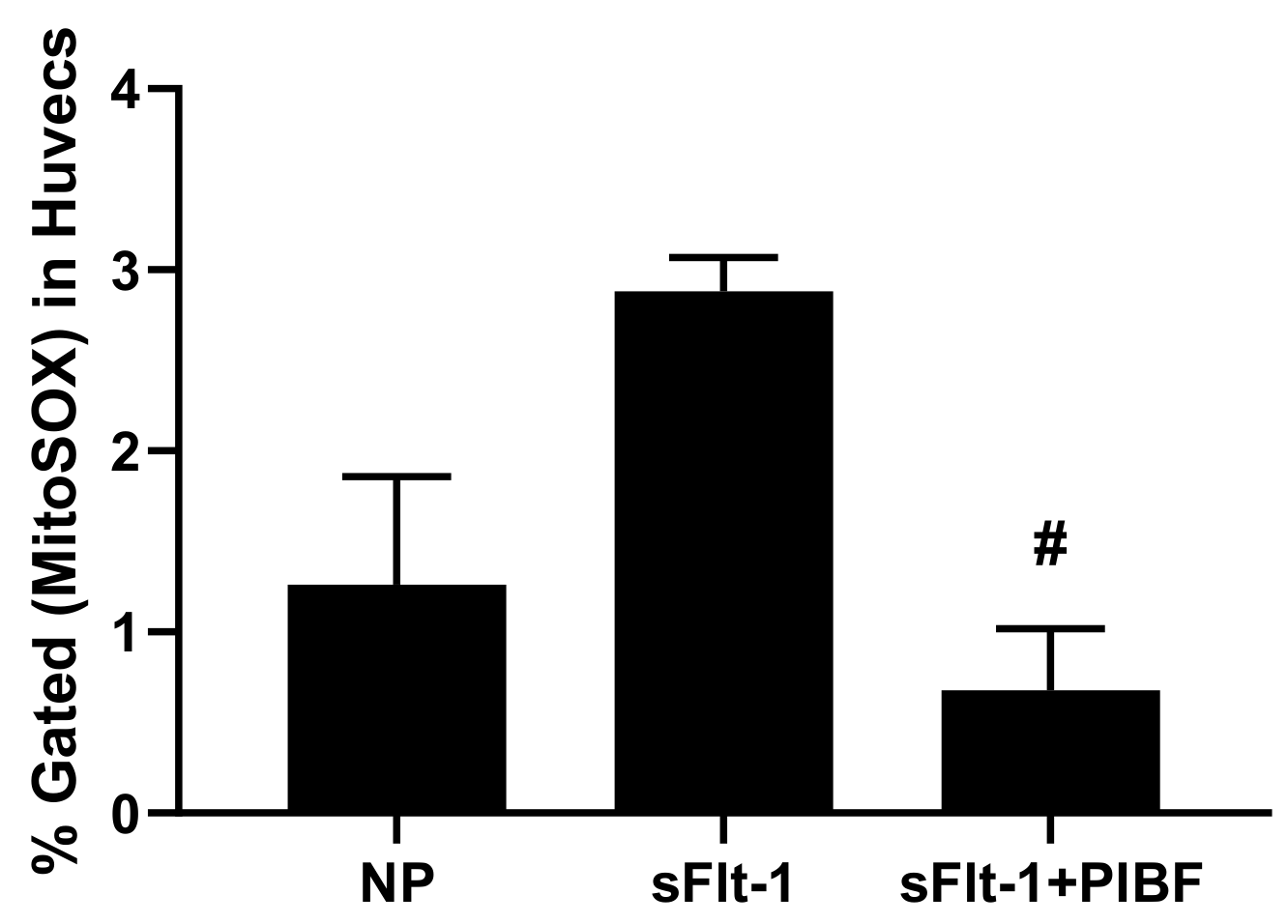
3. Results
Figures, Tables, and Schemes





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Body Weights (g) | Placental Weights (g) | Fetal Weights (g) | |
|---|---|---|---|
| NP | 317 ± 9.32 | 0.57 ± 0.02 | 2.27 ± 0.06 |
| sFlt-1 | 322 ± 8.07 | 0.56 ± 0.01 | 2.31 ± 0.07 |
| sFlt-1+PIBF | 303 ± 6 | 0.54 ± 0.02 | 2.19 ± 0.06 |
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cornelius, D.C.; Cottrell, J.; Amaral, L.M.; LaMarca, B. Inflammatory mediators: A causal link to hypertension during preeclampsia. Br. J. Pharmacol. 2019, 176, 1914–1921. [Google Scholar] [CrossRef]
- Herse, F.; LaMarca, B. Angiotensin II Type 1 Receptor Autoantibody (AT1-AA)-Mediated Pregnancy Hypertension. Am. J. Reprod. Immunol. 2012, 69, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Cífková, R. Hypertension in pregnancy. In Manual of Hypertension of the European Society of Hypertension; CRC Press: Boca Raton, FL, USA, 2021; ISBN 9780367778613. [Google Scholar]
- Roberts, J.M.; Pearson, G.; Cutler, J.; Lindheimer, M. Summary of the NHLBI working group on research on hypertension during pregnancy. Hypertension 2003, 41, 437–445. [Google Scholar] [CrossRef]
- Sibai, B.; Dekker, G.; Kupferminc, M. Pre-eclampsia. Lancet 2005, 365, 785–799. [Google Scholar] [CrossRef]
- Matsubara, K.; Higaki, T.; Matsubara, Y.; Nawa, A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int. J. Mol. Sci. 2015, 16, 4600–4614. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.; Sargent, I.L. Latest advances in understanding preeclampsia. Science 2005, 308, 1592–1594. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar] [PubMed]
- Sandrim, V.C.; Palei, A.C.T.; Luizon, M.; Izidoro-Toledo, T.; Cavalli, R.; Tanus-Santos, J.E. eNOS haplotypes affect the responsiveness to antihypertensive therapy in preeclampsia but not in gestational hypertension. Pharm. J. 2009, 10, 40–45. [Google Scholar] [CrossRef]
- Brewer, J.; Liu, R.; Lu, Y.; Scott, J.; Wallace, K.; Wallukat, G.; Moseley, J.; Herse, F.; Dechend, R.; Martin, J.N.; et al. Endothelin-1, oxidative stress, and endogenous angiotensin II: Mechanisms of angiotensin II type I receptor autoantibody–enhanced renal and blood pressure response during pregnancy. Hypertension 2013, 62, 886–892. [Google Scholar] [CrossRef]
- Murphy, S.R.; LaMarca, B.B.D.; Parrish, M.; Cockrell, K.; Granger, J.P. Control of soluble fms-like tyrosine-1 (sFlt-1) production response to placental ischemia/hypoxia: Role of tumor necrosis factor-α. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R130–R135. [Google Scholar] [CrossRef]
- Wang, A.; Rana, S.; Karumanchi, S.A. Preeclampsia: The Role of Angiogenic Factors in Its Pathogenesis. Physiology 2009, 24, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.M.; Cunningham, M.W., Jr.; Cornelius, D.C.; LaMarca, B. Preeclampsia: Long-term consequences for vascular health. Vasc. Health Risk Manag. 2015, 11, 403. [Google Scholar]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Tam, K.B.T.; Lamarca, B.; Arany, M.; Cockrell, K.; Fournier, L.; Murphy, S.; Martin, J.N.; Granger, J.P. Role of reactive oxygen species during hypertension in response to chronic antiangiogenic factor (sFlt-1) excess in pregnant rats. Am. J. Hypertens. 2011, 24, 110–113. [Google Scholar] [CrossRef][Green Version]
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative stress in preeclampsia and placental diseases. Int. J. Mol. Sci. 2018, 19, 1496. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Sultana, Z.; Maiti, K.; Aitken, J.; Morris, J.; Dedman, L.; Smith, R. Oxidative stress, placental ageing-related pathologies and adverse pregnancy outcomes. Am. J. Reprod. Immunol. 2017, 77, e12653. [Google Scholar] [CrossRef]
- Eirin, A.; Lerman, A.; Lerman, L.O. Mitochondrial injury and dysfunction in hypertension-induced cardiac damage. Eur. Heart J. 2014, 35, 3258–3266. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Vaka, V.R.; McMaster, K.M.; Cunningham, M.W., Jr.; Ibrahim, T.; Hazlewood, R.; Usry, N.; Cornelius, D.C.; Amaral, L.M.; Lamarca, B. Role of mitochondrial dysfunction and reactive oxygen species in mediating hypertension in the reduced uterine perfusion pressure rat model of preeclampsia. Hypertension 2018, 72, 703–711. [Google Scholar] [CrossRef]
- Cunningham, M.W.; Jayaram, A.; Deer, E.; Amaral, L.M.; Vaka, V.R.; Ibrahim, T.; Cornelius, D.C.; Lamarca, B. Tumor necro-sis factor alpha (TNF-α) blockade improves natural killer cell (NK) activation, hypertension, and mitochondrial oxidative stress in a preclinical rat model of preeclampsia. Hypertens. Pregnancy 2020, 39, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, A.; Deer, E.; Amaral, L.M.; Campbell, N.; Vaka, V.R.; Cunningham, M.; Ibrahim, T.; Cornelius, D.C.; LaMarca, B.B. The role of tumor necrosis factor in triggering activation of natural killer cell, multi-organ mitochondrial dysfunction and hypertension during pregnancy. Pregnancy Hypertens. 2021, 24, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.; Reeve, K.E.; Amaral, L.M.; Vaka, V.R.; Franks, M.; Campbell, N.; Fitzgerald, S.; Herrock, O.T.; Ibrahim, T.; Cornelius, D.C.; et al. CD4+ T cells cause renal and placental mitochondrial oxidative stress as mechanisms of hypertension in response to placental ischemia. Am. J. Physiol. Physiol. 2021, 320, F47–F54. [Google Scholar] [CrossRef]
- Raghupathy, R.; Kalinka, J. Cytokine imbalance in pregnancy complications and its modulation. Front. Biosci. 2008, 13, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Szekeres-Bartho, J.; Autran, B.; Debre, P.; Andreu, G.; Denver, L.; Chaouat, G. Immunoregulatory effects of a suppressor factor from healthy pregnant women’s lymphocytes after progesterone induction. Cell. Immunol. 1989, 122, 281–294. [Google Scholar] [CrossRef]
- Szekeres-Bartho, J.; Faust, Z.; Varga, P.; Szereday, L.; Kelemen, K. The immunological pregnancy protective effect of progesterone is manifested via controlling cytokine production. Am. J. Reprod. Immunol. 1996, 35, 348–351. [Google Scholar] [CrossRef]
- Szekeres-Bartho, J.; Barakonyi, A.; Par, G.; Polgar, B.; Palkovics, T.; Szereday, L. Progesterone as an immunomodulatory molecule. Int. Immunopharmacol. 2001, 1, 1037–1048. [Google Scholar] [CrossRef]
- Kiprono, L.V.; Wallace, K.; Moseley, J.; Martin, J., Jr.; LaMarca, B. Progesterone blunts vascular endothelial cell secretion of endothelin-1 in response to placental ischemia. Am. J. Obstet. Gynecol. 2013, 209, 44.e1–44.e6. [Google Scholar] [CrossRef]
- Cottrell, J.N.; Witcher, A.C.; Comley, K.M.; Cunningham, M.W., Jr.; Ibrahim, T.; Cornelius, D.C.; LaMarca, B.D.; Amaral, L.M. Progesterone-induced blocking factor improves blood pressure, inflammation, and pup weight in response to reduced uterine perfusion pressure (RUPP). Am. J. Physiol. Integr. Comp. Physiol. 2021, 320, R719–R727. [Google Scholar] [CrossRef]
- Baltajian, K.; Hecht, J.L.; Wenger, J.B.; Salahuddin, S.; Verlohren, S.; Perschel, F.H.; Zsengeller, Z.K.; Thadhani, R.; Karumanchi, S.A.; Rana, S. Placental lesions of vascular insufficiency are associated with anti-angiogenic state in women with preeclampsia. Hypertens. Pregnancy 2014, 33, 427–439. [Google Scholar] [CrossRef]
- Rebelato, H.J.; Esquisatto, M.A.M.; Moraes, C.; Amaral, M.E.C.; Catisti, R. Gestational protein restriction induces alterations in placental morphology and mitochondrial function in rats during late pregnancy. J. Mol. Histol. 2013, 44, 629–637. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.B.D.; Cockrell, K.; Sullivan, E.; Bennett, W.; Granger, J.P. Role of Endothelin in Mediating Tumor Necrosis Factor-Induced Hypertension in Pregnant Rats. Hypertension 2005, 46, 82–86. [Google Scholar] [CrossRef]
- Novotny, S.; Wallace, K.; Herse, F.; Moseley, J.; Darby, M.; Health, J.; Gill, J.; Wallukat, G.; Martin, J.N.; Dechend, R.; et al. CD4+ T cells play a critical role in mediating hypertension in response to placental ischemia. J. Hypertens. Open Access 2013, 2, 14873. [Google Scholar]
- LaMarca, B.; Speed, J.; Fournier, L.; Babcock, S.A.; Berry, H.; Cockrell, K.; Granger, J.P. Hypertension in response to chronic reductions in uterine perfusion in pregnant rats: Effect of tumor necrosis factor-α blockade. Hypertension 2008, 52, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- LaMarca, B.; Wallace, K.; Herse, F.; Wallukat, G.; Martin, J.N., Jr.; Weimer, A.; Dechend, R. Hypertension in response to placental ischemia during pregnancy: Role of B lymphocytes. Hypertension 2011, 57, 865–871. [Google Scholar] [CrossRef]
- Cornelius, D.C.; Hogg, J.P.; Scott, J.; Wallace, K.; Herse, F.; Moseley, J.; Wallukat, G.; Dechend, R.; Lamarca, B. Administration of interleukin-17 soluble receptor C suppresses TH17 cells, oxidative stress, and hypertension in response to placental ischemia during pregnancy. Hypertension 2013, 62, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.M.; Cornelius, D.C.; Harmon, A.; Moseley, J.; Martin, J.N., Jr.; LaMarca, B. 17-hydroxyprogesterone caproate significantly improves clinical characteristics of preeclampsia in the reduced uterine perfusion pressure rat model. Hypertension 2015, 65, 225–231. [Google Scholar] [CrossRef]
- Veillon, E.W., Jr.; Keiser, S.D.; Parrish, M.R.; Bennett, W.; Cokrell, K.; Ray, L.F.; Granger, J.P.; Martin, J.N.; Lamarca, B. 17-Hydroxyprogesterone blunts the hypertensive response associated with reductions in uterine perfusion pressure in pregnant rats. Am. J. Obstet. Gynecol. 2009, 201, 324.e1–324.e6. [Google Scholar] [CrossRef]
- Raijmakers, M.T.; Dechend, R.; Poston, L. Oxidative stress and preeclampsia: Rationale for antioxidant clinical trials. Hypertension 2004, 44, 374–380. [Google Scholar] [CrossRef]
- Rodrigo, R.; González, J.; Paoletto, F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens. Res. 2011, 34, 431–440. [Google Scholar] [CrossRef]
- McCarthy, C.; Kenny, L.C. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci. Rep. 2016, 6, 32683. [Google Scholar] [CrossRef]
- Sánchez-Aranguren, L.C.; Espinosa-González, C.T.; González-Ortiz, L.M.; Sanabria-Barrera, S.M.; Riaño-Medina, C.E.; Nuñez, A.F.; Ahmed, A.; Vasquez-Vivar, J.; López, M. Soluble Fms-like tyrosine kinase-1 alters cellular metabolism and mitochondrial bioenergetics in preeclampsia. Front. Physiol. 2018, 9, 83. [Google Scholar] [CrossRef]
- Zhai, Y.; Liu, Y.; Qi, Y.; Long, X.; Gao, J.; Yao, X.; Chen, Y.; Wang, X.; Lu, S.; Zhao, Z. The soluble VEGF receptor sFlt-1 contributes to endothelial dysfunction in IgA nephropathy. PLoS ONE 2020, 15, e0234492. [Google Scholar] [CrossRef]
- Amaral, L.M.; Wallace, K.; Owens, M.; Lamarca, B. Pathophysiology and Current Clinical Management of Preeclampsia. Curr. Hypertens. Rep. 2017, 19, 61. [Google Scholar] [CrossRef]
- Granger, J.P.; Alexander, B.T.; Llinás, M.T.; Bennett, W.A.; Khalil, R.A. Pathophysiology of Hypertension During Preeclampsia Linking Placental Ischemia With Endothelial Dysfunction. Hypertension 2001, 38, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Granger, J.P. Inflammatory cytokines, vascular function, and hypertension. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2004, 286, R989–R990. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, B. Endothelial dysfunction. An important mediator in the pathophysiology of hypertension during pre-eclampsia. Minerva Ginecol. 2012, 64, 309–320. [Google Scholar] [PubMed]
- Brennan, L.J.; Morton, J.S.; Davidge, S.T. Vascular Dysfunction in Preeclampsia. Microcirculation 2014, 21, 4–14. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deer, E.; Jones, J.; Cornelius, D.C.; Comley, K.; Herrock, O.; Campbell, N.; Fitzgerald, S.; Ibrahim, T.; LaMarca, B.; Amaral, L.M. Progesterone Induced Blocking Factor Reduces Hypertension and Placental Mitochondrial Dysfunction in Response to sFlt-1 during Pregnancy. Cells 2021, 10, 2817. https://doi.org/10.3390/cells10112817
Deer E, Jones J, Cornelius DC, Comley K, Herrock O, Campbell N, Fitzgerald S, Ibrahim T, LaMarca B, Amaral LM. Progesterone Induced Blocking Factor Reduces Hypertension and Placental Mitochondrial Dysfunction in Response to sFlt-1 during Pregnancy. Cells. 2021; 10(11):2817. https://doi.org/10.3390/cells10112817
Chicago/Turabian StyleDeer, Evangeline, Jalisa Jones, Denise C. Cornelius, Kyleigh Comley, Owen Herrock, Nathan Campbell, Sarah Fitzgerald, Tarek Ibrahim, Babbette LaMarca, and Lorena M. Amaral. 2021. "Progesterone Induced Blocking Factor Reduces Hypertension and Placental Mitochondrial Dysfunction in Response to sFlt-1 during Pregnancy" Cells 10, no. 11: 2817. https://doi.org/10.3390/cells10112817
APA StyleDeer, E., Jones, J., Cornelius, D. C., Comley, K., Herrock, O., Campbell, N., Fitzgerald, S., Ibrahim, T., LaMarca, B., & Amaral, L. M. (2021). Progesterone Induced Blocking Factor Reduces Hypertension and Placental Mitochondrial Dysfunction in Response to sFlt-1 during Pregnancy. Cells, 10(11), 2817. https://doi.org/10.3390/cells10112817