
Activation of mTORC1 by Free Fatty Acids Suppresses LAMP2 and Autophagy Function via ER Stress in Alcohol-Related Liver Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Human Liver Samples
2.3. Chronic Alcohol Feeding and Treatments
2.4. Western Blot
2.5. Cell Culture and Treatments
2.6. Plasmids Transfection
2.7. Histopathology and Immunohistochemistry
2.8. Immunofluorescence
2.9. Plasma ALT/AST Levels
2.10. RNA Isolation and Real-Time PCR
2.11. Quantification of FFAs and TG
2.12. Lactate Dehydrogenase (LDH) Assay
2.13. Statistical Analysis
3. Results
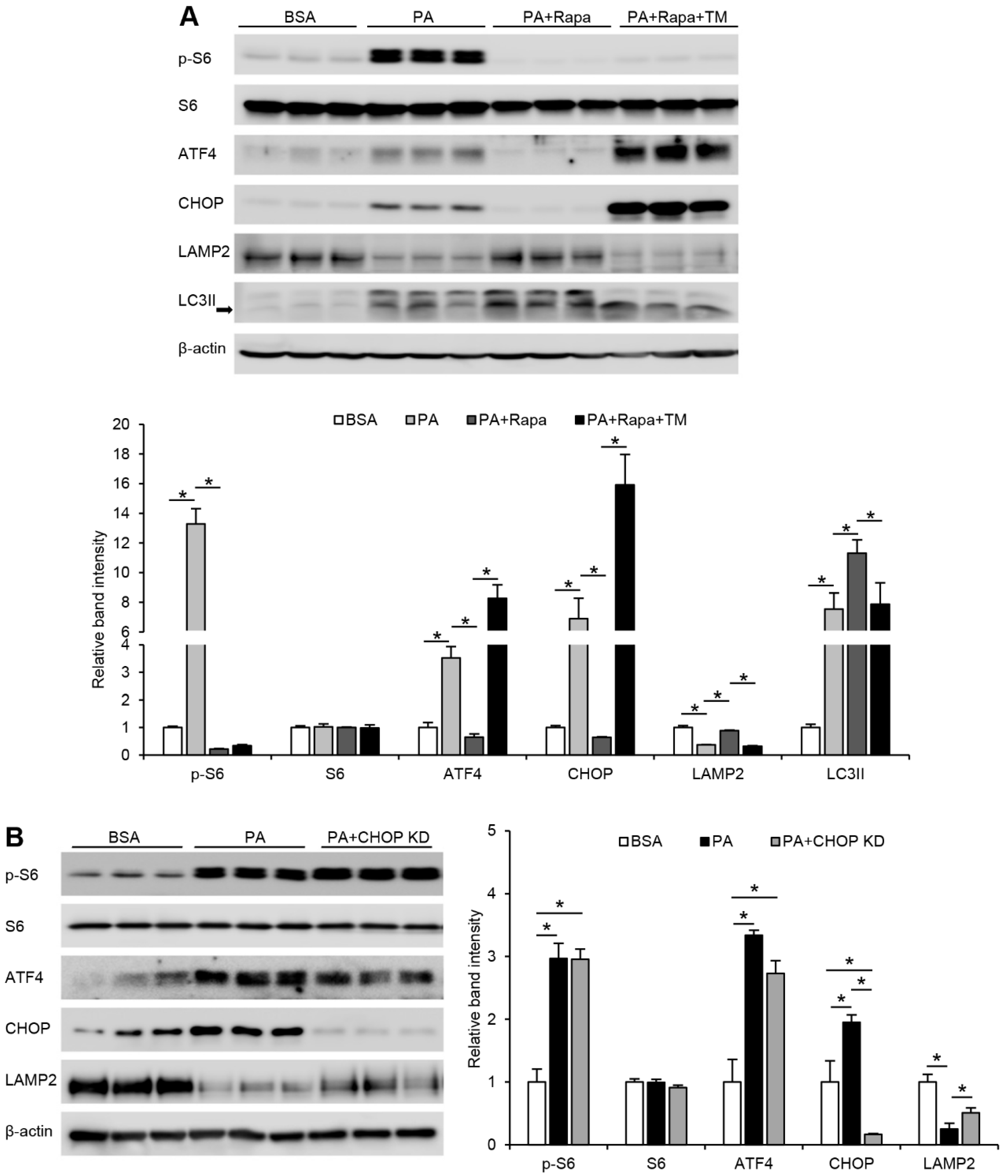
3.1. FFA-induced ER Stress, LAMP2 Reduction, and Impaired Autophagy Flux Is mTORC1-Dependent in Hepatocytes
3.2. mTORC1-Induced ER Stress Targets LAMP2 via ATF4-CHOP Pathway under FFA Treatment in Hepatocytes
3.3. Inhibition of mTORC1 Signaling by Dietary Rapamycin Supplementation Ameliorates Alcohol-Induced ER Stress and Improves Autophagy in Mice
3.4. Inhibition of mTORC1 Signaling by Dietary Rapamycin Supplementation Attenuates Alcohol-Induced Liver Injury in Mice
3.5. Activation of mTORC1 and Accumulation of CHOP in the Liver of Patients with SAH
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, B.; Bataller, R. Alcoholic Liver Disease: Pathogenesis and New Therapeutic Targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Shen, F.; Sherban, A.; Nocon, A.; Li, Y.; Wang, H.; Xu, M.J.; Rui, X.; Han, J.; Jiang, B.; et al. DEP domain-containing mTOR-interacting protein suppresses lipogenesis and ameliorates hepatic steatosis and acute-on-chronic liver injury in alcoholic liver disease. Hepatology 2018, 68, 496–514. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Reviews. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef]
- Wei, X.; Shi, X.; Zhong, W.; Zhao, Y.; Tang, Y.; Sun, W.; Yin, X.; Bogdanov, B.; Kim, S.; McClain, C.; et al. Chronic alcohol exposure disturbs lipid homeostasis at the adipose tissue-liver axis in mice: Analysis of triacylglycerols using high-resolution mass spectrometry in combination with in vivo metabolite deuterium labeling. PLoS ONE 2013, 8, e55382. [Google Scholar] [CrossRef]
- Zhong, W.; Zhao, Y.; Tang, Y.; Wei, X.; Shi, X.; Sun, W.; Sun, X.; Yin, X.; Sun, X.; Kim, S.; et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: Role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am. J. Pathol. 2012, 180, 998–1007. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Zhong, W.; Hao, L.; Dong, H.; Sun, X.; Yue, R.; Li, T.; Zhou, Z. Fatty Acids Inhibit LAMP2-Mediated Autophagy Flux via Activating ER Stress Pathway in Alcohol-Related Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Babuta, M.; Furi, I.; Bala, S.; Bukong, T.N.; Lowe, P.; Catalano, D.; Calenda, C.; Kodys, K.; Szabo, G. Dysregulated Autophagy and Lysosome Function Are Linked to Exosome Production by Micro-RNA 155 in Alcoholic Liver Disease. Hepatology 2019, 70, 2123–2141. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Tanaka, Y.; Kume, S.; Morita, Y.; Chin-Kanasaki, M.; Araki, H.; Isshiki, K.; Araki, S.; Koya, D.; Haneda, M.; et al. Fatty acids are novel nutrient factors to regulate mTORC1 lysosomal localization and apoptosis in podocytes. Biochim. Biophys. Acta 2014, 1842, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Madak-Erdogan, Z.; Band, S.; Zhao, Y.C.; Smith, B.P.; Kulkoyluoglu-Cotul, E.; Zuo, Q.; Santaliz Casiano, A.; Wrobel, K.; Rossi, G.; Smith, R.L.; et al. Free Fatty Acids Rewire Cancer Metabolism in Obesity-Associated Breast Cancer via Estrogen Receptor and mTOR Signaling. Cancer Res. 2019, 79, 2494–2510. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Ochoa, E.; Sánchez-Alegría, K.; Gómez-Inclán, C.; Ferrera, P.; Arias, C. Palmitic acid stimulates energy metabolism and inhibits insulin/PI3K/AKT signaling in differentiated human neuroblastoma cells: The role of mTOR activation and mitochondrial ROS production. Neurochem. Int. 2017, 110, 75–83. [Google Scholar] [CrossRef]
- Zhong, W.; Wei, X.; Hao, L.; Lin, T.D.; Yue, R.; Sun, X.; Guo, W.; Dong, H.; Li, T.; Ahmadi, A.R.; et al. Paneth Cell Dysfunction Mediates Alcohol-related Steatohepatitis Through Promoting Bacterial Translocation in Mice: Role of Zinc Deficiency. Hepatol. (Baltim. Md.) 2020, 71, 1575–1591. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Zhong, W.; Zhang, W.; Zhou, Z. Defect of mitochondrial respiratory chain is a mechanism of ROS overproduction in a rat model of alcoholic liver disease: Role of zinc deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G205–G214. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Cáceres, M.P.; Cereceda, K.; Hernández, S.; Li, Y.; Narro, C.; Rivera, P.; Silva, P.; Ávalos, Y.; Jara, C.; Burgos, P.; et al. Palmitic acid reduces the autophagic flux in hypothalamic neurons by impairing autophagosome-lysosome fusion and endolysosomal dynamics. Mol. Cell. Oncol. 2020, 7, 1789418. [Google Scholar] [CrossRef]
- González-Rodríguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [Green Version]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Zhong, W.; Dong, H.; Guo, W.; Sun, X.; Zhang, W.; Yue, R.; Li, T.; Griffiths, A.; Ahmadi, A.R.; et al. ATF4 activation promotes hepatic mitochondrial dysfunction by repressing NRF1-TFAM signalling in alcoholic steatohepatitis. Gut 2020. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Giles, A.; Nakamura, K.; Lee, J.W.; Hou, X.; Donmez, G.; Li, J.; Luo, Z.; Walsh, K.; et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 1664–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Q.; Hu, Z.; Zhang, F.; Cui, A.; Chen, X.; Jiang, H.; Gao, J.; Chen, X.; Han, Y.; Liang, Q.; et al. Fibroblast growth factor 21 improves hepatic insulin sensitivity by inhibiting mammalian target of rapamycin complex 1 in mice. Hepatology 2016, 64, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, Y.; Song, Q.; Griffiths, A.; Song, Z. mTORC1-IRE1α pathway activation contributes to palmitate-elicited triglyceride secretion and cell death in hepatocytes. Exp. Biol. Med. (Maywood) 2020, 245, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Peng, Z.; Zeng, N.; Silva, J.; He, L.; Chen, J.; Debebe, A.; Stiles, E.X.; Chen, C.-Y.; Stiles, B.L. Chronic Exposure to Palmitic Acid Downregulates AKT in Beta-Cells through Activation of mTOR. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, X.; Ni, H.M.; Ding, W.X. Insufficient autophagy: A novel autophagic flux scenario uncovered by impaired liver TFEB-mediated lysosomal biogenesis from chronic alcohol-drinking mice. Autophagy 2018, 14, 1646–1648. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, W.; Zhong, W.; Hao, L.; Sun, X.; Zhou, Z. Activation of mTORC1 by Free Fatty Acids Suppresses LAMP2 and Autophagy Function via ER Stress in Alcohol-Related Liver Disease. Cells 2021, 10, 2730. https://doi.org/10.3390/cells10102730
Guo W, Zhong W, Hao L, Sun X, Zhou Z. Activation of mTORC1 by Free Fatty Acids Suppresses LAMP2 and Autophagy Function via ER Stress in Alcohol-Related Liver Disease. Cells. 2021; 10(10):2730. https://doi.org/10.3390/cells10102730
Chicago/Turabian StyleGuo, Wei, Wei Zhong, Liuyi Hao, Xinguo Sun, and Zhanxiang Zhou. 2021. "Activation of mTORC1 by Free Fatty Acids Suppresses LAMP2 and Autophagy Function via ER Stress in Alcohol-Related Liver Disease" Cells 10, no. 10: 2730. https://doi.org/10.3390/cells10102730
APA StyleGuo, W., Zhong, W., Hao, L., Sun, X., & Zhou, Z. (2021). Activation of mTORC1 by Free Fatty Acids Suppresses LAMP2 and Autophagy Function via ER Stress in Alcohol-Related Liver Disease. Cells, 10(10), 2730. https://doi.org/10.3390/cells10102730