
Alteration of STIM1/Orai1-Mediated SOCE in Skeletal Muscle: Impact in Genetic Muscle Diseases and Beyond

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Molecular Components of SOCE
2.1. Store-Operated-Calcium Channels
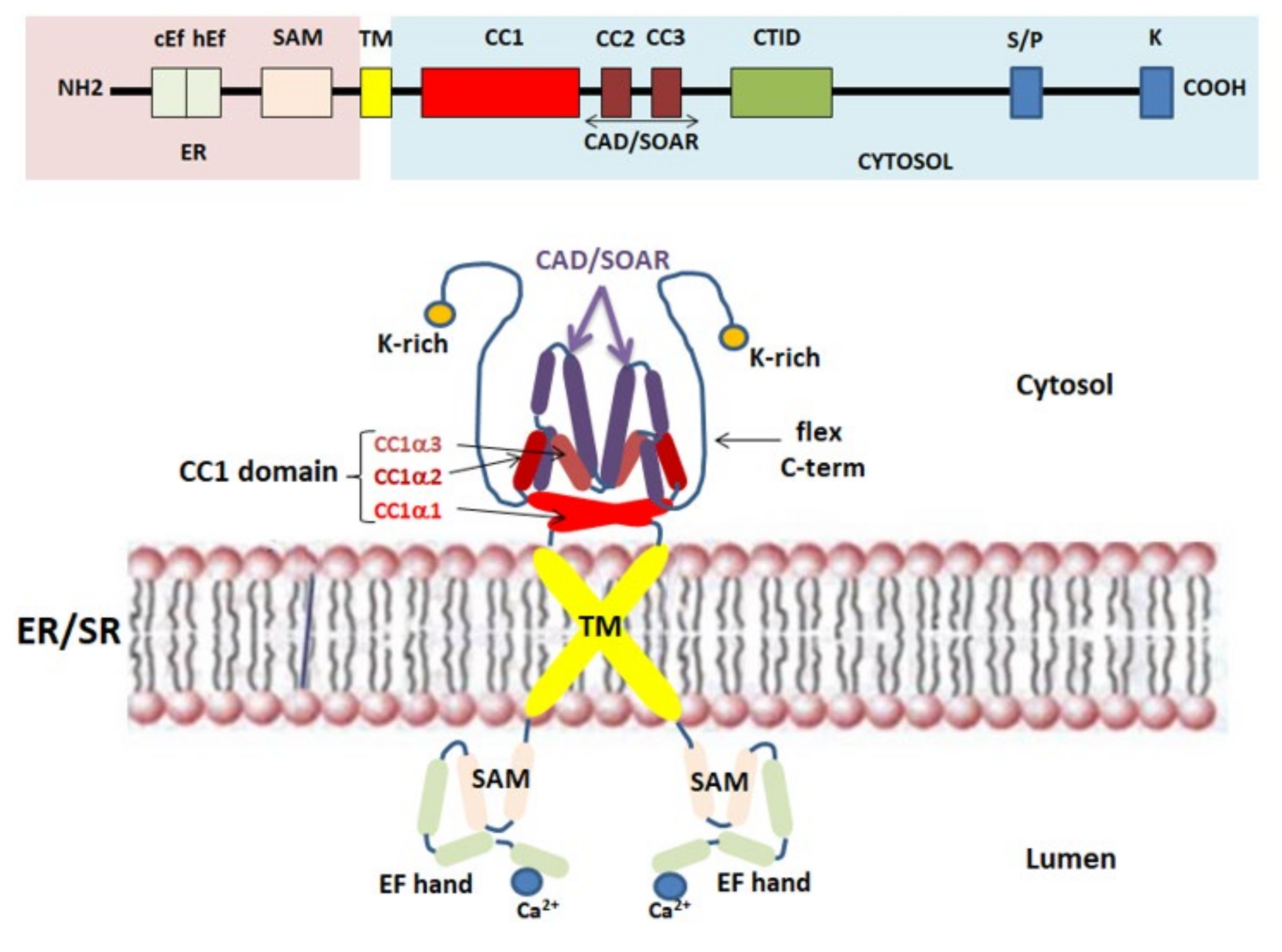
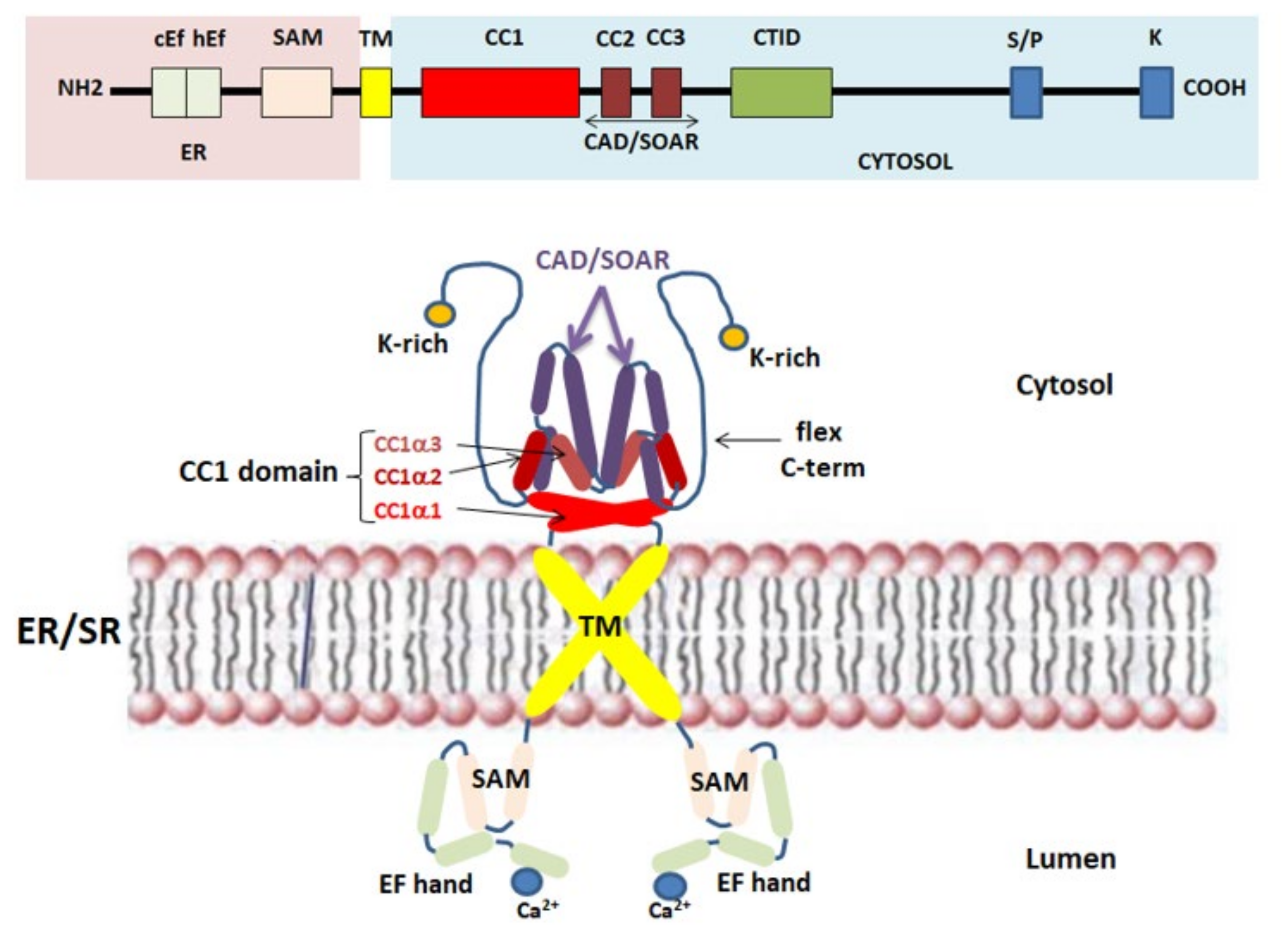
2.2. STIM1 Protein: The Ca2+ Sensor for SOCE
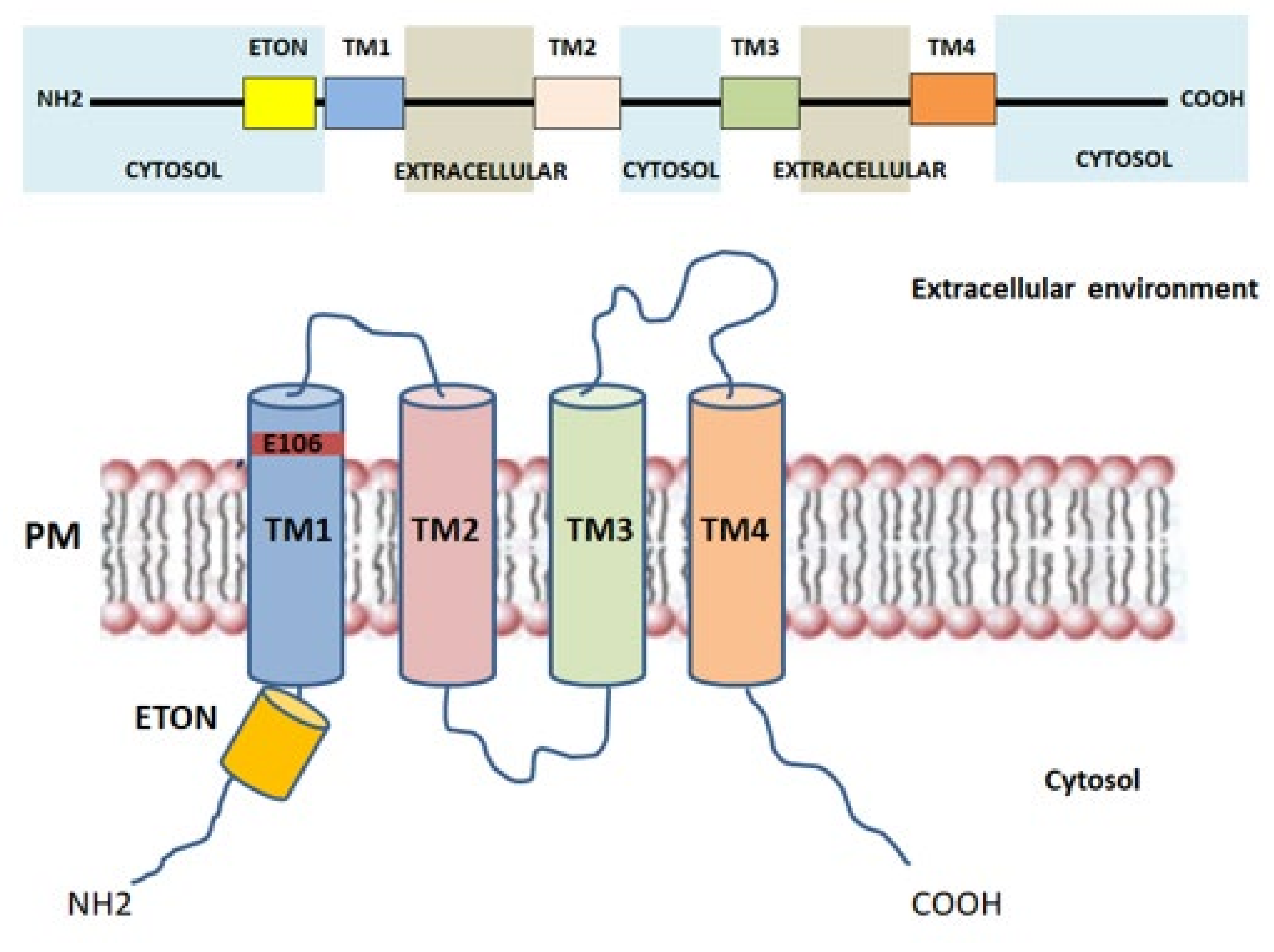
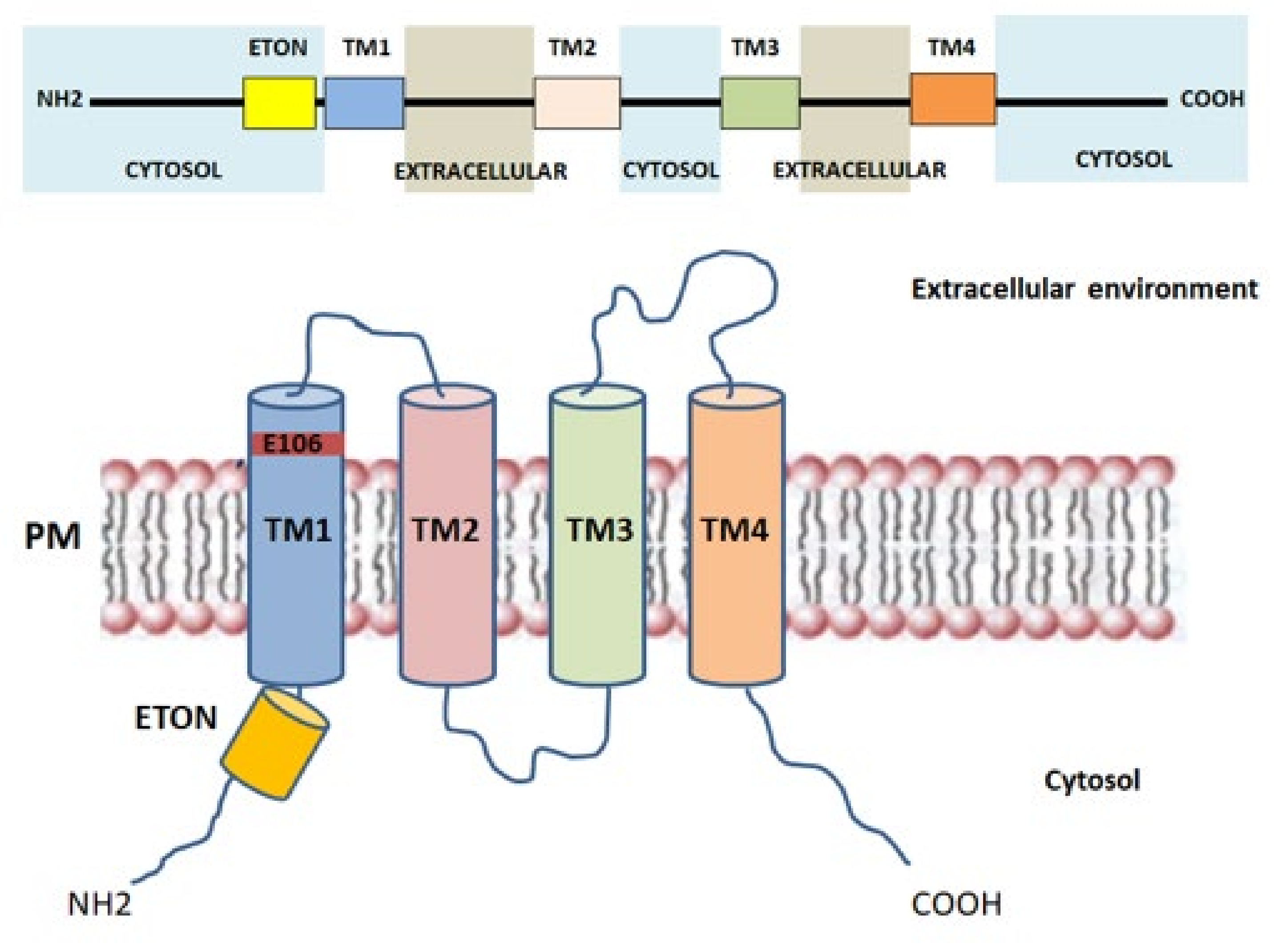
2.3. Orai1: The Key Component of CRAC Current
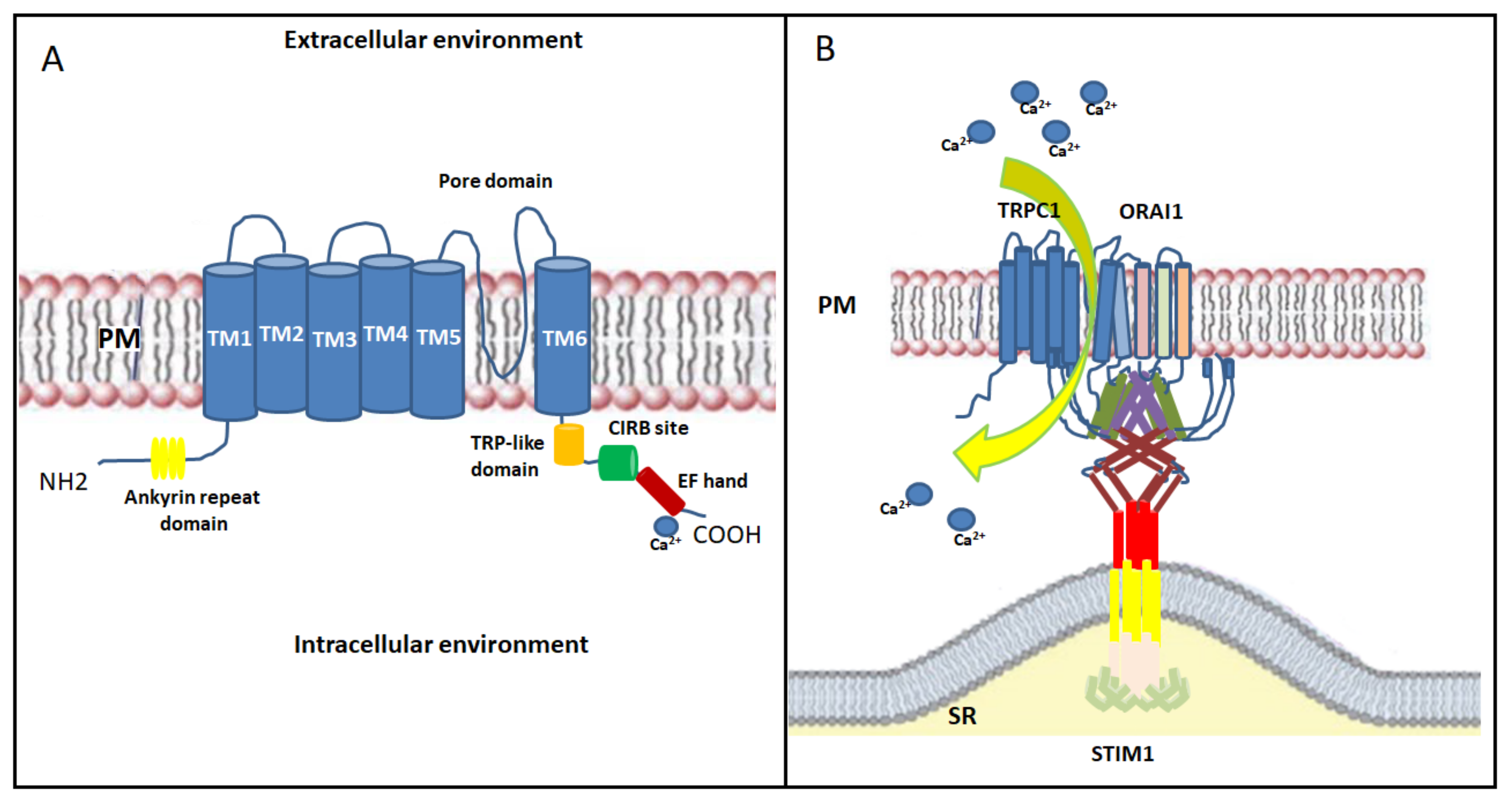
2.4. TRP Channels
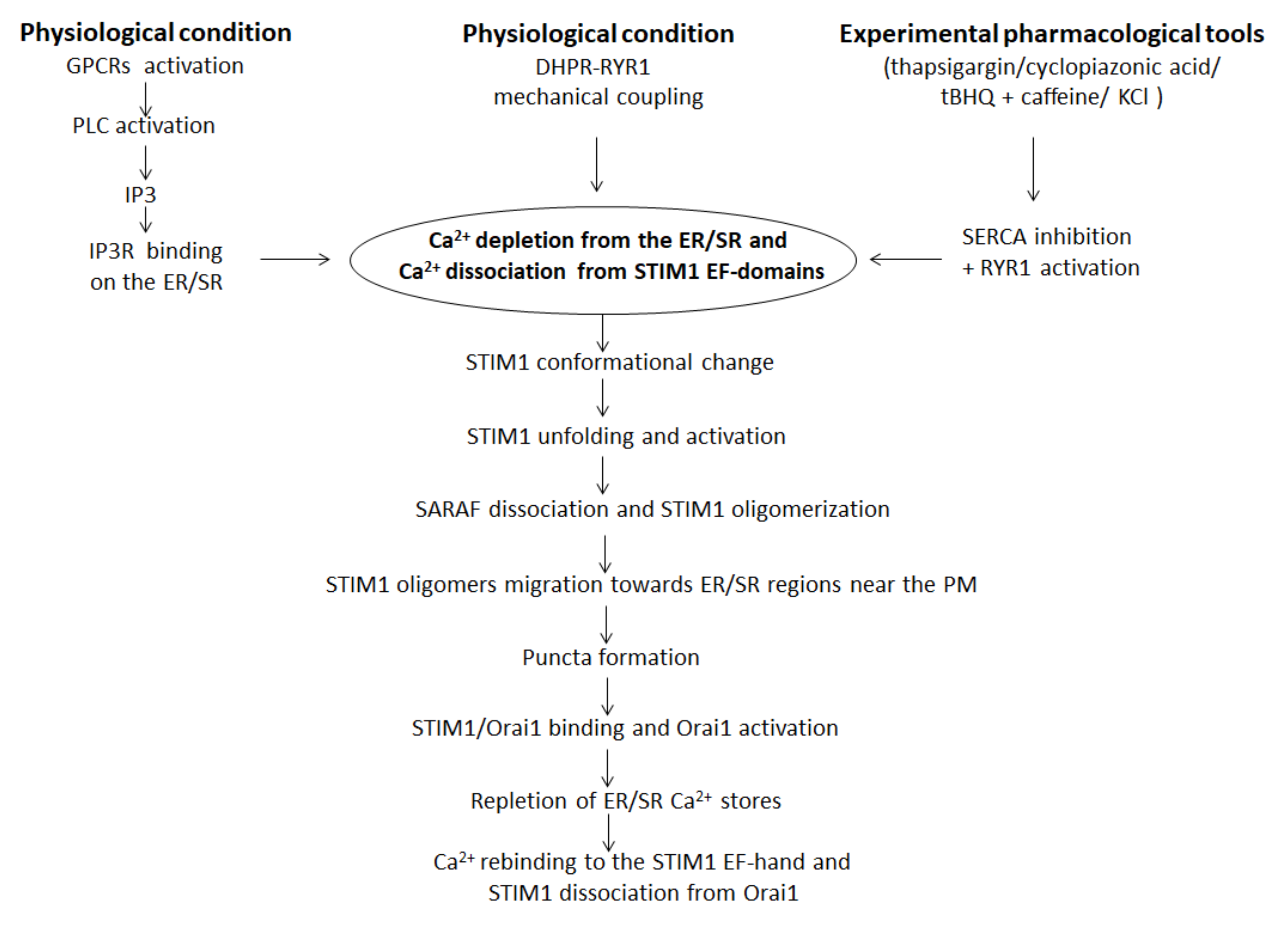
3. SOCE Mechanism in Skeletal Muscle: An Overview
4. STIM1/Orai1-Mediated SOCE Alteration and Skeletal Muscle Diseases
4.1. STIM1/Orai1-Mediated SOCE Alteration in Genetic Skeletal Muscle Disorders
4.2. SOCE Dysfunction in Duchenne Muscular Dystrophy
4.3. SOCE Dysfunction in Skeletal Muscle Wasting Disorders: Cachexia and Sarcopenia
4.4. SOCE Dysfunction in Other Skeletal Muscle Pathological Conditions
5. Therapeutic Perspectives for Counteracting SOCE-Related Skeletal Muscle Diseases
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAD/SOAR domain | CRAC activation domain/STIM1–Orai1 activating region domain |
| CC domains | Conserved cytosolic coiled-coil domains |
| CCE | Capacitive calcium entry |
| cEFh | Canonical EF-hand |
| CIRB | Calmodulin/inositol 1,4,5-trisphosphate receptor-binding |
| CRAC | Ca2+-release-activated-Ca2+ channel |
| CTID | C-terminal inhibitory domain |
| DGC | Dystrophin glycoprotein complex |
| DHPR | Dihydropyridine receptor |
| DMD | Duchenne muscular dystrophy |
| EC coupling | Excitation–contraction coupling |
| ECCE | Excitation-coupled Ca2+ entry |
| ER/SR | Endoplasmic/sarcoplasmic reticulum |
| ERK1/2 | Extracellular signal-regulated kinase 1 and 2 |
| ETON | Extended transmembrane Orai1 N-terminal |
| GHS | Growth hormone secretagogues |
| GPCRs | Plasma membrane G protein-coupled receptors |
| IP3 | Inositol 1,4,5-triphosphate |
| IP3R | Inositol 1,4,5-triphosphate receptor |
| MAP kinases | Mitogen-activated protein kinases |
| MEF2 | Myocyte enhancer factor-2 |
| ncEFh | Non-canonical EF-hand |
| nNOS | Nitric oxide synthase |
| PIP2 | Phosphatidylinositol biphosphate |
| PLC | Phospholipase C |
| PM | Plasma membrane |
| PMCAs | Plasma membrane calcium ATPases |
| RYR1 | Ryanodine receptor type 1 |
| SAM | Sterile motif |
| SARAF | SOCE-associated regulatory factor |
| SCID | Severe combined immunodeficiency |
| SERCA | Sarco-/endoplasmic reticular calcium ATPase |
| SOAR | STIM1–Orai1 activating region |
| SOCCs | Store-operated-calcium channels |
| SOCE | Store-operated Ca2+ entry |
| STIM1 | Stromal-interacting molecule-1 |
| STRMK | Stormorken Syndrome |
| TAM | Tubular aggregate myopathy |
| TAs | Tubular aggregates |
| tBHQ | 2,5-di-(tert-butyl)-1,4-benzohydroquinone |
| TM | Transmembrane helices |
| TRPCs | Transient receptor potential canonical channels |
References
- Gehlert, S.; Bloch, W.; Suhr, F. Ca2+ dependent regulations and signaling in skeletal muscle: From electro-mechanical coupling. Int. J. Mol. Sci. 2015, 16, 1066–1095. [Google Scholar] [CrossRef] [PubMed]
- Böhm, J.; Bulla, M.; Urquhart, J.E.; Malfatti, E.; Williams, S.G.; O’Sullivan, J.; Szlauer, A.; Koch, C.; Baranello, G.; Mora, M.; et al. ORAI1 Mutations with Distinct Channel Gating Defects in Tubular Aggregate Myopathy. Hum. Mutat. 2017, 38, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Silva-Rojas, R.; Laporte, J.; Böhm, J. STIM1/ ORAI1 Loss-of-Function and Gain-of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases. Front. Physiol. 2020, 11, 604941. [Google Scholar] [CrossRef]
- Conte, E.; Pannunzio, A.; Imbrici, P.; Camerino, G.M.; Maggi, L.; Mora, M.; Gibertini, S.; Cappellari, O.; De Luca, A.; Coluccia, M.; et al. Gain-of-Function STIM1 L96V Mutation Causes Myogenesis Alteration in Muscle Cells From a Patient Affected by Tubular Aggregate Myopathy. Front. Cell Dev. Biol. 2021, 9, 635063. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Moloughney, J.G.; Zhang, S.; Komazaki, S.; Weisleder, N. Orai1 mediates exacerbated Ca(2+) entry in dystrophic skeletal muscle. PLoS ONE 2012, 7, e49862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraysse, B.; Liantonio, A.; Cetrone, M.; Burdi, R.; Pierno, S.; Frigeri, A.; Pisoni, M.; Camerino, C.; De Luca, A. The alteration of calcium homeostasis in adult dystrophic mdx muscle fibers is worsened by a chronic exercise in vivo. Neurobiol. Dis. 2004, 17, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Capogrosso, R.F.; Mantuano, P.; Uaesoontrachoon, K.; Cozzoli, A.; Giustino, A.; Dow, T.; Srinivassane, S.; Filipovic, M.; Bell, C.; Vandermeulen, J.; et al. Ryanodine channel complex stabilizer compound S48168/ARM210 as a disease modifier in dystrophin-deficient mdx mice: Proof-of-concept study and independent validation of efficacy. FASEB J. 2018, 32, 1025–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, E.; Camerino, G.M.; Mele, A.; De Bellis, M.; Pierno, S.; Rana, F.; Fonzino, A.; Caloiero, R.; Rizzi, L.; Bresciani, E.; et al. Growth hormone secretagogues prevent dysregulation of skeletal muscle calcium homeostasis in a rat model of cisplatin-induced cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 386–404. [Google Scholar] [CrossRef] [Green Version]
- Weisleder, N.; Brotto, M.; Komazaki, S.; Pan, Z.; Zhao, X.; Nosek, T.; Parness, J.; Takeshima, H.; Ma, J. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J. Cell Biol. 2006, 174, 639–645. [Google Scholar] [CrossRef]
- Fraysse, B.; Desaphy, J.F.; Rolland, J.F.; Pierno, S.; Liantonio, A.; Giannuzzi, V.; Camerino, C.; Didonna, M.P.; Cocchi, D.; De Luca, A.; et al. Fiber type-related changes in rat skeletal muscle calcium homeostasis during aging and restoration by growth hormone. Neurobiol. Dis. 2006, 21, 372–380. [Google Scholar] [CrossRef]
- Romero-Suarez, S.; Shen, J.; Brotto, L.; Hall, T.; Mo, C.; Valdivia, H.H.; Andresen, J.; Wacker, M.; Nosek, T.M.; Qu, C.K.; et al. Muscle-specific inositide phosphatase (MIP/MTMR14) is reduced with age and its loss accelerates skeletal muscle aging process by altering calcium homeostasis. Aging 2010, 2, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, A.M.; Zhao, X.; Weisleder, N.; Brotto, L.S.; Bougoin, S.; Nosek, T.M.; Reid, M.; Hardin, B.; Pan, Z.; Ma, J.; et al. Store-operated Ca(2+) entry (SOCE) contributes to normal skeletal muscle contractility in young but not in aged skeletal muscle. Aging 2011, 3, 621–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. The plasma membrane calcium pump in health and disease. FEBS J. 2013, 280, 5385–5397. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, C. How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim. Biophys. Acta 2009, 1793, 943–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, I.Y.; Ehrlich, B.E. Signaling in Muscle Contraction. Cold Spring Harb. Perspect. Biol. 2015, 7, a006023. [Google Scholar] [CrossRef]
- Cherednichenko, G.; Hurne, A.M.; Fessenden, J.D.; Lee, E.H.; Allen, P.D.; Beam, K.G.; Pessah, I.N. Conformational activation of Ca2+ entry by depolarization of skeletal myotubes. Proc. Natl. Acad. Sci. USA 2004, 101, 15793–15798. [Google Scholar] [CrossRef] [Green Version]
- Dirksen, R.T. Checking your SOCCs and feet: The molecular mechanisms of Ca2+ entry in skeletal muscle. J. Physiol. 2009, 587 Pt 13, 3139–3147. [Google Scholar] [CrossRef]
- Bannister, R.A.; Pessah, I.N.; Beam, K.G. The skeletal L-type Ca(2+) current is a major contributor to excitation-coupled Ca(2+) entry. J. Gen. Physiol. 2009, 133, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Jousset, H.; Frieden, M.; Demaurex, N. STIM1 knockdown reveals that store-operated Ca2+ channels located close to sarco/endoplasmic Ca2+ ATPases (SERCA) pumps silently refill the endoplasmic reticulum. J. Biol. Chem. 2007, 282, 11456–11464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Vig, M.; Beck, A.; Billingsley, J.M.; Lis, A.; Parvez, S.; Peinelt, C.; Koomoa, D.L.; Soboloff, J.; Gill, D.L.; Fleig, A.; et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 2006, 16, 2073–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Zhou, Y.; Wong, H.C.; Chen, Y.; Chen, Y.; Wang, S.; Castiblanco, A.; Liu, A.; Yang, J.J. A single EF-hand isolated from STIM1 forms dimer in the absence and presence of Ca2+. FEBS J. 2009, 276, 5589–5597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.J.; Hyun, C.; Woo, J.S.; Park, C.S.; Kim, D.H.; Lee, E.H. Stromal interaction molecule 1 (STIM1) regulates sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 1a (SERCA1a) in skeletal muscle. Pflugers Arch. 2014, 466, 987–1001. [Google Scholar] [CrossRef]
- Ong, H.L.; Cheng, K.T.; Liu, X.; Bandyopadhyay, B.C.; Paria, B.C.; Soboloff, J.; Pani, B.; Gwack, Y.; Srikanth, S.; Singh, B.B.; et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 2007, 282, 9105–9116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathopulos, P.; Li, G.; Plevin, M.; Ames, J.; Ikura, M. Stored Ca2+ depletion-induced oligomerization of STIM1 via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 2006, 281, 35855–35862. [Google Scholar] [CrossRef] [Green Version]
- Stathopulos, P.B.; Zheng, L.; Li, G.Y.; Plevin, M.J.; Ikura, M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008, 135, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.J.; Woo, J.S.; Hwang, J.H.; Hyun, C.; Cho, C.H.; Kim, D.H.; Lee, E.H. STIM1 negatively regulates Ca(2)(+) release from the sarcoplasmic reticulum in skeletal myotubes. Biochem. J. 2013, 453, 187–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, M.; Lewis, R.S. Numbers count: How STIM and Orai stoichiometry affect store-operated calcium entry. Cell Calcium 2019, 79, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Dyrda, A.; Koenig, S.; Frieden, M. STIM1 long and STIM1 gate differently TRPC1 during store-operated calcium entry. Cell Calcium 2020, 86, 102134. [Google Scholar] [CrossRef]
- Covington, E.D.; Wu, M.M.; Lewis, R.S. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol. Biol. Cell. 2010, 21, 1897–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Novello, M.J.; Zhu, J.; Feng, Q.; Ikura, M.; Stathopulos, P.B. Structural elements of stromal interaction molecule function. Cell Calcium 2018, 73, 88–94. [Google Scholar] [CrossRef]
- Derler, I.; Jardin, I.; Romanin, C. Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Cell Physiol. 2016, 310, C643–C662. [Google Scholar] [CrossRef] [Green Version]
- Jing, J.; He, L.; Sun, A.; Quintana, A.; Ding, Y.; Ma, G.; Tan, P.; Liang, X.; Zheng, X.; Chen, L.; et al. Proteomic mapping of ER–PM junctions identifies STIMATE as a regulator of Ca2+ influx. Nat. Cell Biol. 2015, 17, 1339–1347. [Google Scholar] [CrossRef] [Green Version]
- Quintana, A.; Rajanikanth, V.; Farber-Katz, S.; Gudlur, A.; Zhang, C.; Jing, J.; Zhou, Y.; Rao, A.; Hogan, P.G. TMEM110 regulates the maintenance and remodeling of mammalian ER-plasma membrane junctions competent for STIM-ORAI signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E7083–E7092. [Google Scholar] [CrossRef] [Green Version]
- Berlansky, S.; Humer, C.; Sallinger, M.; Frischauf, I. More Than Just Simple Interaction between STIM and Orai Proteins: CRAC Channel Function Enabled by a Network of Interactions with Regulatory Proteins. Int. J. Mol. Sci. 2021, 22, 471. [Google Scholar] [CrossRef]
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 2011, 194, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.N.; Murphy, R.M.; Cully, T.R.; von Wegner, F.; Friedrich, O.; Launikonis, B.S. Ultra-rapid activation and deactivation of store-operated Ca2+ entry in skeletal muscle. Cell Calcium 2010, 47, 458–467. [Google Scholar] [CrossRef]
- Launikonis, B.S.; Stephenson, D.G.; Friedrich, O. Rapid Ca2+flux through the transverse tubular membrane, activated by individual action potentials in mammalian skeletal muscle: Action potential activated Ca2+flux. J. Physiol. 2009, 587 Pt 10, 2299–2312. [Google Scholar] [CrossRef]
- Antigny, F.; Sabourin, J.; Sauc, S.; Bernheim, L.; Koenig, S.; Frieden, M. TRPC1 and TRPC4 channels functionally interact with STIM1L to promote myogenesis and maintain fast repetitive Ca(2+) release in human myotubes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 806–813. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Ikura, M. Structure and function of endoplasmic reticulum STIM calcium sensors. Curr. Top. Membr. 2013, 71, 59–93. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.S.; Hwang, S.Y.; Smyth, J.T.; Fukushima, M.; Boyles, R.R.; Putney, J.W., Jr. STIM1 is a calcium sensor specialized for digital signaling. Curr. Biol. 2009, 19, 1724–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, H.L.; de Souza, L.B.; Zheng, C.; Cheng, K.T.; Liu, X.; Goldsmith, C.M.; Feske, S.; Ambudkar, I.S. STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions. Sci. Signal. 2015, 8, ra3. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.R.; Lee, K.J.; Huang, M.; Kim, J.O.; Kim, D.H.; Cho, C.H.; Lee, E.H. STIM2 regulates both intracellular Ca(2+) distribution and Ca(2+) movement in skeletal myotubes. Sci. Rep. 2017, 7, 17936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjo, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol. 2015, 209, 653–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef] [Green Version]
- Wei-Lapierre, L.; Carrell, E.M.; Boncompagni, S.; Protasi, F.; Dirksen, R.T. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. 2013, 4, 2805. [Google Scholar] [CrossRef] [Green Version]
- Sztretye, M.; Geyer, N.; Vincze, J.; Al-Gaadi, D.; Olah, T.; Szentesi, P.; Kis, G.; Antal, M.; Balatoni, I.; Csernoch, L.; et al. SOCE Is Important for Maintaining Sarcoplasmic Calcium Content and Release in Skeletal Muscle Fibers. Biophys. J. 2017, 113, 2250–2496. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J.W. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J. Cell Sci. 2012, 125 Pt 18, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feske, S.; Prakriya, M.; Rao, A.; Lewis, R. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J. Exp. Med. 2005, 202, 651–662. [Google Scholar] [CrossRef]
- Soboloff, J.; Spassova, M.; Tang, X.; Hewavitharana, T.; Xu, W.; Gill, D. Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 2006, 281, 20661–20665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Navarro-Borelly, L.; McNally, B.; Prakriya, M. Orai1 mutations alter ion permeation and Ca2+-dependent fast inactivation of CRAC channels: Evidence for coupling of permeation and gating. J. Gen. Physiol. 2007, 130, 525–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothberg, B.S.; Wang, Y.; Gill, D.L. Orai channel pore properties and gating by STIM: Implications from the Orai crystal structure. Sci. Signal. 2013, 6, pe9. [Google Scholar] [CrossRef] [Green Version]
- Amcheslavsky, A.; Wood, M.L.; Yeromin, A.V.; Parker, I.; Freites, J.A.; Tobias, D.J.; Cahalan, M.D. Molecular biophysics of Orai store-operated Ca2+ channels. Biophys. J. 2015, 108, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Ramachandran, S.; Oh-Hora, M.; Rao, A.; Hogan, P.G. Pore architecture of the ORAI1 store-operated calcium channel. Proc. Natl. Acad. Sci. USA 2010, 107, 4896–4901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derler, I.; Plenk, P.; Fahrner, M.; Muik, M.; Jardin, I.; Schindl, R.; Gruber, H.J.; Groschner, K.; Romanin, C. The extended transmembrane Orai1 N-terminal (ETON) region combines binding interface and gate for Orai1 activation by STIM1. J. Biol. Chem. 2013, 288, 29025–29034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calloway, N.; Holowka, D.; Baird, B. A basic sequence in STIM1 promotes Ca2+ influx by interacting with the C-terminal acidic coiled coil of Orai1. Biochemistry 2010, 49, 1067–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butorac, C.; Muik, M.; Derler, I.; Stadlbauer, M.; Lunz, V.; Krizova, A.; Lindinger, S.; Schober, R.; Frischauf, I.; Bhardwaj, R.; et al. A novel STIM1-Orai1 gating interface essential for CRAC channel activation. Cell Calcium 2019, 79, 57–67. [Google Scholar] [CrossRef]
- Fahrner, M.; Pandey, S.K.; Muik, M.; Traxler, L.; Butorac, C.; Stadlbauer, M.; Zayats, V.; Krizova, A.; Plenk, P.; Frischauf, I.; et al. Communication between N terminus and loop2 tunes Orai activation. J. Biol. Chem. 2018, 293, 1271–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derler, I.; Butorac, C.; Krizova, A.; Stadlbauer, M.; Muik, M.; Fahrner, M.; Frischauf, I.; Romanin, C. Authentic CRAC channel activityrequires STIM1 and the conserved portion of the Orai N terminus. J. Biol. Chem. 2018, 293, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- Ahn, M.K.; Lee, K.J.; Cai, C.; Huang, M.; Cho, C.H.; Ma, J.; Lee, E.H. Mitsugumin 53 regulates extracellular Ca(2+) entry and intracellular Ca(2+) release via Orai1 and RyR1 in skeletal muscle. Sci. Rep. 2016, 6, 36909. [Google Scholar] [CrossRef] [Green Version]
- Hoth, M.; Niemeyer, B.A. The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr. Top. Membr. 2013, 71, 237–271. [Google Scholar] [CrossRef]
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The native ORAI channel trio underlies the diversity of Ca 2+ signaling events. Nat. Commun. 2020, 11, 2444. [Google Scholar] [CrossRef]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef]
- Kiviluoto, S.; Decuypere, J.P.; De Smedt, H.; Missiaen, L.; Parys, J.B.; Bultynck, G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet Muscle 2011, 1, 16. [Google Scholar] [CrossRef] [Green Version]
- Gees, M.; Colsoul, B.; Nilius, B. The Role of Transient Receptor Potential Cation Channels in Ca2+ Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.K.; Yeung, S.S.; Au, S.W.; Lam, L.S.; Dai, Z.Q.; Li, Y.H.; Yeung, E.W. Expression and association of TRPC1 with TRPC3 during skeletal myogenesis. Muscle Nerve 2011, 44, 358–365. [Google Scholar] [CrossRef]
- Kunert-Keil, C.; Bisping, F.; Krüger, J.; Brinkmeier, H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genom. 2006, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Hellmich, U.A.; Gaudet, R. Structural biology of TRP channels. Handb. Exp. Pharmacol. 2014, 223, 963–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, M.; Zanou, N.; Van Schoor, M.; Gailly, P. TRPC1 regulates skeletal myoblast migration and differentiation. J. Cell Sci. 2008, 121, 3951–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antigny, F.; Koenig, S.; Bernheim, L.; Frieden, M. During post-natal human myogenesis, normal myotube size requires TRPC1- and TRPC4-mediated Ca2+ entry. J. Cell Sci. 2013, 126 Pt 11, 2525–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanou, N.; Schakman, O.; Louis, P.; Ruegg, U.T.; Dietrich, A.; Birnbaumer, L.; Gailly, P. Trpc1 ion channel modulates phosphatidylinositol 3-kinase/Akt pathway during myoblast differentiation and muscle regeneration. J. Biol. Chem. 2012, 287, 14524–14534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, W.; Yuan, J.P.; Kim, M.S.; Choi, Y.J.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell. 2008, 32, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Zeng, W.; Yuan, J.P.; Shin, D.M.; Worley, P.F.; Muallem, S. Native store-operated Ca2+ influx requires the channel function of orai1 and TRPC1. J. Biol. Chem. 2009, 284, 9733–9741. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Ambudkar, I.S. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 2008, 283, 12935–12940. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Erxleben, C.; Yildirim, E.; Abramowitz, J.; Armstrong, D.L.; Birnbaumer, L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 4682–4687. [Google Scholar] [CrossRef] [Green Version]
- Horinouchi, T.; Higashi, T.; Higa, T.; Terada, K.; Mai, Y.; Aoyagi, H.; Hatate, C.; Nepal, P.; Horiguchi, M.; Harada, T.; et al. Different binding property of STIM1 and its novel splice variant STIM1L to Orai1, TRPC3, and TRPC6 channels. Biochem. Biophys. Res. Commun. 2012, 428, 252–258. [Google Scholar] [CrossRef]
- Stiber, J.; Hawkins, A.; Zhang, Z.S.; Wang, S.; Burch, J.; Graham, V.; Rosenberg, P. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008, 10, 688–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Kurebayashi, N.; Ogawa, Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J. Physiol. 2001, 533, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; van Rossum, D.B.; Patterson, R.L.; Ma, H.T.; Gill, D.L. The cellular and molecular basis of store-operated calcium entry. Nat. Cell Biol. 2002, 4, E263–E272. [Google Scholar] [CrossRef] [PubMed]
- Des Georges, A.; Clarke, O.B.; Zalk, R.; Yuan, Q.; Condon, K.J.; Grassucci, R.A.; Hendrickson, W.A.; Marks, A.R.; Frank, J. Structural Basis for Gating and Activation of RyR1. Cell 2016, 167, 145–157.e17. [Google Scholar] [CrossRef] [Green Version]
- Thastrup, O.; Cullen, P.J.; Drøbak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef] [Green Version]
- Putney, J.W., Jr. Pharmacology of capacitative calcium entry. Mol. Interv. 2001, 1, 84–94. [Google Scholar] [PubMed]
- Lopez, E.; Frischauf, I.; Jardin, I.; Derler, I.; Muik, M.; Cantonero, C.; Salido, G.M.; Smani, T.; Rosado, J.A.; Redondo, P.C. STIM1 phosphorylation at Y 316 modulates its interaction with SARAF and the activation of SOCE and I CRAC. J. Cell Sci. 2019, 132, jcs226019. [Google Scholar] [CrossRef] [Green Version]
- Luik, R.M.; Wang, B.; Prakriya, M.; Wu, M.M.; Lewis, R.S. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 2008, 454, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Shim, A.H.; Tirado-Lee, L.; Prakriya, M. Structural and functional mechanisms of CRAC channel regulation. J. Mol. Biol. 2015, 427, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Luik, R.M.; Wu, M.M.; Buchanan, J.; Lewis, R.S. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006, 174, 815–825. [Google Scholar] [CrossRef]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Cai, X.; Nwokonko, R.M.; Loktionova, N.A.; Wang, Y.; Gill, D.L. The STIM-Orai coupling interface and gating of the Orai1 channel. Cell Calcium 2017, 63, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.T.; Manji, S.S.; Parker, N.J.; Hancock, M.S.; Van Stekelenburg, L.; Eid, J.P.; Senior, P.V.; Kazenwadel, J.S.; Shandala, T.; Saint, R.; et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: Coding for a novel class of transmembrane proteins. Biochem. J. 2001, 357 Pt 3, 673–685. [Google Scholar] [CrossRef]
- Yazbeck, P.; Tauseef, M.; Kruse, K.; Amin, M.R.; Sheikh, R.; Feske, S.; Komarova, Y.; Mehta, D. STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca 2+ Entry. Sci. Rep. 2017, 7, 42758. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.N.; Friedrich, O.; Cully, T.R.; von Wegner, F.; Murphy, R.M.; Launikonis, B.S. Upregulation of store-operated Ca2+ entry in dystrophic mdx mouse muscle. Am. J. Physiol. Cell Physiol. 2010, 299, C42–C50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoover, P.J.; Lewis, R.S. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2011, 108, 13299–13304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 2963. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, X.; Wang, X.; Loktionova, N.A.; Cai, X.; Nwokonko, R.M.; Vrana, E.; Wang, Y.; Rothberg, B.S.; Gill, D.L. STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat. Commun. 2015, 6, 8395. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Nwokonko, R.M.; Cai, X.; Loktionova, N.A.; Abdulqadir, R.; Xin, P.; Niemeyer, B.A.; Wang, Y.; Trebak, M.; Gill, D.L. Cross-linking of Orai1 channels by STIM proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E3398–E3407. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Thillaiappan, N.B.; Taylor, C.W. The store-operated Ca 2+ entry complex comprises a small cluster of STIM1 associated with one Orai1 channel. Proc. Natl. Acad. Sci. USA 2021, 118, e2010789118. [Google Scholar] [CrossRef]
- Manjarrés, I.M.; Alonso, M.T.; García-Sancho, J. Calcium entry-calcium refilling (CECR) coupling between store-operated Ca2+ entry and sarco/endoplasmic reticulum Ca2+-ATPase. Cell Calcium 2011, 49, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.W.; Frieden, M.; Demaurex, N. Local cytosolic Ca2+ elevations are required for stromal interaction molecule 1 (STIM1) de-oligomerization and termination of store-operated Ca2+ entry. J. Biol. Chem. 2011, 286, 36448–36459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, R.S. Store-operated calcium channels: New perspectives on mechanism and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003970. [Google Scholar] [CrossRef] [Green Version]
- Trebak, M.; Zhang, W.; Ruhle, B.; Henkel, M.M.; González-Cobos, J.C.; Motiani, R.K.; Stolwijk, J.A.; Newton, R.L.; Zhang, X. What role for store-operated Ca2+ entry in muscle? Microcirculation 2013, 20, 330–336. [Google Scholar] [CrossRef] [Green Version]
- Carrell, E.M.; Coppola, A.R.; McBride, H.J.; Dirksen, R.T. Orai1 enhances muscle endurance by promoting fatigue-resistant type I fiber content but not through acute store-operated Ca2+ entry. FASEB J. 2016, 30, 4109–4119. [Google Scholar] [CrossRef] [Green Version]
- Darbellay, B.; Arnaudeau, S.; Konig, S.; Jousset, H.; Bader, C.; Demaurex, N.; Bernheim, L. STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J. Biol. Chem. 2009, 284, 5370–5380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boncompagni, S.; Michelucci, A.; Pietrangelo, L.; Dirksen, R.T.; Protasi, F. Exercise-dependent formation of new junctions that promote STIM1-Orai1 assembly in skeletal muscle. Sci. Rep. 2017, 7, 14286. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; McCarl, C.A.; Papolos, A.; Khalil, S.; Luthy, K.; Hivroz, C.; LeDeist, F.; Rieux-Laucat, F.; Rechavi, G.; Rao, A.; et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N. Engl. J. Med. 2009, 360, 1971–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, X.; Choi, R.H.; Launikonis, B.S. Store-operated Ca2+ entry is activated by every action potential in skeletal muscle. Commun. Biol. 2018, 1, 31. [Google Scholar] [CrossRef] [PubMed]
- Morin, G.; Biancalana, V.; Echaniz-Laguna, A.; Noury, J.B.; Lornage, X.; Moggio, M.; Ripolone, M.; Violano, R.; Marcorelles, P.; Maréchal, D.; et al. Tubular Aggregate Myopathy and Stormorken Syndrome: Mutation Spectrum and Genotype/Phenotype Correlation. Hum. Mutat. 2020, 41, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; García-Castañeda, M.; Boncompagni, S.; Dirksen, R.T. Role of STIM1/ORAI1-mediated store-operated Ca2+ entry in skeletal muscle physiology and disease. Cell Calcium 2018, 76, 101–115. [Google Scholar] [CrossRef] [PubMed]
- McCarl, C.A.; Picard, C.; Khalil, S.; Kawasaki, T.; Rother, J.; Papolos, A.; Kutok, J.; Hivroz, C.; Ledeist, F.; Plogmann, K.; et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clin. Immunol. 2009, 124, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Choi, M.; Richardson, A.S.; Reid, B.M.; Seymen, F.; Yildirim, M.; Tuna, E.; Gençay, K.; Simmer, J.P.; Hu, J.C. STIM1 and SLC24A4 are critical for enamel maturation. J. Dent. Res. 2014, 93 (Suppl. 7), 94S–100S. [Google Scholar] [CrossRef] [Green Version]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.N.; et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef] [Green Version]
- Böhm, J.; Chevessier, F.; Maues De Paula, A.; Koch, C.; Attarian, S.; Feger, C.; Hantaï, D.; Laforêt, P.; Ghorab, K.; Vallat, J.M.; et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 2013, 92, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Böhm, J.; Laporte, J. Gain-of-function mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and Stormorken syndrome. Cell Calcium 2018, 76, 1–9. [Google Scholar] [CrossRef]
- Walter, M.C.; Rossius, M.; Zitzelsberger, M.; Vorgerd, M.; Muller-Felber, W.; Ertl-Wagner, B.; Zhang, Y.; Brinkmeier, H.; Senderek, J.; Schoser, B. 50 years to diagnosis: Autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation. Neuromuscul. Disord. 2015, 25, 577–584. [Google Scholar] [CrossRef]
- Hedberg, C.; Niceta, M.; Fattori, F.; Lindvall, B.; Ciolfi, A.; D’Amico, A.; Tasca, G.; Petrini, S.; Tulinius, M.; Tartaglia, M.; et al. Childhood onset tubular aggregate myopathy associated with de novo STIM1 mutations. J. Neurol. 2014, 261, 870–876. [Google Scholar] [CrossRef]
- Endo, Y.; Noguchi, S.; Hara, Y.; Hayashi, Y.K.; Motomura, K.; Miyatake, S.; Murakami, N.; Tanaka, S.; Yamashita, S.; Kizu, R. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca2+ channels. Hum. Mol. Genet. 2015, 24, 637–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, E.; Burki, U.; Marini-Bettolo, C.; Neri, M.; Scotton, C.; Hudson, J.; Bertoli, M.; Evangelista, T.; Vroling, B.; Polvikoski, T. Complex phenotypes associated with STIM1 mutations in both coiled coil and EF-hand domains. Neuromuscul. Disord. 2017, 27, 861–872. [Google Scholar] [CrossRef]
- Morin, G.; Bruechle, N.O.; Singh, A.R.; Knopp, C.; Jedraszak, G.; Elbracht, M.; Bremond-Gignac, D.; Hartmann, K.; Sevestre, H.; Deutz, P.; et al. Gain-of-Function Mutation in STIM1 (P.R304W) Is Associated with Stormorken Syndrome. Hum. Mutat. 2014, 35, 1221–1232. [Google Scholar] [CrossRef]
- Borsani, O.; Piga, D.; Costa, S.; Govoni, A.; Magri, F.; Artoni, A.; Cinnante, C.M.; Fagiolari, G.; Ciscato, P.; Moggio, M.; et al. Stormorken Syndrome Caused by a p.R304W STIM1 Mutation: The First Italian Patient and a Review of the Literature. Front. Neurol. 2018, 9, 859. [Google Scholar] [CrossRef] [PubMed]
- Sura, A.; Jacher, J.; Neil, E.; McFadden, K.; Walkovich, K.; Hannibal, M. Chronic Thrombocytopenia as the Initial Manifestation of STIM1-Related Disorders. Pediatrics 2020, 145, e20192081. [Google Scholar] [CrossRef]
- Bulla, M.; Gyimesi, G.; Kim, J.H.; Bhardwaj, R.; Hediger, M.A.; Frieden, M.; Demaurex, N. ORAI1 channel gating and selectivity is differentially altered by natural mutations in the first or third transmembrane domain. J. Physiol. 2019, 597, 561–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordero-Sanchez, C.; Riva, B.; Reano, S.; Clemente, N.; Zaggia, I.; Ruffinatti, F.A.; Potenzieri, A.; Pirali, T.; Raffa, S.; Sangaletti, S.; et al. A luminal EF-hand mutation in STIM1 in mice causes the clinical hallmarks of tubular aggregate myopathy. Dis. Model. Mech. 2020, 13, dmm041111. [Google Scholar] [CrossRef] [Green Version]
- Silva-Rojas, R.; Treves, S.; Jacobs, H.; Kessler, P.; Messaddeq, N.; Laporte, J.; Böhm, J. STIM1 over-activation generates a multi-systemic phenotype affecting the skeletal muscle, spleen, eye, skin, bones and immune system in mice. Hum. Mol. Genet. 2019, 28, 1579–1593. [Google Scholar] [CrossRef]
- Silva-Rojas, R.; Charles, A.L.; Djeddi, S.; Geny, B.; Laporte, J.; Böhm, J. Pathophysiological Effects of Overactive STIM1 on Murine Muscle Function and Structure. Cells 2021, 10, 1730. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. 1987. Biotechnology 1992, 24, 457–466. [Google Scholar] [PubMed]
- Lynch, G.S.; Rafael, J.A.; Chamberlain, J.S.; Faulkner, J.A. Contraction-induced injury to single permeabilized muscle fibers from mdx, transgenic mdx, and control mice. Am. J. Physiol. Cell Physiol. 2000, 279, C1290–C1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Kodippili, K.; Yue, Y.; Hakim, C.H.; Wasala, L.; Pan, X.; Zhang, K.; Yang, N.N.; Duan, D.; Lai, Y. Dystrophin contains multiple independent membrane-binding domains. Hum. Mol. Genet. 2016, 25, 3647–3653. [Google Scholar] [CrossRef] [Green Version]
- Mallouk, N.; Jacquemond, V.; Allard, B. Elevated subsarcolemmal Ca2+ in mdx mouse skeletal muscle fibers detected with Ca2+-activated K+ channels. Proc. Natl. Acad. Sci. USA 2000, 97, 4950–4955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdi, R.; Rolland, J.F.; Fraysse, B.; Litvinova, K.; Cozzoli, A.; Giannuzzi, V.; Liantonio, A.; Camerino, G.M.; Sblendorio, V.; Capogrosso, R.F.; et al. Multiple pathological events in exercised dystrophic mdx mice are targeted by pentoxifylline: Outcome of a large array of in vivo and ex vivo tests. J. Appl. Physiol. 2009, 106, 1311–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolland, J.F.; De Luca, A.; Burdi, R.; Andreetta, F.; Confalonieri, P.; Conte Camerino, D. Overactivity of exercise-sensitive cation channels and their impaired modulation by IGF-1 in mdx native muscle fibers: Beneficial effect of pentoxifylline. Neurobiol. Dis. 2006, 24, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Shkryl, V.M.; Martins, A.S.; Ullrich, N.D.; Nowycky, M.C.; Niggli, E.; Shirokova, N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 2009, 458, 915–928. [Google Scholar] [CrossRef]
- Murthy, S.E.; Dubin, A.E.; Patapoutian, A. Piezos thrive under pressure: Mechanically activated ion channels in health and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 771–783. [Google Scholar] [CrossRef]
- Robert, V.; Massimino, M.L.; Tosello, V.; Marsault, R.; Cantini, M.; Sorrentino, V.; Pozzan, T. Alteration in calcium handling at the subcellular level in mdx myotubes. J. Biol. Chem. 2001, 276, 4647–4651. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Nico, B.; Liantonio, A.; Didonna, M.P.; Fraysse, B.; Pierno, S.; Burdi, R.; Mangieri, D.; Rolland, J.F.; Camerino, C.; et al. A multidisciplinary evaluation of the effectiveness of cyclosporine a in dystrophic mdx mice. Am. J. Pathol. 2005, 166, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Gervasio, O.L.; Whitehead, N.P.; Yeung, E.W.; Phillips, W.D.; Allen, D.G. TRPC1 binds to caveolin-3 and is regulated by Src kinase—Role in Duchenne muscular dystrophy. J. Cell Sci. 2008, 121, 2246–2255. [Google Scholar] [CrossRef] [Green Version]
- Boittin, F.X.; Shapovalov, G.; Hirn, C.; Ruegg, U.T. Phospholipase A2-derived lysophosphatidylcholine triggers Ca2+ entry in dystrophic skeletal muscle fibers. Biochem. Biophys. Res. Commun. 2010, 391, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Goonasekera, S.A.; Davis, J.; Kwong, J.Q.; Accornero, F.; Wei-LaPierre, L.; Sargent, M.A.; Dirksen, R.T.; Molkentin, J.D. Enhanced Ca2+ influx from STIM1-Orai1 induces muscle pathology in mouse models of muscular dystrophy. Hum. Mol. Genet. 2014, 23, 3706–3715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millay, D.P.; Goonasekera, S.A.; Sargent, M.A.; Maillet, M.; Aronow, B.J.; Molkentin, J.D. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 19023–19028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandebrouck, C.; Martin, D.; Colson-Van Schoor, M.; Debaix, H.; Gailly, P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J. Cell Biol. 2002, 158, 1089–1096. [Google Scholar] [CrossRef]
- Sakuma, K.; Aoi, W.; Yamaguchi, A. Molecular mechanism of sarcopenia and cachexia: Recent research advances. Pflug. Arch. 2017, 469, 573–591. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer. 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Da Rocha, I.M.G.; Marcadenti, A.; de Medeiros, G.O.C.; Bezerra, R.A.; de Rego, J.F.M.; Gonzalez, M.C.; Trussardi Fayh, A.P. Is cachexia associated with chemotherapy toxicities in gastrointestinal cancer patients? A prospective study. J. Cachexia Sarcopenia Muscle 2019, 10, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizawa, T.; Sorimachi, H.; Tomioka, S.; Ishiura, S.; Suzuki, K. Calpain dissociates into subunits in the presence of calcium ions. Biochem. Biophys. Res. Commun. 1995, 208, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Singer, B.D.; Vaughan, D.E. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 2017, 16, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Frontera, W.R.; Suh, D.; Krivickas, L.S.; Hughes, V.A.; Goldstein, R.; Roubenoff, R. Skeletal muscle fiber quality in older men and women. Am. J. Physiol. Cell Physiol. 2000, 279, C611–C618. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef] [Green Version]
- Delbono, O. Molecular mechanisms and therapeutics of the deficit in specific force in ageing skeletal muscle. Biogerontology 2002, 3, 265–270. [Google Scholar] [CrossRef]
- Delbono, O.; O’Rourke, K.S.; Ettinger, W.H. Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J. Membr. Biol. 1995, 148, 211–222. [Google Scholar] [CrossRef]
- Jang, Y.C.; Lustgarten, M.S.; Liu, Y.; Muller, F.L.; Bhattacharya, A.; Liang, H.; Salmon, A.B.; Brooks, S.V.; Larkin, L.; Hayworth, C.R.; et al. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010, 24, 1376–1390. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Weisleder, N.; Thornton, A.; Oppong, Y.; Campbell, R.; Ma, J.; Brotto, M. Compromised storeoperated Ca2+ entry in aged skeletal muscle. Aging Cell 2008, 7, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Zahn, J.M.; Sonu, R.; Vogel, H.; Crane, E.; Mazan-Mamczarz, K.; Rabkin, R.; Davis, R.W.; Becker, K.G.; Owen, A.B.; Kim, S.K. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006, 2, e115. [Google Scholar] [CrossRef]
- Boncompagni, S.; Pecorai, C.; Michelucci, A.; Pietrangelo, L.; Protasi, F. Long-Term Exercise Reduces Formation of Tubular Aggregates and Promotes Maintenance of Ca2+ Entry Units in Aged Muscle. Front. Physiol. 2021, 11, 601057. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.N.; Blackmore, D.G.; Gilbert, D.F.; Murphy, R.M.; Launikonis, B.S. Store-operated calcium entry remains fully functional in aged mouse skeletal muscle despite a decline in STIM1 protein expression. Aging Cell 2011, 10, 675–685. [Google Scholar] [CrossRef]
- Zhao, X.; Yoshida, M.; Brotto, L.; Takeshima, H.; Weisleder, N.; Hirata, Y.; Nosek, T.M.; Ma, J.; Brotto, M. Enhanced resistance to fatigue and altered calcium handling properties of sarcalumenin knockout mice. Physiol. Genom. 2005, 23, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Boncompagni, S.; Pietrangelo, L.; Takano, T.; Protasi, F.; Dirksen, R.T. Pre-assembled Ca2+ entry units and constitutively active Ca2+ entry in skeletal muscle of calsequestrin-1 knockout mice. J. Gen. Physiol. 2020, 152, e202012617. [Google Scholar] [CrossRef]
- Demaurex, N.; Poburko, D.; Frieden, M. Regulation of plasma membrane calcium fluxes by mitochondria. Biochim. Biophys. Acta 2009, 1787, 1383–1394. [Google Scholar] [CrossRef]
- Deak, A.T.; Blass, S.; Khan, M.J.; Groschner, L.N.; Waldeck-Weiermair, M.; Hallstrom, S.; Graier, W.F.; Malli, R. IP3-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. J. Cell Sci. 2014, 127, 2944–2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoth, M.; Fanger, C.M.; Lewis, R.S. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J. Cell Biol. 1997, 137, 633–648. [Google Scholar] [CrossRef] [Green Version]
- Quintana, A.; Schwarz, E.C.; Schwindling, C.; Lipp, P.; Kaestner, L.; Hoth, M. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. J. Biol. Chem. 2006, 281, 40302–40309. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Rahman, T. Unveiling some FDA-approved drugs as inhibitors of the store-operated Ca 2+ entry pathway. Sci. Rep. 2017, 7, 12881. [Google Scholar] [CrossRef] [Green Version]
- Riva, B.; Griglio, A.; Serafini, M.; Cordero-Sanchez, C.; Aprile, S.; Di Paola, R.; Gugliandolo, E.; Alansary, D.; Biocotino, I.; Lim, D.; et al. Pyrtriazoles, a Novel Class of Store-Operated Calcium Entry Modulators: Discovery, Biological Profiling, and in Vivo Proof-of-Concept Efficacy in Acute Pancreatitis. J. Med. Chem. 2018, 61, 9756–9783. [Google Scholar] [CrossRef]
- Le Guilcher, C.; Luyten, T.; Parys, J.B.; Pucheault, M.; Dellis, O. Synthesis and Characterization of Store-Operated Calcium Entry Inhibitors Active in the Submicromolar Range. Int. J. Mol. Sci. 2020, 21, 9777. [Google Scholar] [CrossRef]
- Meizoso-Huesca, A.; Launikonis, B.S. The Orai1 inhibitor BTP2 has multiple effects on Ca2+ handling in skeletal muscle. J. Gen. Physiol. 2021, 153, e202012747. [Google Scholar] [CrossRef]
- Shimizu-Motohashi, Y.; Komaki, H.; Motohashi, N.; Takeda, S.; Yokota, T.; Aoki, Y. Restoring Dystrophin Expression in Duchenne Muscular Dystrophy: Current Status of Therapeutic Approaches. J. Pers. Med. 2019, 9, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillan, H.J. Intermittent glucocorticoid regimes for younger boys with duchenne muscular dystrophy: Balancing efficacy with side effects. Muscle Nerve 2019, 59, 638–639. [Google Scholar] [CrossRef] [PubMed]
- Bello, L.; Gordish-Dressman, H.; Morgenroth, L.P.; Henricson, E.K.; Duong, T.; Hoffman, E.P.; Cnaan, A.; McDonald, C.M. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurology 2015, 85, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Heslop, E.; Turner, C.; Irvin, A.; Muntoni, F.; Straub, V.; Guglieri, M. Gene therapy in Duchenne muscular dystrophy: Identifying and preparing for the challenges ahead. Neuromusc. Dis. 2021, 31, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, F.; Rossi, R.; Falzarano, M.S.; Ferlini, A. Innovative Therapeutic Approaches for Duchenne Muscular Dystrophy. J. Clin. Med. 2021, 10, 820. [Google Scholar] [CrossRef]
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 2005, 562 Pt 2, 367–380. [Google Scholar] [CrossRef]
- Cereda, E.; Turri, A.; Klersy, C.; Cappello, S.; Ferrari, A.; Filippi, A.R.; Brugnatelli, S.; Caraccia, M.; Chiellino, S.; Borioli, V.; et al. Whey protein isolate supplementation improves body composition, muscle strength, and treatment tolerance in malnourished advanced cancer patients undergoing chemotherapy. Cancer Med. 2019, 8, 6923–6932. [Google Scholar] [CrossRef]
- Anderson, L.J.; Albrecht, E.D.; Garcia, J.M. Update on management of cancer-related cachexia. Curr. Oncol. Rep. 2017, 19, 3. [Google Scholar] [CrossRef]
- Pierno, S.; De Luca, A.; Desaphy, J.F.; Fraysse, B.; Liantonio, A.; Didonna, M.P.; Lograno, M.; Cocchi, D.; Smith, R.G.; Conte Camerino, D. Growth hormone secretagogues modulate the electrical and contractile properties of rat skeletal muscle through a ghrelin-specific receptor. Br. J. Pharmacol. 2003, 139, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.A.; Splenser, A.; Guillory, B.; Luo, J.; Mendiratta, M.; Belinova, B.; Halder, T.; Zhang, G.; Li, Y.P.; Garcia, J.M. Ghrelin prevents tumour- and cisplatin-induced muscle wasting: Characterization of multiple mechanisms involved. J. Cachexia Sarcopenia Muscle 2015, 6, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Anker, S.D. Treatment of cachexia: An overview of recent developments. Int. J. Cardiol. 2015, 184, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Skeletal Muscle Diseases | SOCE Activity | Underlying Mechanism | REF |
|---|---|---|---|
| CRAC channelopathy: SCID, autoimmunity, ectodermal dysplasia, defects in sweat gland function and dental enamel formation, and muscle hypotonia | Reduced SOCE | Genetic Orai1/STIM1 mutations that lead to a reduced Orai1/STIM1 functionality. | [3,21,115,116,117] |
| Tubular Aggregate Myopathy (TAM) Stormorken Syndrome | Increased SOCE | Genetic Orai1/STIM1 mutations causing the production of a constitutively active protein. | [2,3,4,118,120,123,126,127,128] |
| Duchenne Muscular Dystrophy | Increased SOCE | Orai1 upregulation or STIM1 and/or TRPCs overexpression. | [6,7,99,136,141,143,145,146,147] |
| Sarcopenia | Reduced SOCE | Decrease in DHPR expression and consequent uncoupling between DHPR and RYR1 proteins. STIM1/Orai1 expression was unchanged. | [12,13,159,160,162] |
| Cachexia | Reduced SOCE | STIM1/Orai1 downregulation. | [8,152] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conte, E.; Imbrici, P.; Mantuano, P.; Coppola, M.A.; Camerino, G.M.; De Luca, A.; Liantonio, A. Alteration of STIM1/Orai1-Mediated SOCE in Skeletal Muscle: Impact in Genetic Muscle Diseases and Beyond. Cells 2021, 10, 2722. https://doi.org/10.3390/cells10102722
Conte E, Imbrici P, Mantuano P, Coppola MA, Camerino GM, De Luca A, Liantonio A. Alteration of STIM1/Orai1-Mediated SOCE in Skeletal Muscle: Impact in Genetic Muscle Diseases and Beyond. Cells. 2021; 10(10):2722. https://doi.org/10.3390/cells10102722
Chicago/Turabian StyleConte, Elena, Paola Imbrici, Paola Mantuano, Maria Antonietta Coppola, Giulia Maria Camerino, Annamaria De Luca, and Antonella Liantonio. 2021. "Alteration of STIM1/Orai1-Mediated SOCE in Skeletal Muscle: Impact in Genetic Muscle Diseases and Beyond" Cells 10, no. 10: 2722. https://doi.org/10.3390/cells10102722
APA StyleConte, E., Imbrici, P., Mantuano, P., Coppola, M. A., Camerino, G. M., De Luca, A., & Liantonio, A. (2021). Alteration of STIM1/Orai1-Mediated SOCE in Skeletal Muscle: Impact in Genetic Muscle Diseases and Beyond. Cells, 10(10), 2722. https://doi.org/10.3390/cells10102722