
An Oil Hyper-Accumulator Mutant Highlights Peroxisomal ATP Import as a Regulatory Step for Fatty Acid Metabolism in Aurantiochytrium limacinum

 , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
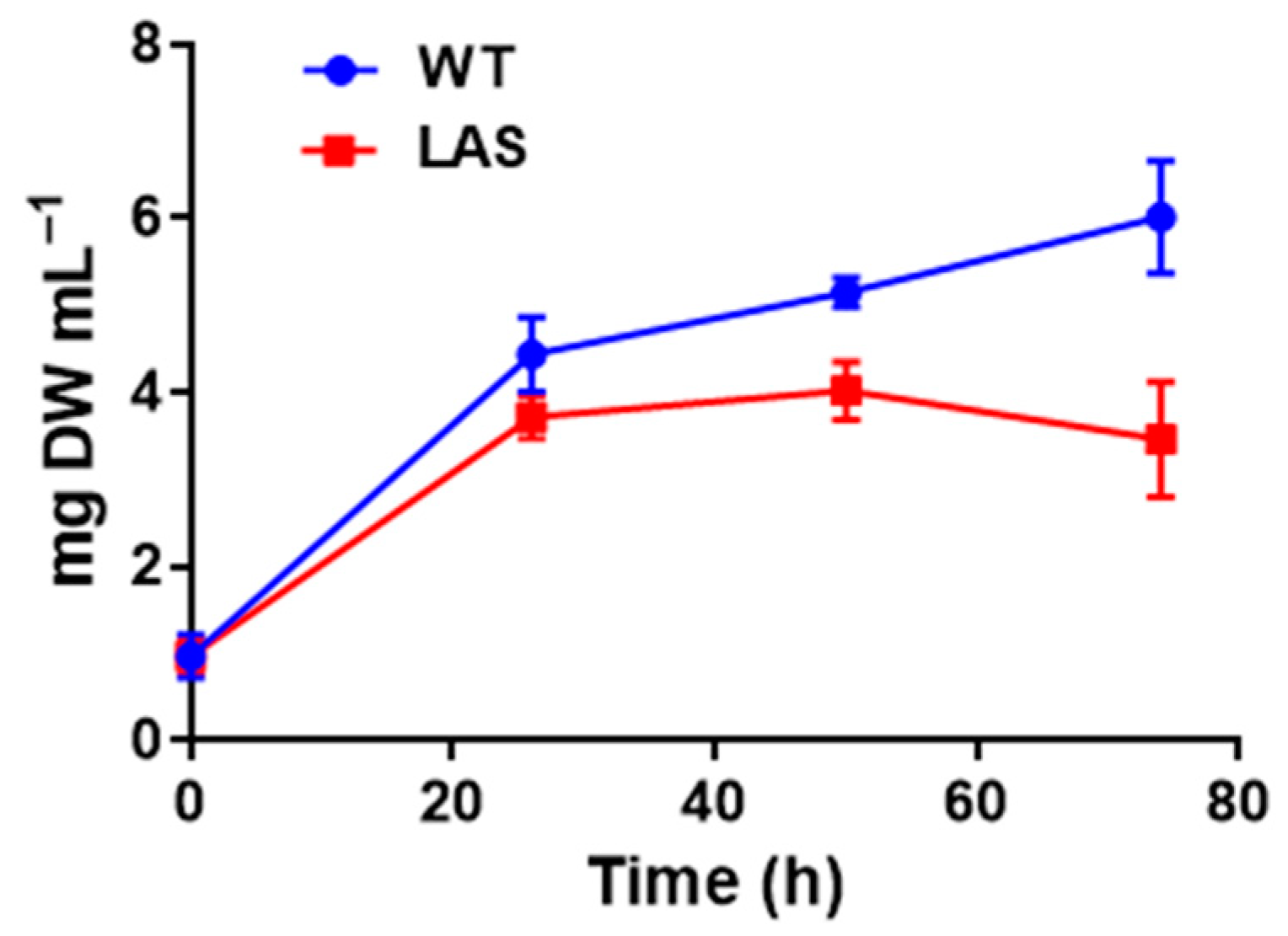
2.1. Aurantiochytrium limacinum Strain, Culture Media, and Growth Experiments
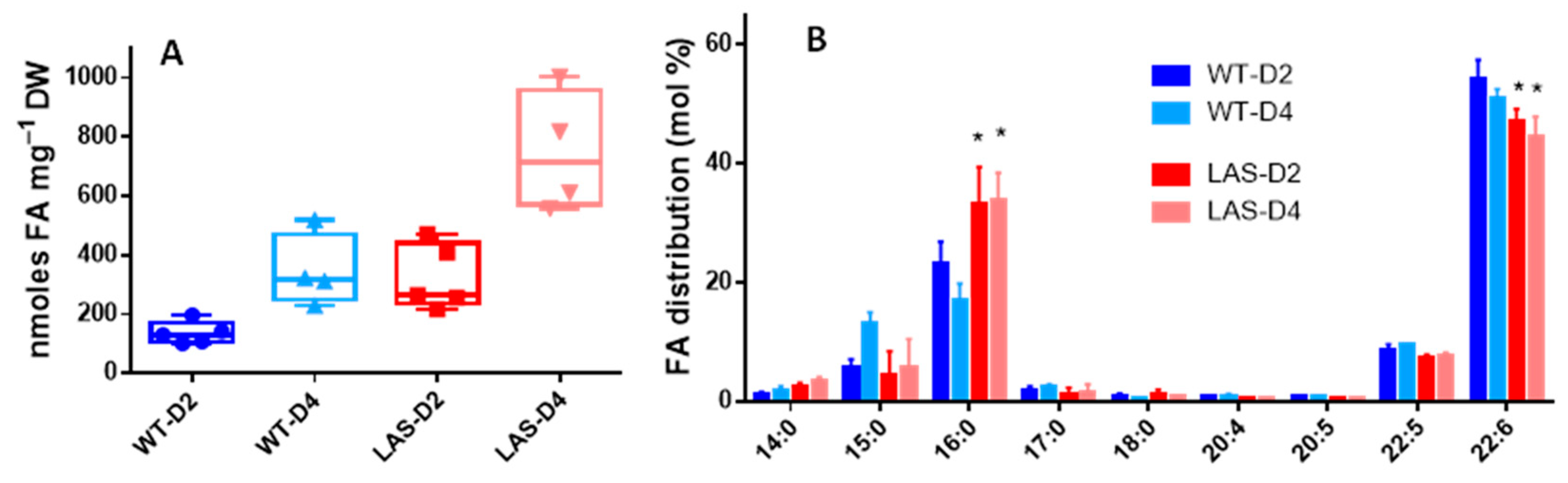
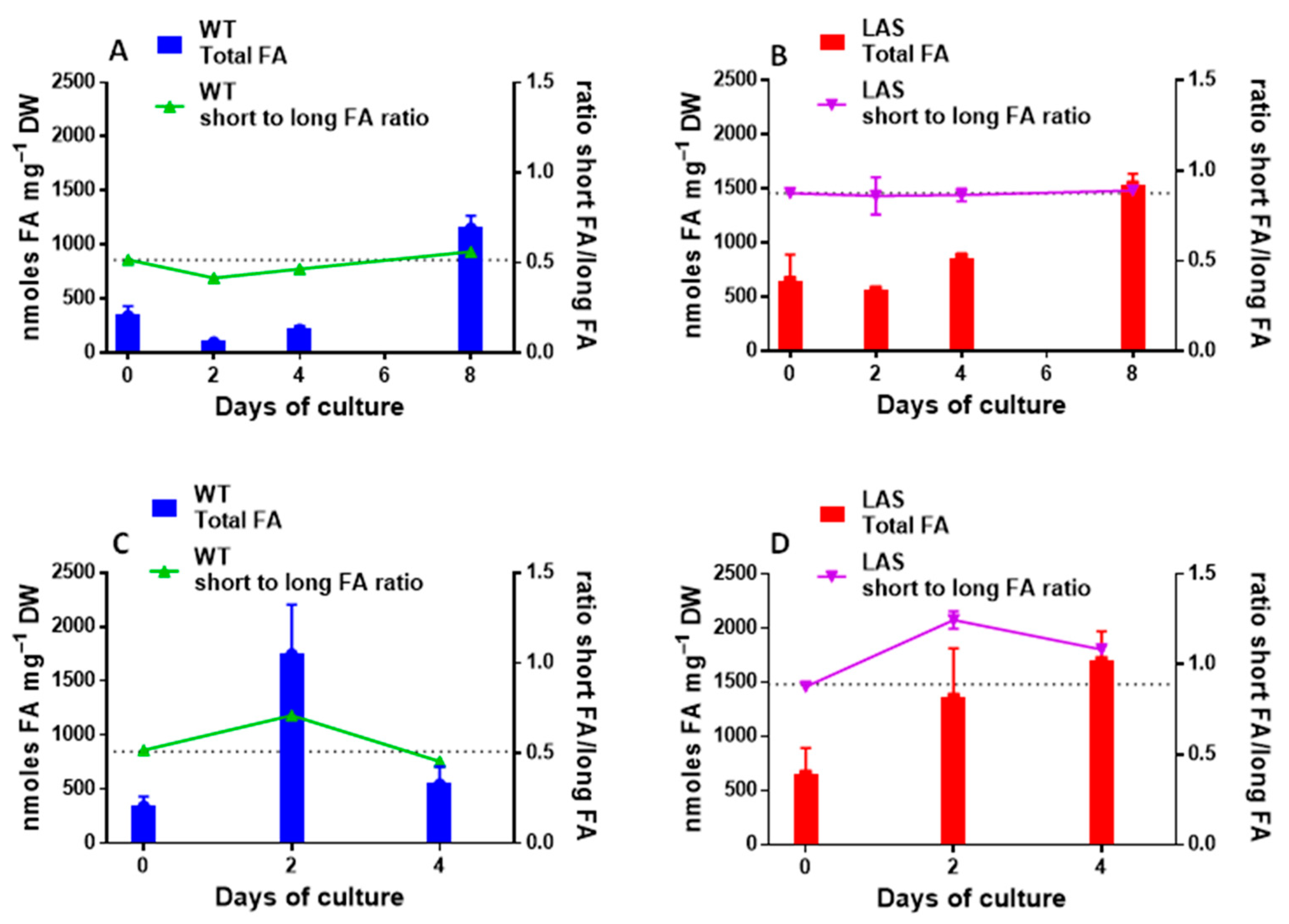
2.2. Lipid Analyses
2.3. Genomic, Transcriptomic, and qRT-PCR Analyses
2.4. Yeast Cultures and Transformations
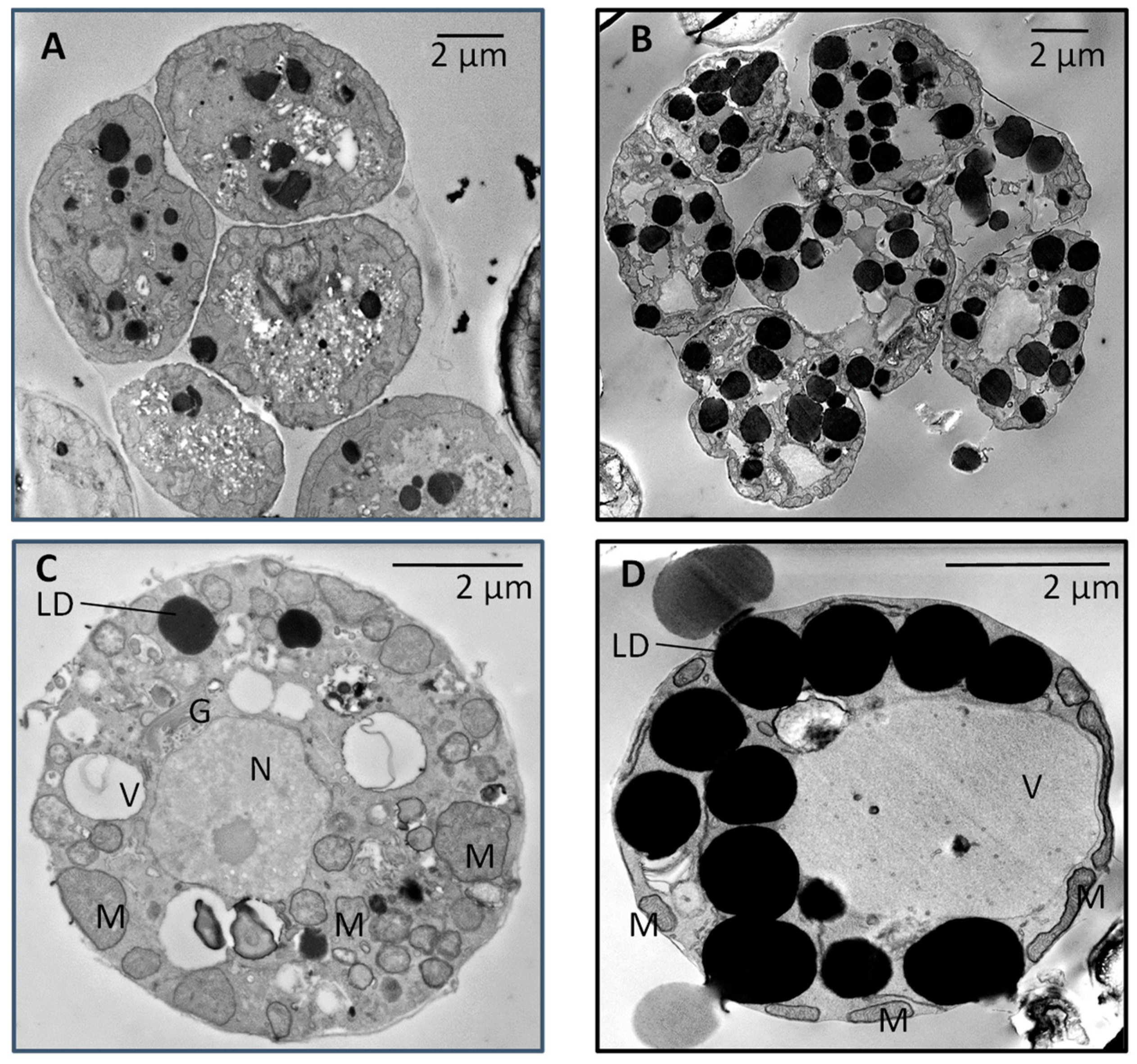
2.5. Sample Preparation for Electronic Microscopy
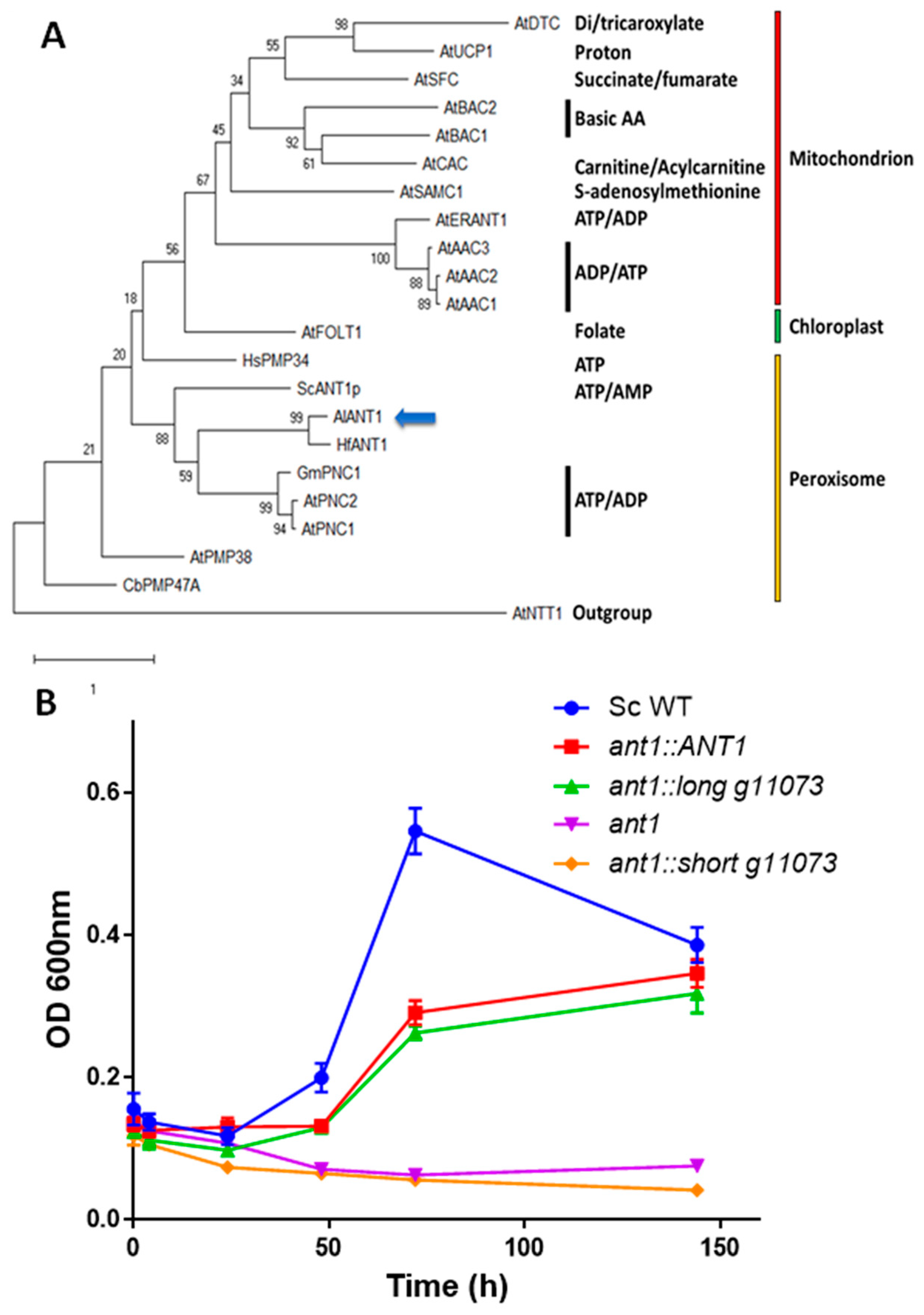
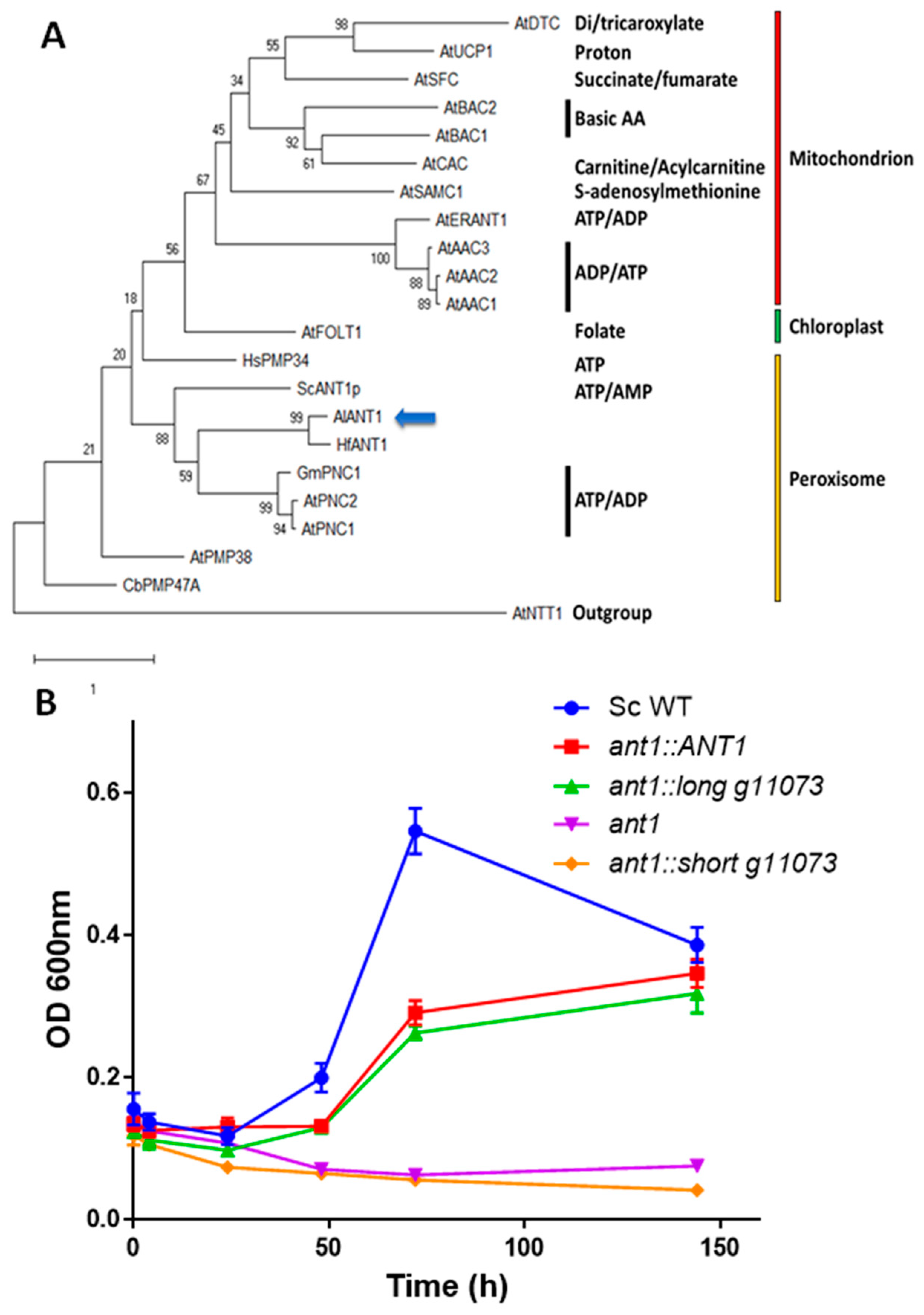
2.6. Phylogenetic Analyses
3. Results
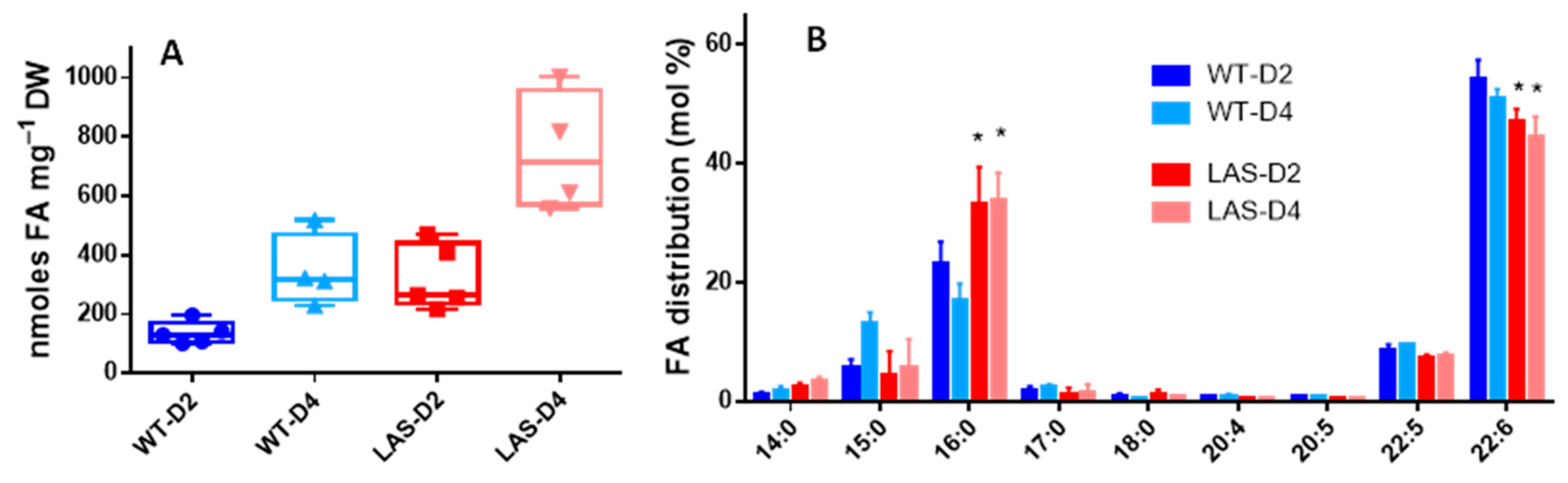
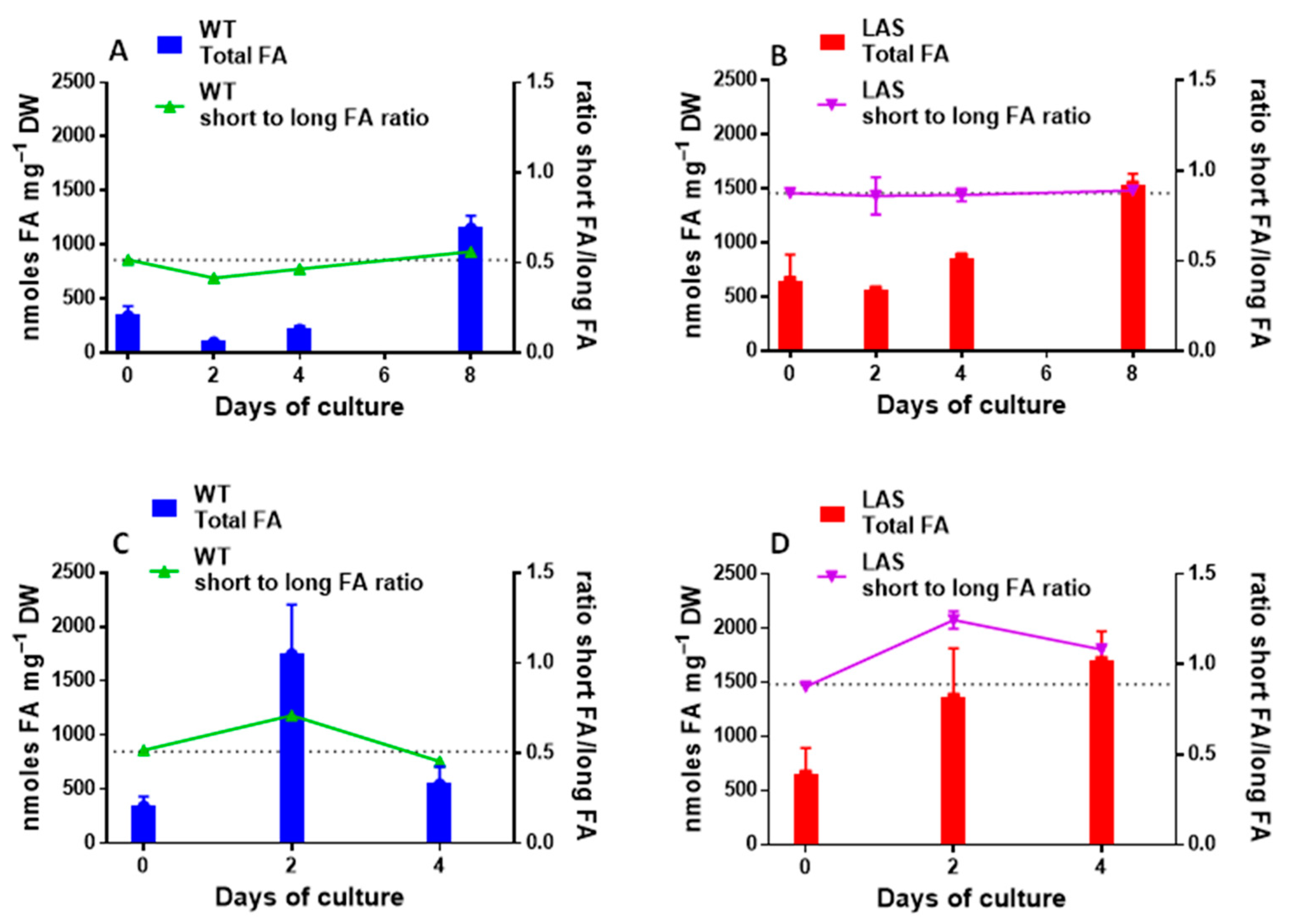
3.1. Growth and Fatty Acid Content of LAS and WT
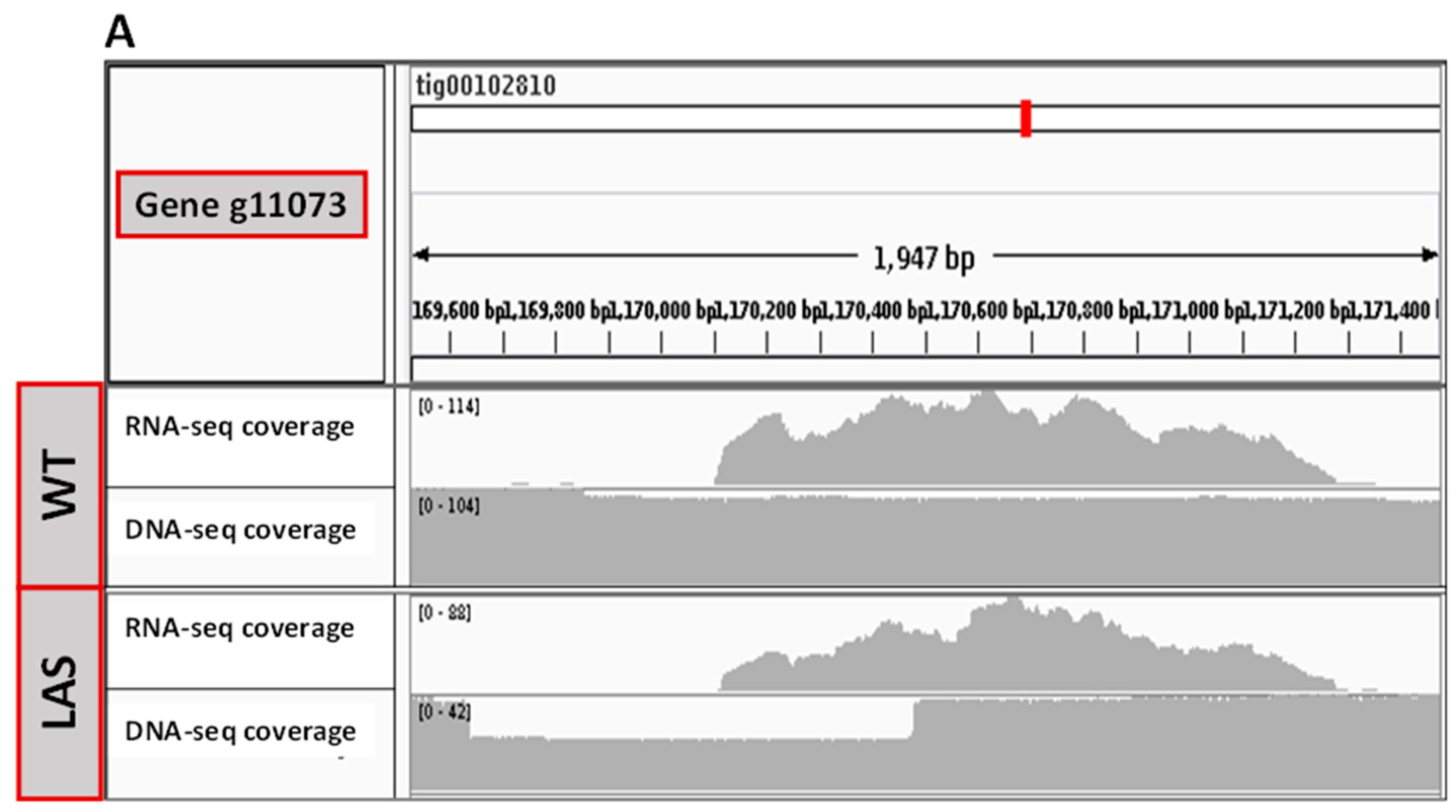
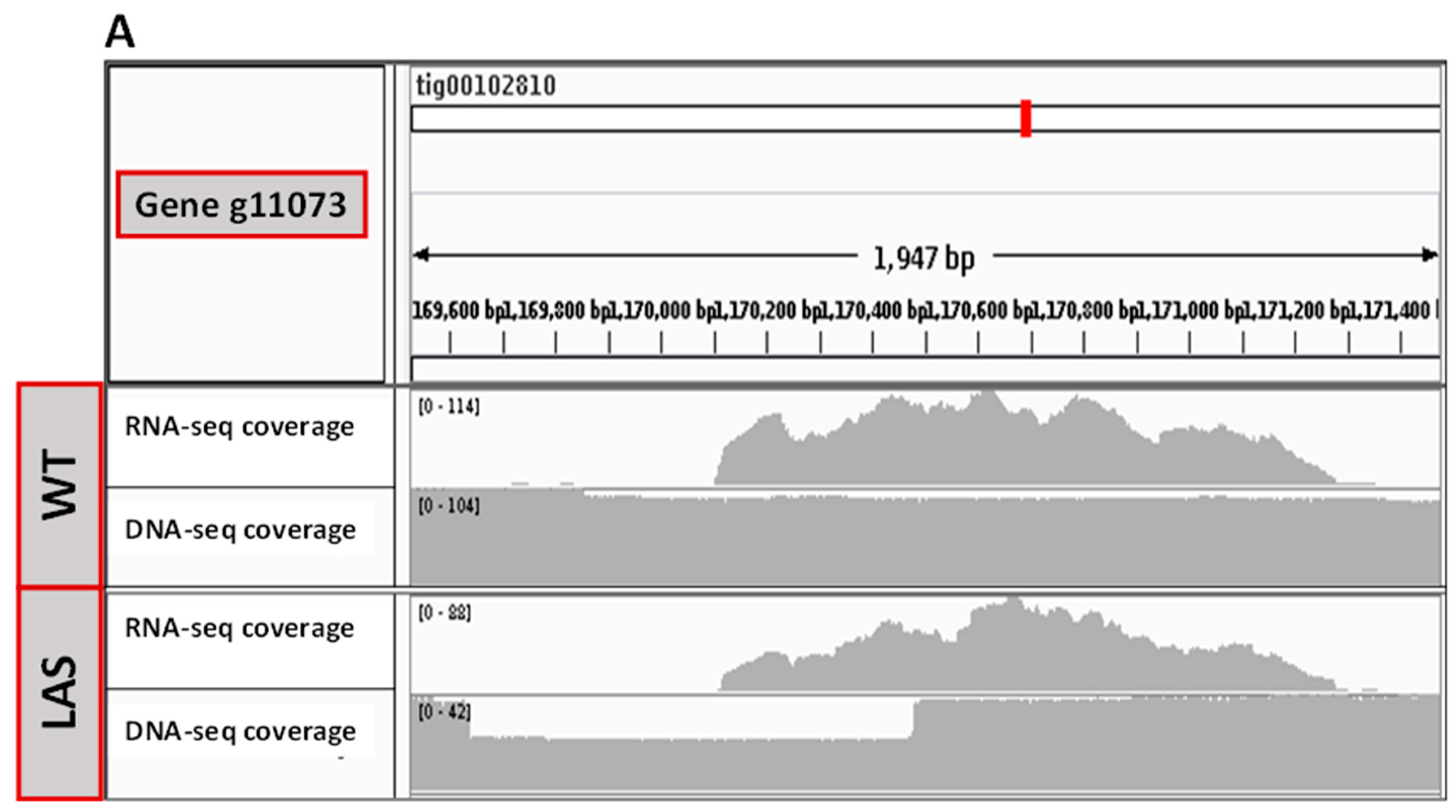
3.2. Transcriptomic and Genomic Analyses of LAS
3.3. Functional Characterization of g11073
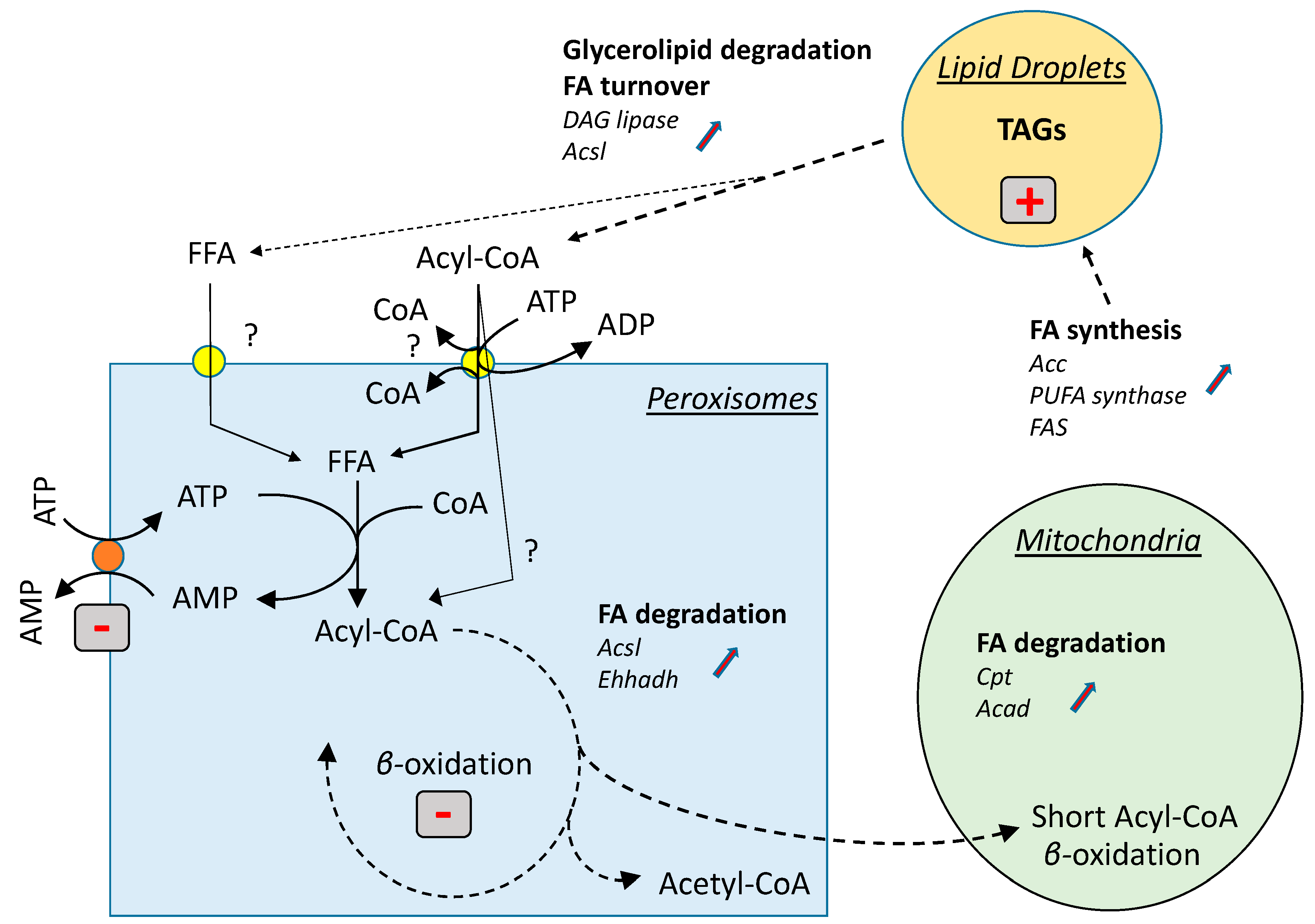
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Genomic and Transcriptomic Approach
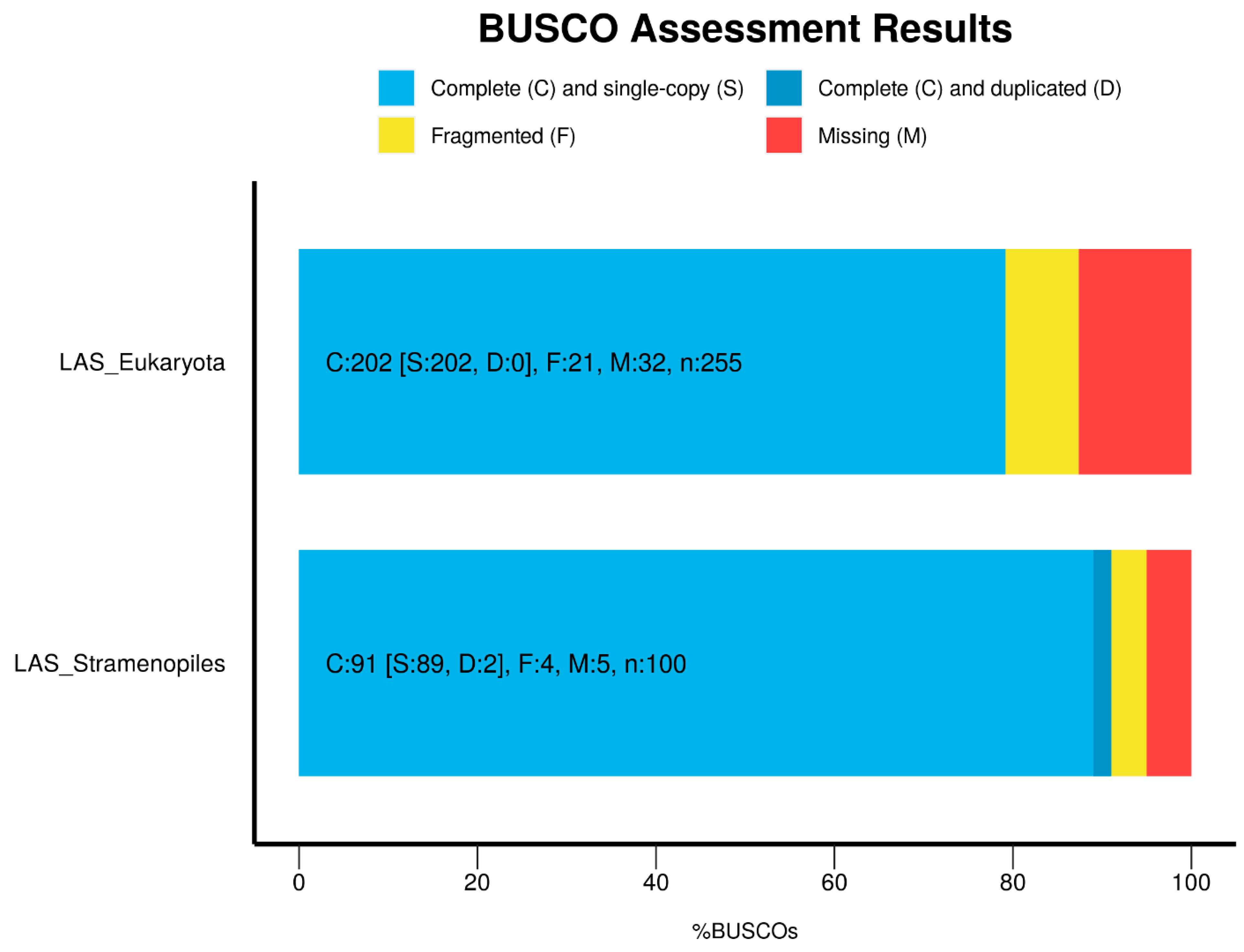
Appendix A.2. Genome Assembly

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PacBio Sequel I | |
|---|---|
| Sequenced Bases | 5,967,660,711 bp |
| Number of Reads | 676,120 |
| Sequencing Layout | Single End Long Reads |
| Max Read Length | 70,183 bp |
| Read N50 | 15,440 bp |
| Estimate Genome Coverage | 97× |
| Aurantiochytrium limacinum LAS | |
|---|---|
| Number of Scaffolds | 147 |
| Genome Size | 59,284,731 bp |
| Number of Contigs larger than 50 Kbp | 106 |
| N50 | 1,222,801 bp |
| L50 | 16 |
| Largest Contig | 3,306,648 bp |
| GC Content | 45.28% |

Appendix A.3. RNA Sequencing
| Sample ID | Raw Reads | Trimmed Reads |
|---|---|---|
| CCAP_4062/1_A | 43,636,868 | 39,160,710 |
| CCAP_4062/1_B | 28,851,896 | 25,815,902 |
| CCAP_4062/1_C | 33,658,698 | 29,801,748 |
| LAS_4A | 32,324,384 | 28,722,048 |
| LAS_4B | 31,644,014 | 28,589,096 |
| LAS_4C | 41,736,058 | 38,014,092 |
| Sample ID | Uniquely Mapped (%) | Multi-Mapped (%) |
|---|---|---|
| CCAP_4062/1_A | 91.05% | 2.06% |
| CCAP_4062/1_B | 91.24% | 2.02% |
| CCAP_4062/1_C | 90.81% | 2.14% |
| LAS_4A | 94.23% | 1.76% |
| LAS_4B | 93.58% | 1.80% |
| LAS_4C | 94.06% | 1.77% |
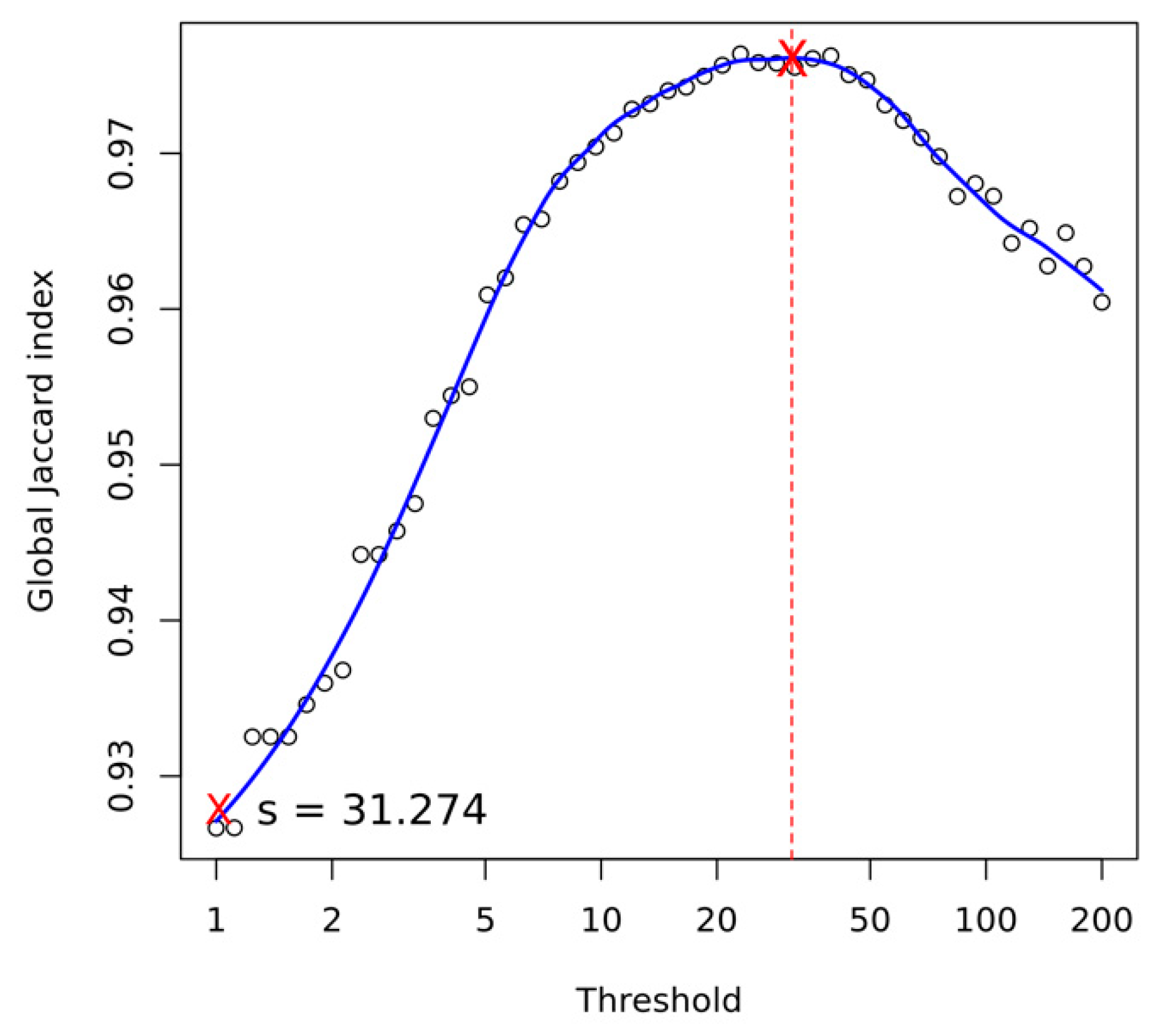
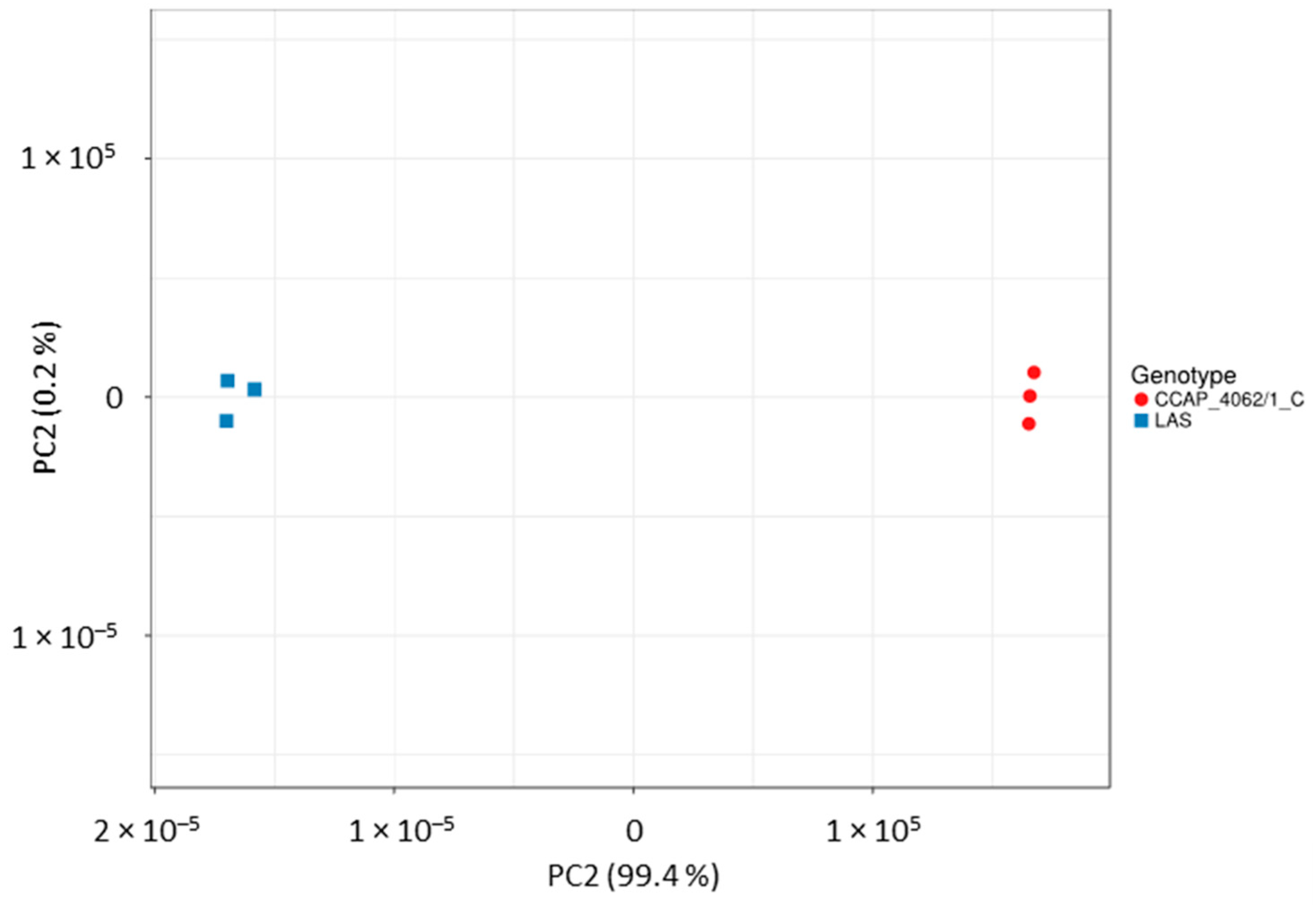
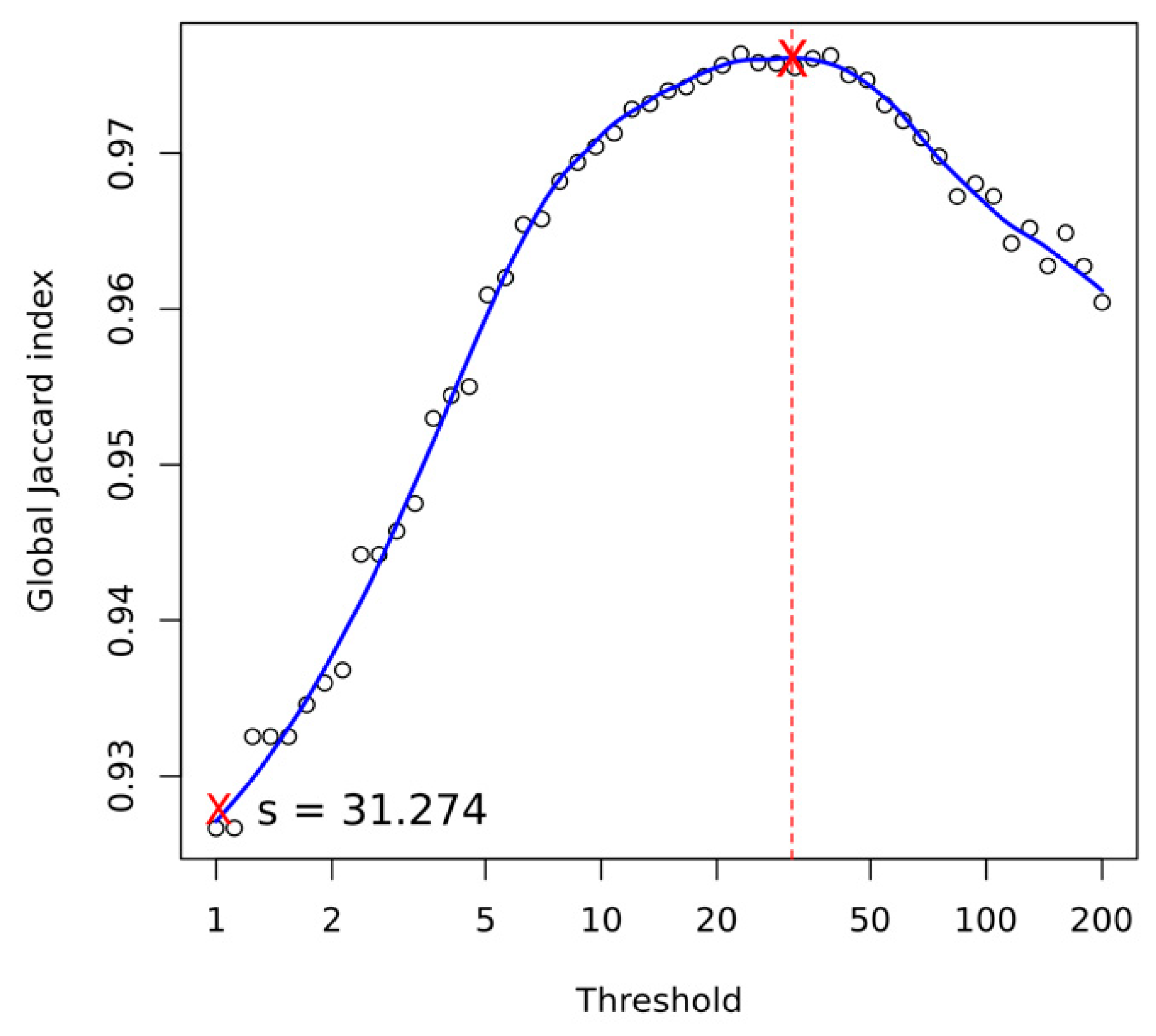
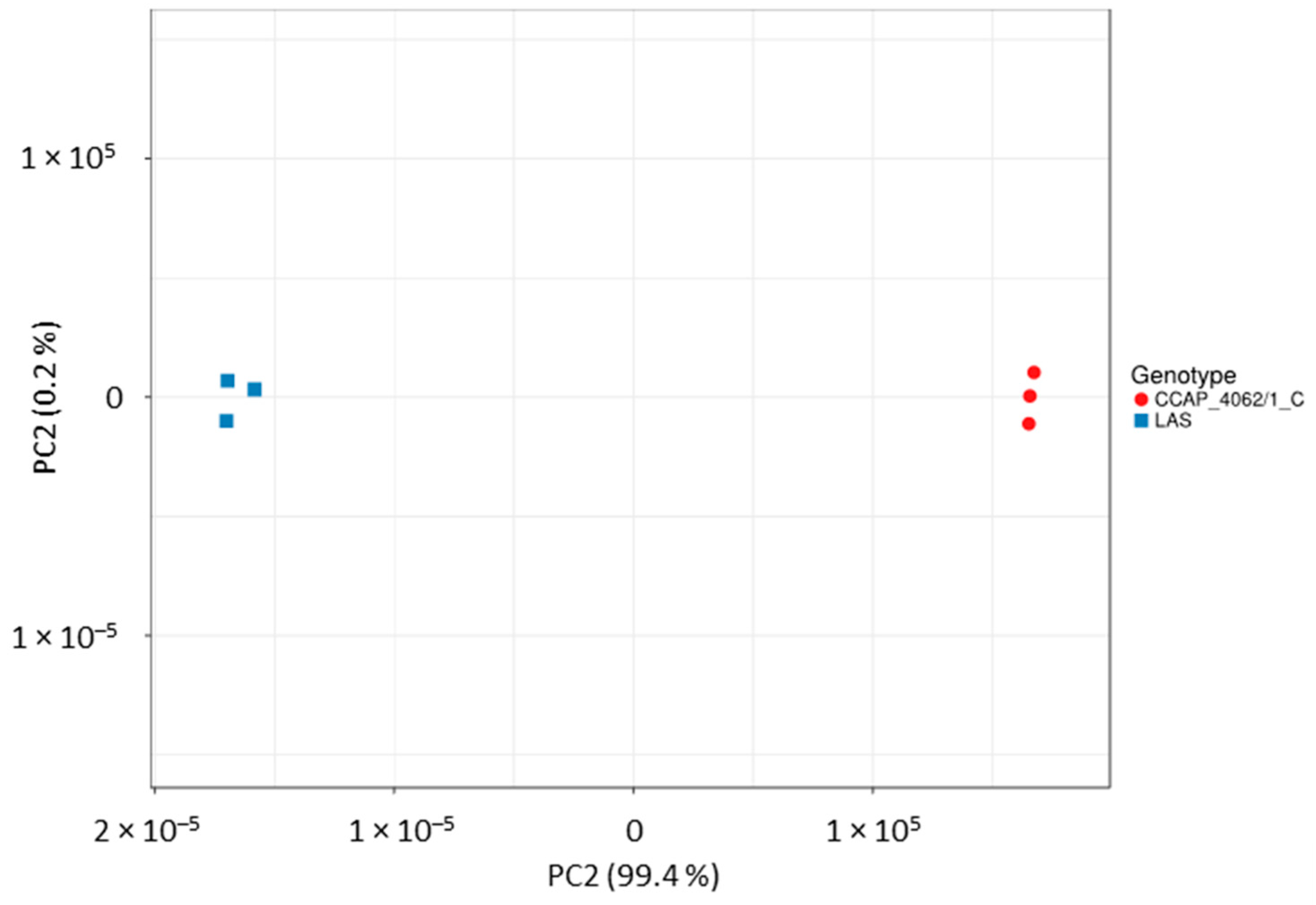
Appendix A.4. Structural Variant Calling
| Statistics | LAS | CCAP_4062/1 |
|---|---|---|
| Percentage of Mapping | 99.66% | 90.76% |
| Average Mapping Quality | 47.73 | 50.33 |
| Average Coverage | 30 | 92 |
Appendix A.5. Experimental Design, Materials, and Methods
Appendix A.5.1. The Strains and Maintenance Culture Conditions
Appendix A.5.2. Cloning and Genetic Transformation
Appendix A.5.3. Nucleic Acid Extraction and Sequencing
Appendix A.6. Bioinformatics Methods
Appendix A.6.1. Genome Assembly
Appendix A.6.2. RNA-seq Analysis
Appendix A.6.3. Structural Variant Calling
References
- Massana, R.; Pernice, M.C.; Bunge, J.A.; del Campo, J. Sequence diversity and novelty of natural assemblages of picoeukaryotes from the Indian Ocean. ISME J. 2010, 5, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, X.; Liu, X.; Jiao, N.; Wang, G. Abundance and Novel Lineages of Thraustochytrids in Hawaiian Waters. Microb. Ecol. 2013, 66, 823–830. [Google Scholar] [CrossRef]
- Raghukumar, S. Ecology of the marine protists, the Labyrinthulomycetes (Thraustochytrids and Labyrinthulids). Eur. J. Protistol. 2002, 38, 127–145. [Google Scholar] [CrossRef]
- Raghukumar, S.; Damare, V.S. Increasing evidence for the important role of Labyrinthulomycetes in marine ecosystems. Bot. Mar. 2011, 54, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Aasen, I.M.; Ertesvåg, H.; Heggeset, T.M.B.; Liu, B.; Brautaset, T.; Vadstein, O.; Ellingsen, T.E. Thraustochytrids as production organisms for docosahexaenoic acid (DHA), squalene, and carotenoids. Appl. Microbiol. Biotechnol. 2016, 100, 4309–4321. [Google Scholar] [CrossRef] [PubMed]
- Morabito, C.; Bournaud, C.; Maës, C.; Schuler, M.; Cigliano, R.A.; Dellero, Y.; Maréchal, E.; Amato, A.; Rébeillé, F. The lipid metabolism in thraustochytrids. Prog. Lipid Res. 2019, 76, 101007. [Google Scholar] [CrossRef] [PubMed]
- Bowles, R.D.; Hunt, A.E.; Bremer, G.B.; Duchars, M.G.; Eaton, R.A. Long-chain n-3 polyunsaturated fatty acid production by members of the marine protistan group the thraustochytrids: Screening of isolates and optimisation of docosahexaenoic acid production. J. Biotechnol. 1999, 70, 193–202. [Google Scholar] [CrossRef]
- Fan, K.-W.; Jiang, Y.; Faan, Y.-W.; Chen, F. Lipid Characterization of Mangrove Thraustochytrid—Schizochytrium mangrovei. J. Agric. Food Chem. 2007, 55, 2906–2910. [Google Scholar] [CrossRef] [PubMed]
- Raghukumar, S. Thraustochytrid Marine Protists: Production of PUFAs and Other Emerging Technologies. Mar. Biotechnol. 2008, 10, 631–640. [Google Scholar] [CrossRef]
- Manikan, V.; Nazir, M.Y.M.; Kalil, M.S.; Isa, M.H.M.; Kader, A.J.A.; Yusoff, W.M.W.; Hamid, A.A. A new strain of docosahexaenoic acid producing microalga from Malaysian coastal waters. Algal Res. 2015, 9, 40–47. [Google Scholar] [CrossRef]
- Dellero, Y.; Cagnac, O.; Rose, S.; Seddiki, K.; Cussac, M.; Morabito, C.; Lupette, J.; Cigliano, R.A.; Sanseverino, W.; Kuntz, M.; et al. Proposal of a new thraustochytrid genus Hondaea gen. nov and comparison of its lipid dynamics with the closely related pseudo-cryptic genus Aurantiochytrium. Algal Res. Biomass Biofuels Bioprod. 2018, 35, 125–141. [Google Scholar] [CrossRef]
- Cunnane, S.C. Problems with essential fatty acids: Time for a new paradigm? Prog. Lipid Res. 2003, 42, 544–568. [Google Scholar] [CrossRef]
- Innis, S.M. Dietary omega 3 fatty acids and the developing brain. Brain Res. 2008, 1237, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Byreddy, A.R. Thraustochytrids as an alternative source of omega-3 fatty acids, carotenoids and enzymes. Lipid Technol. 2016, 28, 68–70. [Google Scholar] [CrossRef]
- Lippmeier, J.C.; Crawford, K.S.; Owen, C.B.; Rivas, A.A.; Metz, J.G.; Apt, K.E. Characterization of Both Polyunsaturated Fatty Acid Biosynthetic Pathways in Schizochytrium sp. Lipids 2009, 44, 621–630. [Google Scholar] [CrossRef]
- Hauvermale, A.; Kuner, J.; Rosenzweig, B.; Guerra, D.; Diltz, S.; Metz, J.G. Fatty acid production in Schizochytrium sp.: Involvement of a polyunsaturated fatty acid synthase and a type I fatty acid synthase. Lipids 2006, 41, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Meesapyodsuk, D.; Qiu, X. Biosynthetic mechanism of very long chain polyunsaturated fatty acids in Thraustochytrium sp. 26185. J. Lipid Res. 2016, 57, 1854–1864. [Google Scholar] [CrossRef] [Green Version]
- Metz, J.G.; Roessler, P.; Facciotti, D.; Levering, C.; Dittrich, F.; Lassner, M.; Valentine, R.; Lardizabal, K.; Domergue, F.; Yamada, A.; et al. Production of Polyunsaturated Fatty Acids by Polyketide Synthases in Both Prokaryotes and Eukaryotes. Science 2001, 293, 290–293. [Google Scholar] [CrossRef]
- Kobayashi, T.; Sakaguchi, K.; Matsuda, T.; Abe, E.; Hama, Y.; Hayashi, M.; Honda, D.; Okita, Y.; Sugimoto, S.; Okino, N.; et al. Increase of Eicosapentaenoic Acid in Thraustochytrids through Thraustochytrid Ubiquitin Promoter-Driven Expression of a Fatty Acid Δ5 Desaturase Gene. Appl. Environ. Microbiol. 2011, 77, 3870–3876. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, T.; Sakaguchi, K.; Hamaguchi, R.; Kobayashi, T.; Abe, E.; Hama, Y.; Hayashi, M.; Honda, D.; Okita, Y.; Sugimoto, S.; et al. Analysis of Delta 12-fatty acid desaturase function revealed that two distinct pathways are active for the synthesis of PUFAs in T. aureum ATCC 34304. J. Lipid Res. 2012, 53, 1210–1222. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zang, X.; Cao, X.; Wang, Z.; Liu, C.; Sun, D.; Guo, Y.; Zhang, F.; Yang, Q.; Hou, P.; et al. Cloning of the pks3 gene of Aurantiochytrium limacinum and functional study of the 3-ketoacyl-ACP reductase and dehydratase enzyme domains. PLoS ONE 2018, 13, e0208853. [Google Scholar] [CrossRef]
- Li, Z.; Chen, X.; Li, J.; Meng, T.; Wang, L.; Chen, Z.; Shi, Y.; Ling, X.; Luo, W.; Liang, D.; et al. Functions of PKS Genes in Lipid Synthesis of Schizochytrium sp. by Gene Disruption and Metabolomics Analysis. Mar. Biotechnol. 2018, 20, 792–802. [Google Scholar] [CrossRef]
- Ren, L.-J.; Chen, S.-L.; Geng, L.-J.; Ji, X.-J.; Xu, X.; Song, P.; Gao, S.; Huang, H. Exploring the function of acyltransferase and domain replacement in order to change the polyunsaturated fatty acid profile of Schizochytrium sp. Algal Res. 2018, 29, 193–201. [Google Scholar] [CrossRef]
- Nutahara, E.; Abe, E.; Uno, S.; Ishibashi, Y.; Watanabe, T.; Hayashi, M.; Okino, N.; Ito, M. The glycerol-3-phosphate acyltransferase PLAT2 functions in the generation of DHA-rich glycerolipids in Aurantiochytrium limacinum F26-b. PLoS ONE 2019, 14, e0211164. [Google Scholar] [CrossRef] [PubMed]
- Abe, E.; Ikeda, K.; Nutahara, E.; Hayashi, M.; Yamashita, A.; Taguchi, R.; Doi, K.; Honda, D.; Okino, N.; Ito, M. Novel Lysophospholipid Acyltransferase PLAT1 of Aurantiochytrium limacinum F26-b Responsible for Generation of Palmitate-Docosahexaenoate-Phosphatidylcholine and Phosphatidylethanolamine. PLoS ONE 2014, 9, e102377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellero, Y.; Maës, C.; Morabito, C.; Schuler, M.; Bournaud, C.; Cigliano, R.A.; Maréchal, E.; Amato, A.; Rébeillé, F. The zoospores of the thraustochytridAurantiochytrium limacinum: Transcriptional reprogramming and lipid metabolism associated to their specific functions. Environ. Microbiol. 2020, 22, 1901–1916. [Google Scholar] [CrossRef]
- Reddy, J.K.; Hashimoto, T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: An adaptive metabolic system. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2016, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Perez, C.M.T.; Kitahori, T.; Hata, K.; Aoi, M.; Takahashi, H.; Sakuma, T.; Okamura, Y.; Nakashimada, Y.; Yamamoto, T.; et al. Improvement of fatty acid productivity of thraustochytrid, Aurantiochytrium sp. by genome editing. J. Biosci. Bioeng. 2020, 131, 373–380. [Google Scholar] [CrossRef]
- Hao, X.; Luo, L.; Jouhet, J.; Rébeillé, F.; Maréchal, E.; Hu, H.; Pan, Y.; Tan, X.; Chen, Z.; You, L.; et al. Enhanced triacylglycerol production in the diatom Phaeodactylum tricornutum by inactivation of a Hotdog-fold thioesterase gene using TALEN-based targeted mutagenesis 06 Biological Sciences 0601 Biochemistry and Cell Biology. Biotechnol. Biofuels 2018, 11, 312. [Google Scholar] [CrossRef]
- Altpeter, F.; Springer, N.M.; Bartley, L.E.; Blechl, A.E.; Brutnell, T.P.; Citovsky, V.; Conrad, L.J.; Gelvin, S.B.; Jackson, D.P.; Kausch, A.P.; et al. Advancing Crop Transformation in the Era of Genome Editing. Plant. Cell 2016, 28, 1510–1520. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Nannas, N.J.; Fu, F.-F.; Shi, J.; Aspinwall, B.; Parrott, W.A.; Dawe, R.K. Genome-Scale Sequence Disruption Following Biolistic Transformation in Rice and Maize. Plant. Cell 2019, 31, 368–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Guo, W.L.; Wang, X.R.; Zhuang, T.T.; Clarke, J.L.; Liu, B.; Wang, X.L. Unintended consequence of plant transformation: Biolistic transformation caused transpositional activation of an endogenous retrotransposon Tos17 in rice ssp. japonica cv. Matsumae. Plant. Cell Rep. 2009, 28, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Stanley, G.S. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Dellero, Y.; Rose, S.; Metton, C.; Morabito, C.; Lupette, J.; Jouhet, J.; Maréchal, E.; Rébeillé, F.; Amato, A. Ecophysiology and lipid dynamics of a eukaryotic mangrove decomposer. Environ. Microbiol. 2018, 20, 3057–3068. [Google Scholar] [CrossRef] [PubMed]
- Morabito, C.; Aiese Cigliano, R.; Maréchal, E.; Rébeillé, F.; Amato, A. Illumina and PacBio DNA sequencing data, de novo assembly and annotation of the genome of Aurantiochytrium limacinum strain CCAP_4062/1. Data Brief 2020, 31, 105729. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST (c)) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Gietz, R.D.; Schiestl, R.H. Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 38–41. [Google Scholar] [CrossRef]
- Flori, S.; Jouneau, P.-H.; Gallet, B.; Estrozi, L.F.; Moriscot, C.; Schoehn, G.; Finazzi, G.; Falconet, D. Imaging Plastids in 2D and 3D: Confocal and Electron Microscopy. In Plastids: Methods and Protocols; Maréchal, E., Ed.; Springer: New York, NY, USA, 2018; pp. 113–122. [Google Scholar] [CrossRef]
- Arai, Y.; Hayashi, M.; Nishimura, M. Proteomic Identification and Characterization of a Novel Peroxisomal Adenine Nucleotide Transporter Supplying ATP for Fatty Acid beta-Oxidation in Soybean and Arabidopsis. Plant. Cell 2008, 20, 3227–3240. [Google Scholar] [CrossRef] [Green Version]
- Lemoine, F.; Correia, D.; Lefort, V.; Doppelt-Azeroual, O.; Mareuil, F.; Cohen-Boulakia, S.; Gascuel, O. NGPhylogeny.fr: New generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019, 47, W260–W265. [Google Scholar] [CrossRef] [Green Version]
- Emsley, J.; McEwan, P.A.; Gailani, D. Structure and function of factor XI. Blood 2010, 115, 2569–2577. [Google Scholar] [CrossRef] [Green Version]
- Tordai, H.; Bányai, L.; Patthy, L. The PAN module: The N-terminal domains of plasminogen and hepatocyte growth factor are homologous with the apple domains of the prekallikrein family and with a novel domain found in numerous nematode proteins. FEBS Lett. 1999, 461, 63–67. [Google Scholar] [CrossRef]
- Wendland, B.; Emr, S.D. Pan1p, Yeast eps15, Functions as a Multivalent Adaptor That Coordinates Protein–Protein Interactions Essential for Endocytosis. J. Cell Biol. 1998, 141, 71–84. [Google Scholar] [CrossRef] [PubMed]
- van Roermund, C.W.T.; Drissen, R.; van den Berg, M.; Ijlst, L.; Hettema, E.H.; Tabak, H.F.; Waterham, H.R.; Wanders, R.J.A. Identification of a peroxisomal ATP carrier required for medium-chain fatty acid beta-oxidation and normal peroxisome proliferation in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001, 21, 4321–4329. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, L.; Rottensteiner, H.; Girzalsky, W.; Scarcia, P.; Palmieri, F.; Erdmann, R. Identification and functional reconstitution of the peroxisomal adenine nucleotide transporter of S-cerevisiae. Mol. Biol. Cell 2001, 12, 502A. [Google Scholar]
- Nakagawa, T.; Imanaka, T.; Morita, M.; Ishiguro, K.; Yurimoto, H.; Yamashita, A.; Kato, N.; Sakai, Y. Peroxisomal Membrane Protein Pmp47 Is Essential in the Metabolism of Middle-chain Fatty Acid in Yeast Peroxisomes and Is Associated with Peroxisome Proliferation. J. Biol. Chem. 2000, 275, 3455–3461. [Google Scholar] [CrossRef] [Green Version]
- Dulermo, R.; Gamboa-Meléndez, H.; Ledesma-Amaro, R.; Thévenieau, F.; Nicaud, J.-M. Unraveling fatty acid transport and activation mechanisms in Yarrowia lipolytica. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 1202–1217. [Google Scholar] [CrossRef]
- Linka, N.; Theodoulou, F.; Haslam, R.; Linka, M.; Napier, J.; Neuhaus, H.E.; Weber, A. Peroxisomal ATP Import Is Essential for Seedling Development in Arabidopsis thaliana. Plant. Cell 2008, 20, 3241–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armenteros, J.J.A.; Sonderby, C.K.; Sonderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 4049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Roermund, C.W.; Ijlst, L.; Majczak, W.; Waterham, H.R.; Folkerts, H.; Wanders, R.J.; Hellingwerf, K.J. Peroxisomal Fatty Acid Uptake Mechanism in Saccharomyces cerevisiae. J. Biol. Chem. 2012, 287, 20144–20153. [Google Scholar] [CrossRef] [Green Version]
- Charton, L.; Plett, A.; Linka, N. Plant peroxisomal solute transporter proteins. J. Integr. Plant. Biol. 2019, 61, 817–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roermund, C.W.T.; Waterham, H.R.; Ijlst, L.; Wanders, R.J.A. Fatty acid metabolism in Saccharomyces cerevisiae. Experientia 2003, 60, 1838–1851. [Google Scholar] [CrossRef]
- Partier, A.; Gay, G.; Tassy, C.; Beckert, M.; Feuillet, C.; Barret, P. Molecular and FISH analyses of a 53-kbp intact DNA fragment inserted by biolistics in wheat (Triticum aestivum L.) genome. Plant. Cell Rep. 2017, 36, 1547–1559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Phan, B.H.; Wang, K.; Artelt, B.J.; Jiang, J.; Parrott, W.A.; Dawe, R.K. Stable integration of an engineered megabase repeat array into the maize genome. Plant. J. 2012, 70, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Svitashev, S.K.; Pawlowski, W.P.; Makarevitch, I.; Plank, D.W.; Somers, D.A. Complex transgene locus structures implicate multiple mechanisms for plant transgene rearrangement. Plant J. 2002, 32, 433–445. [Google Scholar] [CrossRef] [Green Version]
- Chankova, S.G.; Dimova, E.; Dimitrova, M.; Bryant, P.E. Induction of DNA double-strand breaks by zeocin in Chlamydomonas reinhardtii and the role of increased DNA double-strand breaks rejoining in the formation of an adaptive response. Radiat. Environ. Biophys. 2007, 46, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Makarevitch, I.; Svitashev, S.; Somers, D. Complete sequence analysis of transgene loci from plants transformed via microprojectile bombardment. Plant Mol. Biol. 2003, 52, 421–432. [Google Scholar] [CrossRef]
- Yokoyama, R.; Honda, D. Taxonomic rearrangement of the genus Schizochytrium sensu lato based on morphology, chemotaxonomic characteristics, and 18S rRNA gene phylogeny (Thraustochytriaceae, Labyrinthulomycetes): Emendation for Schizochytrium and erection of Aurantiochytrium and Oblongichytrium gen. nov. Mycoscience 2007, 48, 199–211. [Google Scholar] [CrossRef]
- Xiao, C.-L.; Chen, Y.; Xie, S.-Q.; Chen, K.-N.; Wang, Y.; Han, Y.; Luo, F.; Xie, Z. MECAT: Fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. Nat. Methods 2017, 14, 1072–1074. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptivek-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLOS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Rau, A.; Gallopin, M.; Celeux, G.; Jaffrézic, F. Data-based filtering for replicated high-throughput transcriptome sequencing experiments. Bioinformatics 2013, 29, 2146–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, D.; Vingron, M. SVIM: Structural variant identification using mapped long reads. Bioinformatics 2019, 35, 2907–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge – Accurate paired shotgun read merging via overlap. PLOS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Oikkonen, L.; Lise, S. Making the most of RNA-seq: Pre-processing sequencing data with Opossum for reliable SNP variant detection. Wellcome Open Res. 2017, 2. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef]
- Falda, M.; Toppo, S.; Pescarolo, A.; Lavezzo, E.; Di Camillo, B.; Facchinetti, A.; Cilia, E.; Velasco, R.; Fontana, P. Argot2: A large scale function prediction tool relying on semantic similarity of weighted Gene Ontology terms. BMC Bioinform. 2012, 13, S14. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaccard, P. The distribution of the flora in the alpine zone 1. New Phytol. 1912, 11, 37–50. [Google Scholar] [CrossRef]



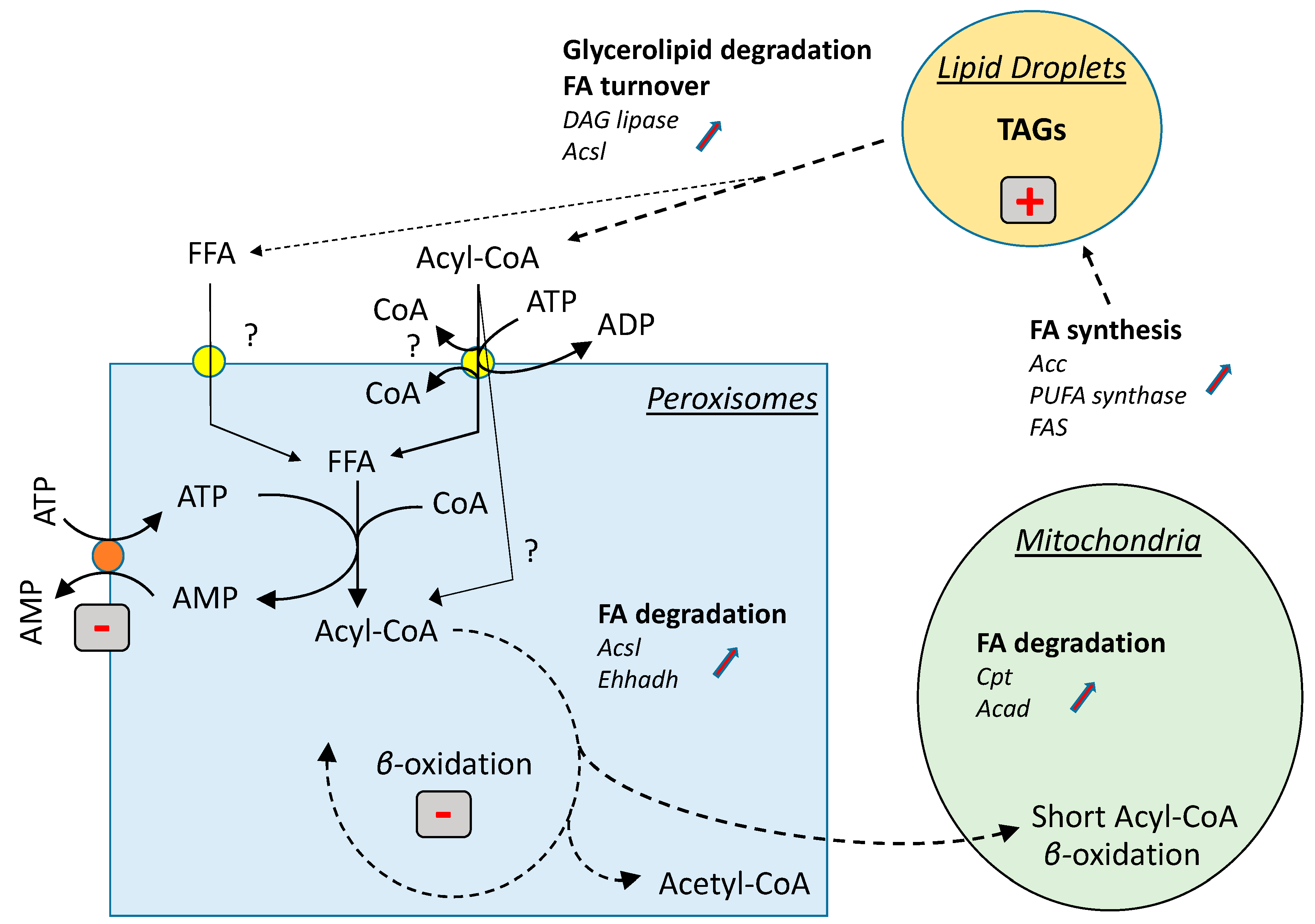
| Name | Symbol | Reference in Aurli1 | Log2FC |
|---|---|---|---|
| Fatty Acid Synthesis | |||
| Acetyl-CoA carboxylase | ACC | gw1.10.632.1 | 1.45 |
| PUFA synthase | PufaA | fgenesh1_pg.14_251 | 2.21 |
| PUFA synthase | PufaB | estExt_fgenesh1_kg.C_140136 | 1.6 |
| PUFA synthase | PufaC | estExt_fgenesh1_kg.C_190026 | 1.38 |
| Fatty Acid Synthase I | Fas1 | e_gw1.21.366.1 | 1.33 |
| Fatty Acid Degradation | |||
| Mitochondrial carnitine/acylcarnitine transporter | CACT | fgenesh1_kg.16_67_isotig06873 | 1.78 |
| carnitine palmitoyl transferase CPT1 | Cpt1 | fgenesh1_kg.6_177_isotig02684 | 1.28 |
| carnitine palmitoyl transferase CPT3 | Cpt2-2 | estExt_fgenesh1_kg.C_60140 | 1.37 |
| Acyl-CoA dehydrogenase | Acad4 | estExt_Genewise1.C_9_t10269 | 1.17 |
| Enoyl-CoA hydratase | Ech6 | gm1.2779_g | 1.58 |
| Bifunctional enzyme | Ehhadh3 | gm1.11390_g | 1.17 |
| Long chain acyl-CoA synthase/ligase | Acsl | e_gw1.11.199.1 | 3.01 |
| Long chain acyl-CoA synthase/ligase | Acsl | e_gw1.2.456.1 | 2.93 |
| Long chain acyl-CoA synthase/ligase | Acsl | estExt_fgenesh1_kg.C_40024 | 2.78 |
| Long chain acyl-CoA synthase/ligase (peroxisomal) | Acsl | estExt_fgenesh1_kg.C_160013 | 2.51 |
| Long chain acyl-CoA synthase/ligase (peroxisomal) | Acsl | estExt_fgenesh1_kg.C_160012 | 2.07 |
| Long chain acyl-CoA synthase/ligase | Acsl | gw1.11.174.1 | 2.06 |
| Long chain acyl-CoA synthase/ligase | Acsl | fgenesh1_pm.5_56 | 1.51 |
| Long chain acyl-CoA synthase/ligase (peroxisomal) | Acsl | e_gw1.16.213.1 | 1.31 |
| Glycerolipid Degradation | |||
| DAG lipase | Dagl6 | fgenesh1_pg.9_214 | 2.46 |
| DAG lipase | Dagl8 | gm1.4322_g | 1.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deragon, E.; Schuler, M.; Aiese Cigliano, R.; Dellero, Y.; Si Larbi, G.; Falconet, D.; Jouhet, J.; Maréchal, E.; Michaud, M.; Amato, A.; et al. An Oil Hyper-Accumulator Mutant Highlights Peroxisomal ATP Import as a Regulatory Step for Fatty Acid Metabolism in Aurantiochytrium limacinum. Cells 2021, 10, 2680. https://doi.org/10.3390/cells10102680
Deragon E, Schuler M, Aiese Cigliano R, Dellero Y, Si Larbi G, Falconet D, Jouhet J, Maréchal E, Michaud M, Amato A, et al. An Oil Hyper-Accumulator Mutant Highlights Peroxisomal ATP Import as a Regulatory Step for Fatty Acid Metabolism in Aurantiochytrium limacinum. Cells. 2021; 10(10):2680. https://doi.org/10.3390/cells10102680
Chicago/Turabian StyleDeragon, Etienne, Martin Schuler, Riccardo Aiese Cigliano, Younès Dellero, Gregory Si Larbi, Denis Falconet, Juliette Jouhet, Eric Maréchal, Morgane Michaud, Alberto Amato, and et al. 2021. "An Oil Hyper-Accumulator Mutant Highlights Peroxisomal ATP Import as a Regulatory Step for Fatty Acid Metabolism in Aurantiochytrium limacinum" Cells 10, no. 10: 2680. https://doi.org/10.3390/cells10102680
APA StyleDeragon, E., Schuler, M., Aiese Cigliano, R., Dellero, Y., Si Larbi, G., Falconet, D., Jouhet, J., Maréchal, E., Michaud, M., Amato, A., & Rébeillé, F. (2021). An Oil Hyper-Accumulator Mutant Highlights Peroxisomal ATP Import as a Regulatory Step for Fatty Acid Metabolism in Aurantiochytrium limacinum. Cells, 10(10), 2680. https://doi.org/10.3390/cells10102680