
Shedding Light on the Possible Link between ADAMTS13 and Vaccine—Induced Thrombotic Thrombocytopenia



{kind=link}
{kind=link}
Abstract
:1. Introduction
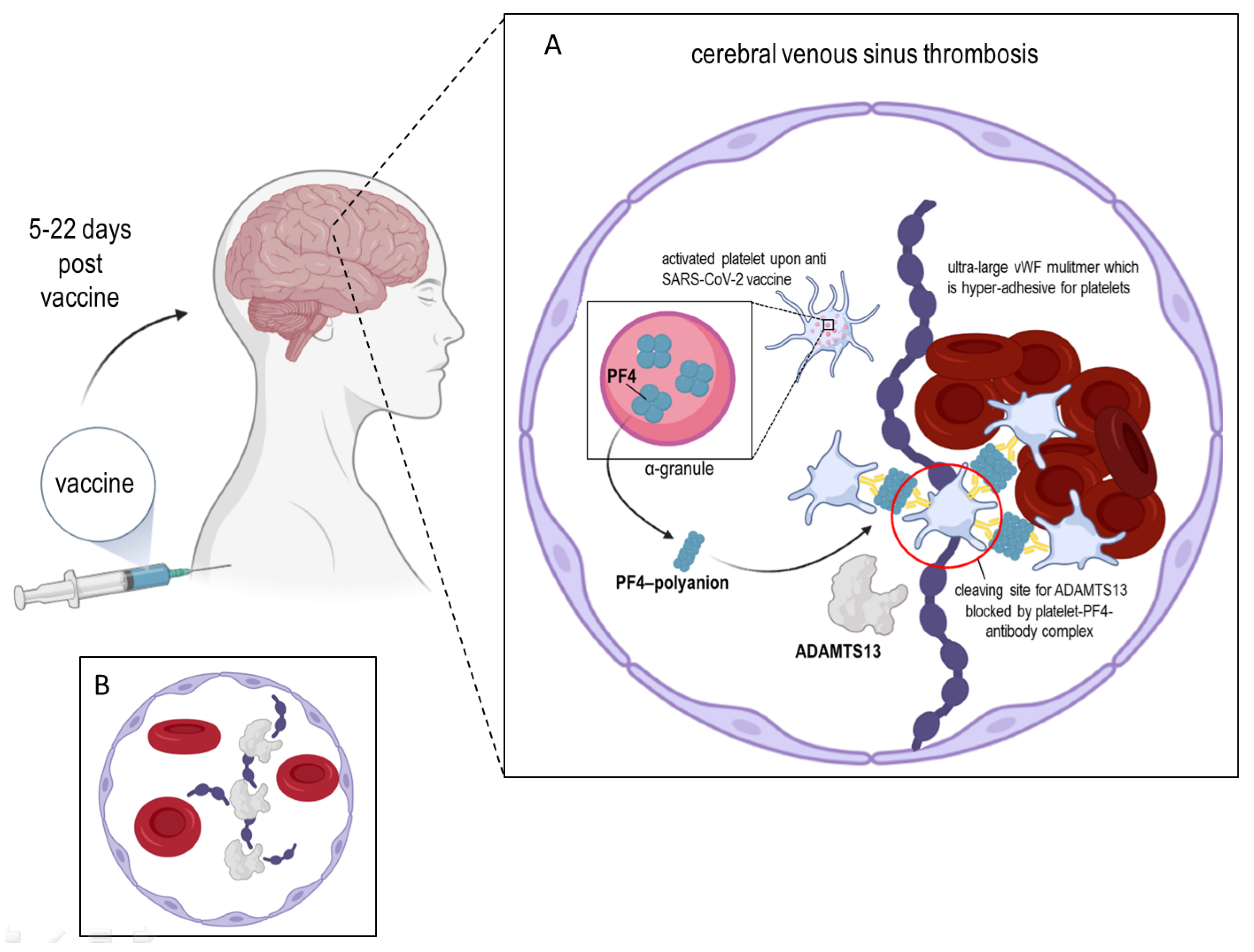
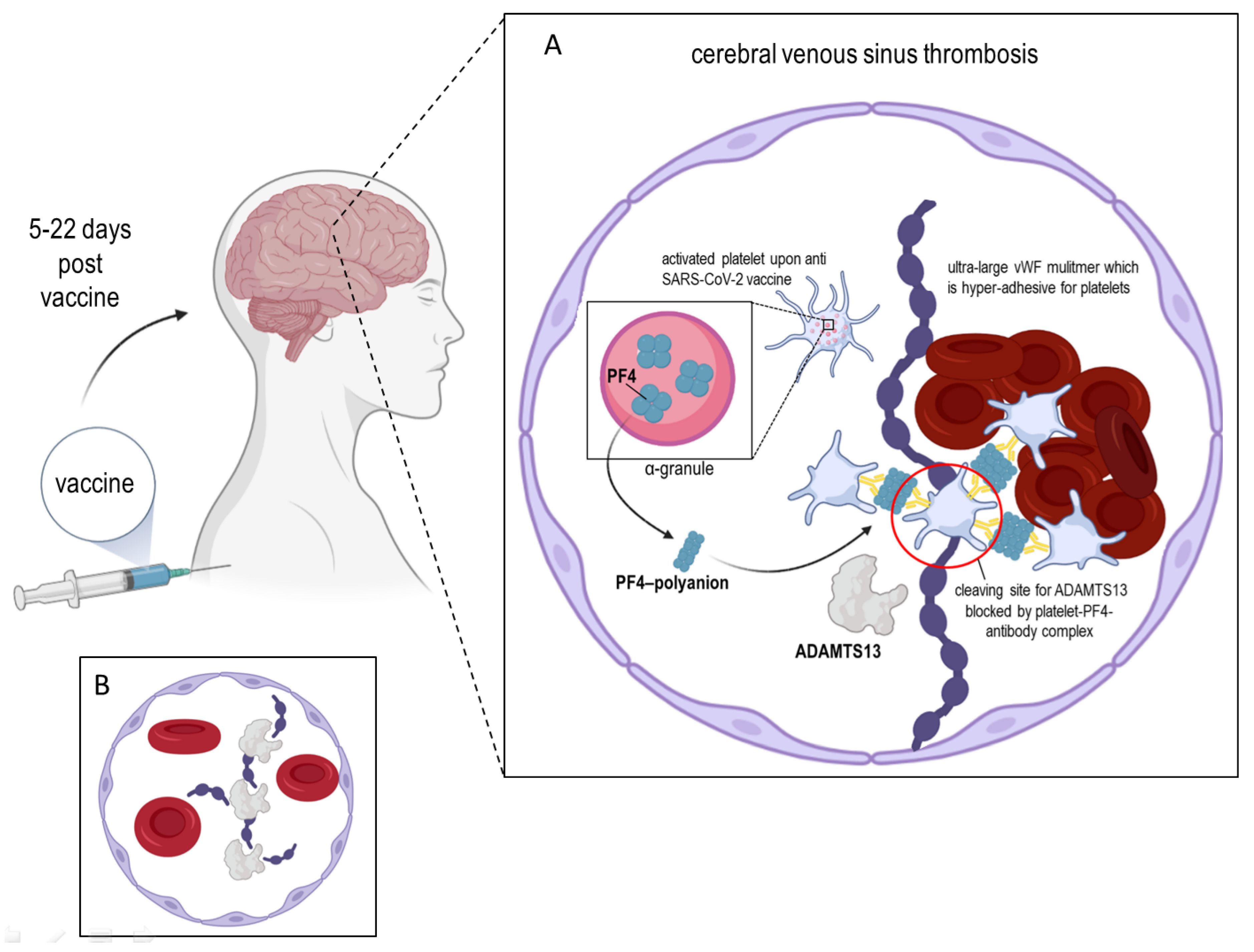
2. ADAMTS13 and the Platelet Factor 4
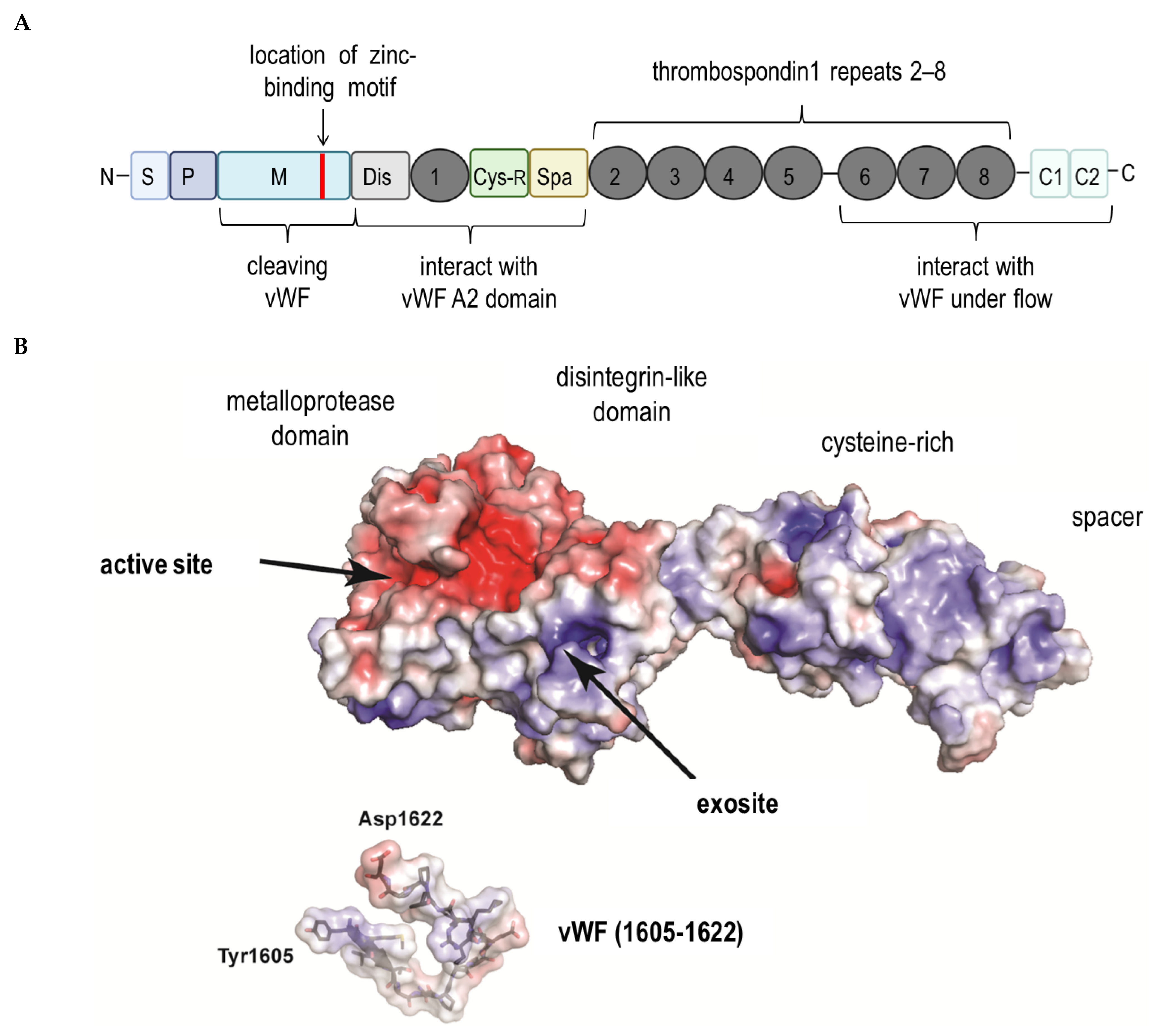
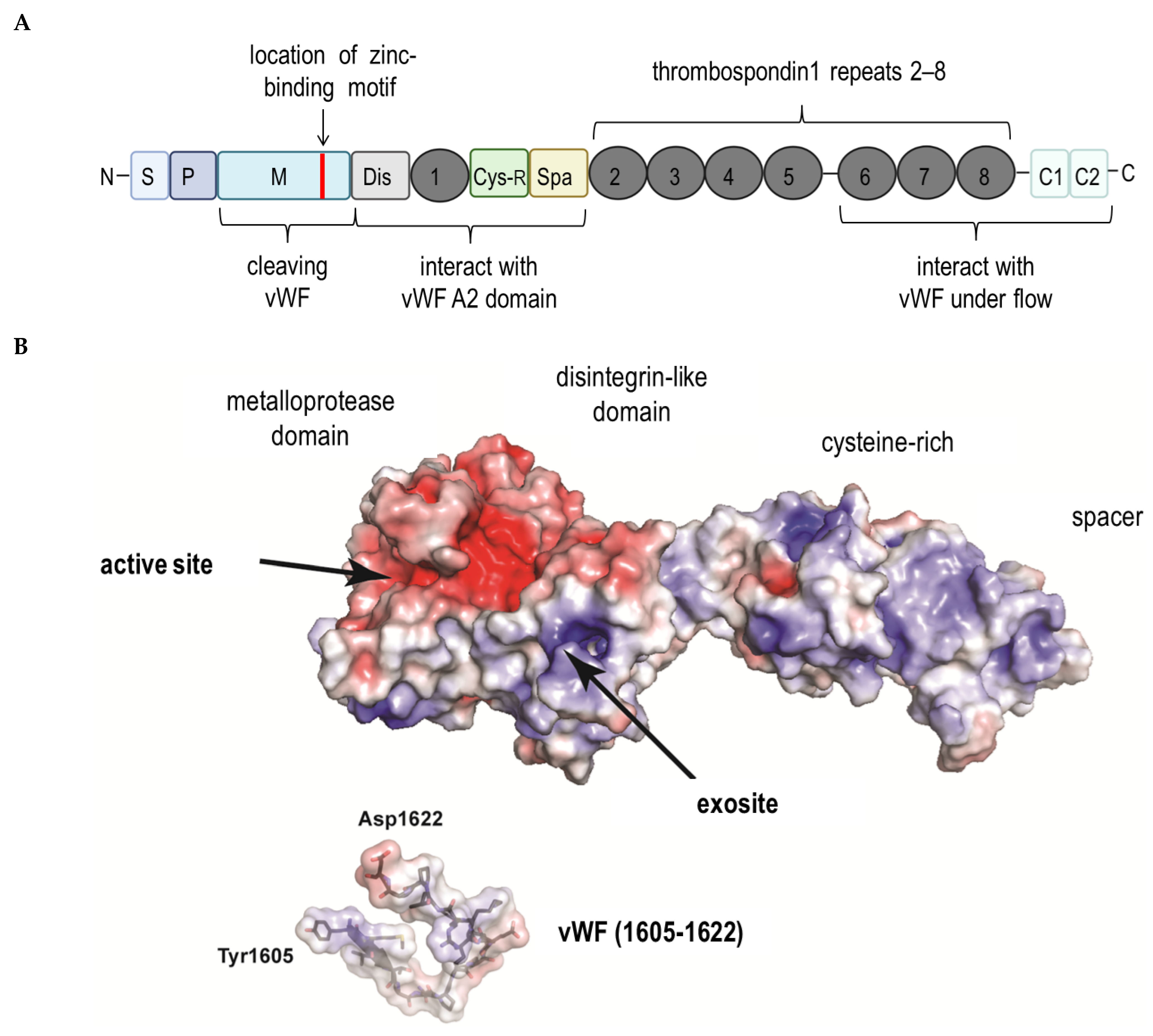
3. ADAMTS13 and the Von Willebrand Factor
4. Conclusions and Final Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ewer, K.; Sebastian, S.; Spencer, A.J.; Gilbert, S.; Hill, A.V.S.; Lambe, T. Chimpanzee adenoviral vectors as vaccines for outbreak pathogens. Hum. Vaccin. Immunother. 2017, 13, 3020–3032. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.H.; Sørvoll, I.H.; Michelsen, A.E.; Munthe, L.A.; Lund-Johansen, F.; Ahlen, M.T.; Wiedmann, M.; Aamodt, A.H.; Skattør, T.H.; Tjønnfjord, G.E.; et al. Thrombosis and Thrombocytopenia after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Singh, D.; Lown, R.; Poles, A.; Solomon, T.; Levi, M.; Goldblatt, D.; Kotoucek, P.; Thomas, W.; Lester, W. Pathologic antibodies to platelet factor 4 after ChAdOx1 nCoV-19 vaccination. N. Engl. J. Med. 2021, 384, 2202–2211. [Google Scholar] [CrossRef]
- Johnston, I.; Sarkar, A.; Hayes, V.; Koma, G.T.; Arepally, G.M.; Chen, J.; Chung, D.W.; López, J.A.; Cines, D.B.; Rauova, L.; et al. Recognition of PF4-VWF complexes by heparin-induced thrombocytopenia antibodies contributes to thrombus propagation. Blood 2020, 135, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, K.; Suzuki, H.; McMullen, B.; Chung, D. Purification of human von Willebrand factor–cleaving protease and its identification as a new member of the metalloproteinase family. Blood 2001, 98, 1662–1666. [Google Scholar] [CrossRef]
- Zheng, X.; Chung, D.; Takayama, T.K.; Majerus, E.M.; Sadler, J.E.; Fujikawa, K. Structure of von Willebrand factor–cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J. Biol. Chem. 2001, 276, 41059–41063. [Google Scholar] [CrossRef] [Green Version]
- Nazy, I.; Elliott, T.D. Arnold DM Platelet factor 4 inhibits ADAMTS13 activity and regulates the multimeric distribution of von Willebrand factor. Br. J. Haematol. 2020, 190, 594–598. [Google Scholar] [CrossRef]
- Poncz, M.; Surrey, S.; LaRocco, P.; Weiss, M.J.; Rappaport, E.F.; Conway, T.M.; Schwartz, E. Cloning and characterization of platelet factor 4 cDNA derived from a human erythroleukemic cell line. Blood 1987, 69, 219–223. [Google Scholar] [CrossRef]
- Schaffner, A.; Rhyn, P.; Schoedon, G.; Schaer, D.J. Regulated expression of platelet factor 4 in human monocytes—Role of PARs as a quantitatively important monocyte activation pathway. J. Leukoc. Biol. 2005, 78, 202–209. [Google Scholar] [CrossRef]
- De Jong, E.K.; de Haas, A.H.; Brouwer, N.; van Weering, H.R.; Hensens, M.; Bechmann, I.; Pratley, P.; Wesseling, E.; Boddeke, H.W.; Biber, K. Expression of CXCL4 in microglia in vitro and in vivo and its possible signaling through CXCR3. J. Neurochem. 2008, 105, 1726–1736. [Google Scholar] [CrossRef]
- Rucinski, B.; Niewiarowski, S.; James, P.; Walz, D.A.; Budzynski, A.Z. Antiheparin proteins secreted by human platelets. purification, characterization, and radioimmunoassay. Blood 1979, 53, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Brandt, E.; Ludwig, A.; Petersen, F.; Flad, H.D. Platelet-derived CXC chemokines: Old players in new games. Immunol. Rev. 2000, 177, 204–216. [Google Scholar] [CrossRef]
- Visentin, G.P.; Ford, S.E.; Scott, J.P.; Aster, R.H. Antibodies from patients with heparin-induced thrombocytopenia/thrombosis are specific for platelet factor 4 complexed with heparin or bound to endothelial cells. J. Clin. Investig. 1994, 93, 81–88. [Google Scholar] [CrossRef]
- Kelton, J.G.; Sheridan, D.; Santos, A.; Smith, J.; Steeves, K.; Smith, C.; Brown, C.; Murphy, W.G. Heparin-induced thrombocytopenia: Laboratory studies. Blood 1988, 72, 925–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauova, L.; Poncz, M.; McKenzie, S.E.; Reilly, M.P.; Arepally, G.; Weisel, J.W.; Nagaswami, C.; Cines, D.B.; Sachais, B.S. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005, 105, 131–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, A.J.; Kaser-Glanzmann, R.; Jakábová, M.; Lüscher, E.F. Characterization of a chondroitin 4 -sulfate proteoglycan carrier for heparin neutralizing activity (platelet factor 4) released from human blood platelets. Biochim. Biophys. Acta 1972, 286, 312–329. [Google Scholar] [CrossRef]
- Holt, J.C.; Niewiarowski, S. Biochemistry of alpha granule proteins. Semin. Hematol. 1985, 22, 151–163. [Google Scholar] [PubMed]
- Wilkinson, J.M.; Ladinig, A.; Bao, H.; Kommadath, A.; Stothard, P.; Lunney, J.K.; Harding, J.C.; Plastow, G.S. Differences in Whole Blood Gene Expression Associated with Infection Time-Course and Extent of Fetal Infection. PLoS ONE 2016, 11, e0153615. [Google Scholar] [CrossRef]
- Kullaya, V.; van derVen, A.; Mpagama, S.; Mmbaga, B.T.; de Groot, P.; Kibiki, G.; de Mast, Q. Platelet-monocyte interaction in Mycobacterium tuberculosis infection. Tuberculosis 2018, 111, 86–93. [Google Scholar] [CrossRef]
- Guo, L.; Feng, K.; Wang, Y.C.; Mei, J.J.; Ning, R.T.; Zheng, H.W.; Wang, J.J.; Worthen, G.S.; Wang, X.; Song, J.; et al. Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection. Mucosal Immunol. 2017, 10, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Aivado, M.; Spentzos, D.; Germing, U.; Alterovitz, G.; Meng, X.Y.; Grall, F.; Giagounidis, A.A.N.; Klement, G.; Steidl, U.; Out, H.H.; et al. Serum proteome profiling detects myelodysplastic syndromes and identifies CXC chemokine ligands 4 and 7 as markers for advanced disease. Proc. Natl. Acad. Sci. USA 2007, 104, 1307–1312. [Google Scholar] [CrossRef] [Green Version]
- Wilson, N.O.; Jain, V.; Roberts, C.E.; Lucchi, N.; Joel, P.K.; Singh, M.P.; Nagpal, A.C.; Dash, A.P.; Udhayakumar, V.; Singh, N.; et al. CXCL4 and CXCL10 predict risk of fatal cerebral malaria. Dis. Markers 2011, 30, 39–49. [Google Scholar] [CrossRef]
- Auerbach, D.J.; Lin, Y.; Miao, H.Y.; Cimbro, R.; DiFiore, M.J.; Gianolini, M.E.; Furci, L.; Biswas, P.; Fauci, A.S.; Lusso, P. Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 9569–9574. [Google Scholar] [CrossRef] [Green Version]
- Domschke, G.; Gleissner, C.A. CXCL4-induced macrophages in human atherosclerosis. Cytokine 2019, 122, 154141. [Google Scholar] [CrossRef] [PubMed]
- Werner, L.; Guzner-Gur, H.; Dotan, I. Involvement of CXCR4/CXCR7/CXCL12 Interactions in Inflammatory bowel disease. Theranostics 2013, 3, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Yeo, L.; Adlard, N.; Biehl, M.; Juarez, M.; Smallie, T.; Snow, M.; Buckley, C.D.; Raza, K.; Filer, A.; Scheel-Toellner, D. Expression of chemokines CXCL4 and CXCL7 by synovial macrophages defines an early stage of rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affandi, A.J.; Silva-Cardoso, S.C.; Garcia, S.; Leijten, E.F.; van Kempen, T.S.; Marut, W.; van Roon, J.A.; Radstake, T.R. CXCL4 is a novel inducer of human Th17 cells and correlates with IL-17 and IL-22 in psoriatic arthritis. Eur. J. Immunol. 2018, 48, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J.E. Pathophysiology of thrombotic thrombocytopenic purpura. Blood 2017, 130, 1181–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrdinová, J.; D’Angelo, S.; Graça, N.A.G.; Ercig, B.; Vanhoorelbeke, K.; Veyradier, A.; Voorberg, J.; Coppo, P. Dissecting the pathophysiology of immune thrombotic thrombocytopenic purpura: Interplay between genes and environmental triggers. Haematologica 2018, 103, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South, K.; Lane, D.A. ADAMTS-13 and von Willebrand factor: A dynamic duo. J. Thromb. Haemost. 2018, 16, 6–18. [Google Scholar] [CrossRef]
- Petri, A.; Kim, H.J.; Xu, Y.; de Groot, R.; Li, C.; Vandenbulcke, A.; Vanhoorelbeke, K.; Emsley, J.; Crawley, J.T.B. Crystal structure and substrate-induced activation of ADAMTS13. Nat. Commun. 2019, 10, 3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelpe, A.S.; Petri, A.; Roose, E.; Pareyn, I.; Deckmyn, H.; de Meyer, S.F.; Crawley, J.T.B.; Vanhoorelbeke, K. Antibodies that conformationally activate ADAMTS13 allosterically enhance metalloprotease domain function. Blood Adv. 2020, 4, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Tatsumi, K.; Matsumoto, M.; Fujimoto, M.; Matsuyama, T.; Ishikawa, M.; Iwamoto, T.-A.; Mori, T.; Wanaka, A.; Fukui, H.; et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood 2005, 106, 922–924. [Google Scholar] [CrossRef] [Green Version]
- Shang, D.; Zheng, X.W.; Niiya, M.; Zheng, X.L. Apical sorting of ADAMTS13 in vascular endothelial cells and Madin-Darby canine kidney cells depends on the CUB domains and their association with lipid rafts. Blood 2006, 108, 2207–2215. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Murata, M.; Matsubara, Y.; Uchida, T.; Ishihara, H.; Shibano, T.; Ashida, S.-I.; Soejima, K.; Okada, Y.; Ikeda, Y. Detection of von Willebrand factor–cleaving protease (ADAMTS-13) in human platelets. Biochem. Biophys. Res. Commun. 2004, 313, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Manea, M.; Kristoffersson, A.; Schneppenheim, R.; Saleem, M.A.; Mathieson, P.W.; Mörgelin, M.; Björk, P.; Holmberg, L.; Karpman, D. Podocytes express ADAMTS13 in normal renal cortex and in patients with thrombotic thrombocytopenic purpura. Br. J. Haematol. 2007, 138, 651–662. [Google Scholar] [CrossRef]
- Tauchi, R.; Imagama, S.; Ohgomori, T.; Natori, T.; Shinjo, R.; Ishiguro, N.; Kadomatsu, K. ADAMTS-13 is produced by glial cells and upregulated after spinal cord injury. Neurosci. Lett. 2012, 517, 1–6. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Capoferri, C.; Canciani, M.T. Plasma levels of von Willebrand factor regulate ADAMTS-13, its major cleaving protease. Br. J. Haematol. 2004, 126, 213–218. [Google Scholar] [CrossRef]
- Shahidi, M. Thrombosis and von Willebrand Factor. Adv. Exp. Med. Biol. 2017, 906, 285–306. [Google Scholar]
- Jin, S.Y.; Skipwith, C.G.; Zheng, X.L. Amino acid residues Arg(659), Arg(660), and Tyr(661) in the spacer domain of ADAMTS13 are critical for cleavage of von Willebrand factor. Blood 2010, 115, 2300–2310. [Google Scholar] [CrossRef]
- Fujimura, Y.; Fukui, H.; Usami, Y.; Titani, K. Domain structure of human von Willebrand factor, and its modulators involved in the platelet adhesion process in vitro. Rinsho Ketsueki 1991, 32, 475–480. [Google Scholar] [PubMed]
- Meyer, D.; Girma, J.P. von Willebrand factor: Structure and function. Thromb. Haemost. 1993, 70, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Hattori, R.; Hamilton, K.K.; Fugate, R.D.; McEver, R.P.; Sims, P.J. Stimulated secretion of endothelial von Willebrand factor is accompanied by rapid redistribution to the cell surface of the intracellular granule membrane protein GMP-140. J. Biol. Chem. 1989, 264, 7768–7771. [Google Scholar] [CrossRef]
- Collins, P.; Wilkie, M.; Razak, K.; Abbot, S.; Harley, S.; Bax, C.; Zaidi, M.; Blake, D.; Cunningham, J.; Newland, A. Cyclosporine and cremaphor modulate von Willebrand factor release from cultured human endothelial cells. Transplantation 1993, 56, 1218–1223. [Google Scholar] [CrossRef]
- Mannucci, P.M. Platelet von Willebrand factor in inherited and acquired bleeding disorders. Proc. Natl. Acad. Sci. USA 1995, 92, 2428–2432. [Google Scholar] [CrossRef] [Green Version]
- Cleator, J.H.; Zhu, W.Q.; Vaughan, D.E.; Hamm, H.E. Differential regulation of endothelial exocytosis of P-selectin and von Willebrand factor by protease-activated receptors and cAMP. Blood 2006, 107, 2736–2744. [Google Scholar] [CrossRef]
- Ruggeri, Z.M. Old concepts and new developments in the study of platelet aggregation. J. Clin. Investig. 2000, 105, 699–701. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, Z.M. The role of von Willebrand factor in thrombus formation. Thromb. Res. 2007, 120, S5–S9. [Google Scholar] [CrossRef] [Green Version]
- Amelot, A.A.; Tagzirt, M.; Ducouret, G.; Kuen, R.L.; Le Bonniec, B.F. Platelet factor 4 (CXCL4) seals blood clots by altering the structure of fibrin. J. Biol. Chem. 2007, 282, 710–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.F.; Moake, J.L.; Nolasco, L.; Bernardo, A.; Arceneaux, W.; Shrimpton, C.N.; Schade, A.J.; McIntire, L.V.; Fujikawa, K.; López, J.A. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002, 100, 4033–4039. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Xiao, J.; Mao, Y.; Zheng, X.L. Carboxyl terminus of ADAMTS13 directly inhibits platelet aggregation and ultra large von Willebrand factor string formation under flow in a free-thiol-dependent manner. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Yeh, H.C.; Zhou, Z.; Choi, H.; Tekeoglu, S.; May, W., III; Wang, C.; Turner, N.; Scheiflinger, F.; Moake, J.L.; Dong, J.-F. Disulfide bond reduction of von Willebrand factor by ADAMTS-13. J. Thromb. Haemost. 2010, 8, 2778–2788. [Google Scholar] [CrossRef]
- Borchiellini, A.; Fijnvandraat, K.; ten Cate, J.W.; Pajkrt, D.; van Deventer, S.J.; Pasterkamp, G.; Meijer-Huizinga, F.; Zwart-Huinink, L.; Voorberg, J.; van Mourik, J.A. Quantitative analysis of von Willebrand factor propeptide release in vivo: Effect of experimental endotoxemia and administration of 1-deamino-8- D-arginine vasopressin in humans. Blood 1996, 88, 2951–2958. [Google Scholar] [CrossRef] [Green Version]
- Hattori, R.; Hamilton, K.K.; McEver, R.P.; Sims, P.J. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J. Biol. Chem. 1989, 264, 9053–9060. [Google Scholar] [CrossRef]
- Huang, J.; Motto, D.G.; Bundle, D.R.; Sadler, J.E. Shiga toxin B subunits induce VWF secretion by human endothelial cells and thrombotic microangiopathy in ADAMTS13-deficient mice. Blood 2010, 116, 3653–3659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannucci, P.M. Desmopressin (DDAVP) in the treatment of bleeding disorders: The first twenty years. Haemophilia 2000, 6, 60–67. [Google Scholar] [CrossRef]
- Goodeve, A.C. The genetic basis of von Willebrand disease. Blood Rev. 2010, 24, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Muster, V.; Gary, T.; Raggam, R.B.; Wölfler, A.; Brodmann, M. Pulmonary embolism and thrombocytopenia following ChAdOx1 vaccination. Lancet 2021, 397, 1842. [Google Scholar] [CrossRef]
- Rzymski, P.; Perek, B.; Flisiak, R. Thrombotic Thrombocytopenia after COVID-19 Vaccination: In Search of the Underlying Mechanism. Vaccines 2021, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Selleng, K.; Mayerle, J.; Palankar, R.; Wesche, J.; Reiche, S.; Aebischer, A.; Warkentin, T.E.; Muenchhoff, M.; Hellmuth, J.C.; et al. Anti-platelet factor 4 antibodies causing VITT do not cross-react with SARS-CoV-2 spike protein. Blood 2021, 138, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szóstek-Mioduchowska, A.; Kordowitzki, P. Shedding Light on the Possible Link between ADAMTS13 and Vaccine—Induced Thrombotic Thrombocytopenia. Cells 2021, 10, 2785. https://doi.org/10.3390/cells10102785
Szóstek-Mioduchowska A, Kordowitzki P. Shedding Light on the Possible Link between ADAMTS13 and Vaccine—Induced Thrombotic Thrombocytopenia. Cells. 2021; 10(10):2785. https://doi.org/10.3390/cells10102785
Chicago/Turabian StyleSzóstek-Mioduchowska, Anna, and Paweł Kordowitzki. 2021. "Shedding Light on the Possible Link between ADAMTS13 and Vaccine—Induced Thrombotic Thrombocytopenia" Cells 10, no. 10: 2785. https://doi.org/10.3390/cells10102785
APA StyleSzóstek-Mioduchowska, A., & Kordowitzki, P. (2021). Shedding Light on the Possible Link between ADAMTS13 and Vaccine—Induced Thrombotic Thrombocytopenia. Cells, 10(10), 2785. https://doi.org/10.3390/cells10102785