
MicroRNAs in the Onset of Schizophrenia


Abstract
:1. Introduction
2. Temporal Dynamics in the Levels of MiRNAs across the Lifespan
3. Temporal Dynamics in the Activity of MiRNAs across the Lifespan
4. Neurobiology during the Age of SCZ Onset
5. Peripheral MiRNAs during Conversion to Psychosis
6. MiR-29 and MiR-132-3p in Adolescent Neurodevelopment and Disease
6.1. The MiR-29 Family and MiR-132-3p Levels Increase with Age and May Be Dysregulated in SCZ
6.2. Shared Roles of MiR-29 and MiR-132-3p: Cortical Ocular Dominance Plasticity and DNMT3A
7. 22q11DS Disrupts the MiRNA Pathway in an Age-Dependent Manner
8. MIR137HG Variance Predicts Variance in the Age at SCZ Onset
9. Possible Mechanisms of MiRNA-Dependent Onset of SCZ
9.1. Direct Mechanism: Aberrations in MiRNAs during Adolescence Disrupt Adolescent Brain Maturation and Cause Disease Onset
9.2. Delayed Mechanism: Aberrations in MiRNAs during Early Development Disrupt Brain Maturation during Adolescence
9.3. Progressive Mechanism: Aberrations in Individual MiRNAs Cause Small Deficits That Accumulate during Development and Become Disruptive during Adolescence
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tandon, R.; Keshavan, M.S.; Nasrallah, H.A. Schizophrenia, “Just the Facts” What we know in 2008. 2. Epidemiology and etiology. Schizophr. Res. 2008, 102, 1–18. [Google Scholar] [CrossRef]
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr. Res. 2009, 110, 1–23. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Kendler, K.S.; Neale, M.C. Schizophrenia as a Complex Trait. Arch. Gen. Psychiatry 2003, 60, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Dean, K.; Murray, R.M. Environmental risk factors for psychosis. Dialogues Clin. Neurosci. 2005, 7, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.Y.; Teoh, S.L.; Wu, D.B.-C.; Kotirum, S.; Chiou, C.-F.; Chaiyakunapruk, N. Global economic burden of schizophrenia: A systematic review. Neuropsychiatr. Dis. Treat. 2016, 12, 357–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, D.O.; Jeffries, C.D.; Jarskog, L.F.; Thomson, J.M.; Woods, K.; Newman, M.A.; Parker, J.S.; Jin, J.; Hammond, S.M. microRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol. 2007, 8, R27. [Google Scholar] [CrossRef] [Green Version]
- Beveridge, N.J.; Gardiner, E.; Carroll, A.P.; Tooney, P.A.; Cairns, M.J. Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol. Psychiatry 2010, 15, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.P.; Bruse, S.E.; David-Rus, R.; Buyske, S.; Brzustowicz, L.M. Altered MicroRNA expression profiles in postmortem brain samples from individuals with schizophrenia and bipolar disorder. Biol. Psychiatry 2011, 69, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.H.; Reimers, M.; Maher, B.; Williamson, V.; McMichael, O.; McClay, J.L.; van den Oord, E.J.C.G.; Riley, B.P.; Kendler, K.S.; Vladimirov, V.I. MicroRNA expression profiling in the prefrontal cortex of individuals affected with schizophrenia and bipolar disorders. Schizophr. Res. 2010, 124, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.H.; Zeier, Z.; Xi, L.; Lanz, T.A.; Deng, S.; Strathmann, J.; Willoughby, D.; Kenny, P.J.; Elsworth, J.D.; Lawrence, M.S.; et al. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc. Natl. Acad. Sci. USA 2012, 109, 3125–3130. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Gao, S.; Lindberg, D.; Panja, D.; Wakabayashi, Y.; Li, K.; Kleinman, J.E.; Zhu, J.; Li, Z. Temporal dynamics of miRNAs in human DLPFC and its association with miRNA dysregulation in schizophrenia. Transl. Psychiatry 2019, 9, 196. [Google Scholar] [CrossRef]
- Beveridge, N.J.; Tooney, P.A.; Carroll, A.P.; Gardiner, E.; Bowden, N.; Scott, R.J.; Tran, N.; Dedova, I.; Cairns, M.J. Dysregulation of miRNA 181b in the temporal cortex in schizophrenia. Hum. Mol. Genet. 2008, 17, 1156–1168. [Google Scholar] [CrossRef]
- Liu, Y.; Chang, X.; Hahn, C.G.; Gur, R.E.; Sleiman, P.A.M.; Hakonarson, H. Non-coding RNA dysregulation in the amygdala region of schizophrenia patients contributes to the pathogenesis of the disease. Transl. Psychiatry 2018, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarelli, D.M.; Beveridge, N.J.; Tooney, P.A.; Cairns, M.J. Upregulation of dicer and MicroRNA expression in the dorsolateral prefrontal cortex Brodmann area 46 in schizophrenia. Biol. Psychiatry 2011, 69, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Hauberg, M.E.; Roussos, P.; Grove, J.; Børglum, A.D.; Mattheisen, M. Analyzing the Role of MicroRNAs in Schizophrenia in the Context of Common Genetic Risk Variants. JAMA Psychiatry 2016, 73, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011, 43, 969–976. [Google Scholar] [CrossRef]
- Ripke, S.; O’Dushlaine, C.; Chambert, K.; Moran, J.L.; Kähler, A.K.; Akterin, S.; Bergen, S.E.; Collins, A.L.; Crowley, J.J.; Fromer, M.; et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013, 45, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.R.; Farh, K.-H.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; Huang, H.; et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Goes, F.S.; McGrath, J.; Avramopoulos, D.; Wolyniec, P.; Pirooznia, M.; Ruczinski, I.; Nestadt, G.; Kenny, E.E.; Vacic, V.; Peters, I.; et al. Genome-wide association study of schizophrenia in Ashkenazi Jews. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2015, 168, 649–659. [Google Scholar] [CrossRef]
- Duan, J.; Shi, J.; Fiorentino, A.; Leites, C.; Chen, X.; Moy, W.; Chen, J.; Alexandrov, B.S.; Usheva, A.; He, D.; et al. A rare functional noncoding variant at the GWAS-Implicated MIR137/MIR2682 locus might confer risk to schizophrenia and bipolar disorder. Am. J. Hum. Genet. 2014, 95, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strazisar, M.; Cammaerts, S.; van der Ven, K.; Forero, D.A.; Lenaerts, A.; Nordin, A.; Almeida-Souza, L.; Genovese, G.; Timmerman, V.; Liekens, A.; et al. MIR137 variants identified in psychiatric patients affect synaptogenesis and neuronal transmission gene sets. Mol. Psychiatry 2014, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hollins, S.L.; Zavitsanou, K.; Walker, F.R.; Cairns, M.J. Alteration of imprinted Dlk1-Dio3 miRNA cluster expression in the entorhinal cortex induced by maternal immune activation and adolescent cannabinoid exposure. Transl. Psychiatry 2014, 4, e452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnica, W.; Merico, D.; Costain, G.; Alfred, S.E.; Wei, J.; Marshall, C.R.; Scherer, S.W.; Bassett, A.S. Copy number variable micrornas in schizophrenia and their neurodevelopmental gene targets. Biol. Psychiatry 2015, 77, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.-P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.; Kim, Y.; Jin, H.; Kim, V.N. The Drosha—DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 3016–3027. [Google Scholar] [CrossRef] [Green Version]
- Landthaler, M.; Yalcin, A.; Tuschl, T. The Human DiGeorge Syndrome Critical Region Gene 8 and Its D. melanogaster Homolog Are Required for miRNA Biogenesis. Curr. Biol. 2004, 14, 2162–2167. [Google Scholar] [CrossRef] [Green Version]
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [Green Version]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Hutvágner, G.; McLachlan, J.; Pasquinelli, A.E.; Bálint, E.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [Green Version]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Zhou, K.; Smith, A.M.; Noland, C.L.; Doudna, J.A. Differential roles of human Dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 2013, 41, 6568–6576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006, 20, 1885–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, M.R.; Mathonnet, G.; Sundermeier, T.; Mathys, H.; Zipprich, J.T.; Svitkin, Y.V.; Rivas, F.; Jinek, M.; Wohlschlegel, J.; Doudna, J.A.; et al. Mammalian miRNA RISC Recruits CAF1 and PABP to Affect PABP-Dependent Deadenylation. Mol. Cell 2009, 35, 868–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassett, A.S.; Chow, E.W.C. Schizophrenia and 22q11. 2 deletion syndrome. Curr. Psychiatry Rep. 2008, 10, 148–157. [Google Scholar] [CrossRef] [Green Version]
- McDonald-McGinn, D.M.; Sullivan, K.; Marino, B.; Swillen, A.; Vortsman, J.; Zackai, E.; Emanuel, B.; Vermeesch, J.; Morrow, B.; Scambler, P.; et al. 22q11.2 Deletion Syndrome. Nat. Rev. Dis. Prim. 2015, 1, 621–626.e1. [Google Scholar] [CrossRef] [Green Version]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef]
- Earls, L.R.; Fricke, R.G.; Yu, J.; Berry, R.B.; Baldwin, L.T.; Zakharenko, S.S. Age-Dependent MicroRNA Control of Synaptic Plasticity in 22q11 Deletion Syndrome and Schizophrenia. J. Neurosci. 2012, 32, 14132–14144. [Google Scholar] [CrossRef] [Green Version]
- Rey, R.; Suaud-Chagny, M.F.; Dorey, J.M.; Teyssier, J.R.; d’Amato, T. Widespread transcriptional disruption of the microRNA biogenesis machinery in brain and peripheral tissues of individuals with schizophrenia. Transl. Psychiatry 2020, 10, 376. [Google Scholar] [CrossRef]
- Gochman, P.; Miller, R.; Rapoport, J.L. Childhood-Onset Schizophrenia: The Challenge of Diagnosis. Curr. Psychiatry Rep. 2011, 13, 321–322. [Google Scholar] [CrossRef] [Green Version]
- Hafner, H.; Maurer, K.; Loffler, W.; Riecher-Rossler, A. The influence of age and sex on the onset of early course of schizophrenia. Br. J. Psychiatry 1993, 162, 80–86. [Google Scholar] [CrossRef]
- Ziats, M.N.; Rennert, O.M. Identification of differentially expressed microRNAs across the developing human brain. Mol. Psychiatry 2014, 19, 848–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somel, M.; Guo, S.; Fu, N.; Yan, Z.; Hu, H.Y.; Xu, Y.; Yuan, Y.; Ning, Z.; Hu, Y.; Menzel, C.; et al. MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome Res. 2010, 20, 1207–1218. [Google Scholar] [CrossRef] [Green Version]
- Somel, M.; Liu, X.; Tang, L.; Yan, Z.; Hu, H.; Guo, S.; Jiang, X.; Zhang, X.; Xu, G.; Xie, G.; et al. MicroRNA-driven developmental remodeling in the brain distinguishes humans from other primates. PLoS Biol. 2011, 9, e1001214. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.P.; Bruse, S.E.; Jornsten, R.; Liu, Y.; Brzustowicz, L.M. Chronological changes in microRNA expression in the developing human brain. PLoS ONE 2013, 8, e60480. [Google Scholar] [CrossRef] [Green Version]
- Beveridge, N.J.; Santarelli, D.M.; Wang, X.; Tooney, P.A.; Webster, M.J.; Weickert, C.S.; Cairns, M.J. Maturation of the human dorsolateral prefrontal cortex coincides with a dynamic shift in microRNA expression. Schizophr. Bull. 2014, 40, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Podolska, A.; Kaczkowski, B.; Kamp Busk, P.; Søkilde, R.; Litman, T.; Fredholm, M.; Cirera, S. MicroRNA expression profiling of the porcine developing brain. PLoS ONE 2011, 6, e14494. [Google Scholar] [CrossRef] [PubMed]
- Krichevsky, A.M.; King, K.S.; Donahue, C.P.; Khrapko, K.; Kosik, K.S. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 2003, 9, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.J.; Chen, G.; Zhao, P.P.; Lu, M.H.; Jian, J.; Liu, M.F.; Yuan, X.B. Transcriptome analysis of microRNAs in developing cerebral cortex of rat. BMC Genom. 2012, 13, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miska, E.A.; Alvarez-Saavedra, E.; Townsend, M.; Yoshii, A.; Sestan, N.; Rakic, P.; Constantine-Paton, M.; Horvitz, H.R. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004, 5, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eda, A.; Takahashi, M.; Fukushima, T.; Hohjoh, H. Alteration of microRNA expression in the process of mouse brain growth. Gene 2011, 485, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Fertuzinhos, S.; Li, M.; Kawasawa, Y.I.; Ivic, V.; Franjic, D.; Singh, D.; Crair, M.; Sestan, N. Laminar and temporal expression dynamics of coding and noncoding RNAs in the mouse neocortex. Cell Rep. 2014, 6, 938–950. [Google Scholar] [CrossRef] [Green Version]
- Swahari, V.; Nakamura, A.; Hollville, E.; Stroud, H.; Simon, J.M.; Ptacek, T.S.; Beck, M.V.; Flowers, C.; Guo, J.; Plestant, C.; et al. MicroRNA-29 is an essential regulator of brain maturation through regulation of CH methylation. Cell Rep. 2021, 35, 108946. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S. The microRNA registry. Nucleic Acids Res. 2004, 32, 109–111. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, 140–144. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; Van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, 154–158. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, 152–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Magee, R.; Telonis, A.G.; Cherlin, T.; Rigoutsos, I.; Londin, E. Assessment of isomiR discrimination using commercial qPCR methods. Non-Coding RNA 2017, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasello, L.; Distefano, R.; Nigita, G.; Croce, C.M. The MicroRNA Family Gets Wider: The IsomiRs Classification and Role. Front. Cell Dev. Biol. 2021, 9, 1–15. [Google Scholar] [CrossRef]
- Llorens, F.; Bañez-Coronel, M.; Pantano, L.; del Río, J.A.; Ferrer, I.; Estivill, X.; Martí, E. A highly expressed miR-101 isomiR is a functional silencing small RNA. BMC Genom. 2013, 14, 104. [Google Scholar] [CrossRef] [Green Version]
- Van der Kwast, R.V.C.T.; Woudenberg, T.; Quax, P.H.A.; Nossent, A.Y. MicroRNA-411 and Its 5′-IsomiR Have Distinct Targets and Functions and Are Differentially Regulated in the Vasculature under Ischemia. Mol. Ther. 2019, 28, 157–170. [Google Scholar] [CrossRef]
- Van der Kwast, R.V.C.T.; Parma, L.; van der Bent, M.L.; van Ingen, E.; Baganha, F.; Peters, H.A.B.; Goossens, E.A.C.; Simons, K.H.; Palmen, M.; de Vries, M.R.; et al. Adenosine-to-Inosine Editing of Vasoactive MicroRNAs Alters Their Targetome and Function in Ischemia. Mol. Ther. Nucleic Acids 2020, 21, 932–953. [Google Scholar] [CrossRef]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu-Gruttadauria, J.; Xiao, Y.; Gebert, L.F.; MacRae, I.J. Beyond the seed: Structural basis for supplementary micro RNA targeting by human Argonaute2. EMBO J. 2019, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Martí, E.; Pantano, L.; Bañez-Coronel, M.; Llorens, F.; Miñones-Moyano, E.; Porta, S.; Sumoy, L.; Ferrer, I.; Estivill, X. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010, 38, 7219–7235. [Google Scholar] [CrossRef] [PubMed]
- Wee, L.M.; Flores-Jasso, C.F.; Salomon, W.E.; Zamore, P.D. Argonaute divides Its RNA guide into domains with distinct functions and RNA-binding properties. Cell 2012, 151, 1055–1067. [Google Scholar] [CrossRef] [Green Version]
- Ameres, S.L.; Horwich, M.D.; Hung, J.-H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA-Directed Trimming and Tailing of Small Silencing RNAs. Science 2010, 328, 1534–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bail, S.; Swerdel, M.; Liu, H.; Jiao, X.; Goff, L.A.; Hart, R.P.; Kiledjian, M. Differential regulation of microRNA stability. Rna 2010, 16, 1032–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Mata, M.; Gaidatzis, D.; Vitanescu, M.; Stadler, M.B.; Wentzel, C.; Scheiffele, P.; Filipowicz, W.; Großhans, H. Potent degradation of neuronal miRNAs induced by highly complementary targets. EMBO Rep. 2015, 16, 500–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingston, E.R.; Bartel, D.P. Global analyses of the dynamics of mammalian microRNA metabolism. Genome Res. 2019, 29, 1777–1790. [Google Scholar] [CrossRef]
- Juvvuna, P.K.; Khandelia, P.; Lee, L.M.; Makeyev, E.V. Argonaute identity defines the length of mature mammalian microRNAs. Nucleic Acids Res. 2012, 40, 6808–6820. [Google Scholar] [CrossRef]
- Nowakowski, T.J.; Rani, N.; Golkaram, M.; Zhou, H.R.; Alvarado, B.; Huch, K.; West, J.A.; Leyrat, A.; Pollen, A.A.; Kriegstein, A.R.; et al. Regulation of cell-type-specific transcriptomes by microRNA networks during human brain development. Nat. Neurosci. 2018, 21, 1784–1792. [Google Scholar] [CrossRef]
- Boudreau, R.L.; Jiang, P.; Gilmore, B.L.; Spengler, R.M.; Tirabassi, R.; Nelson, J.A.; Ross, C.A.; Xing, Y.; Davidson, B.L. Transcriptome-wide discovery of microRNA binding sites in Human Brain. Neuron 2014, 81, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Sangiao-Alvarellos, S.; Manfredi-Lozano, M.; Ruiz-Pino, F.; Navarro, V.M.; Sánchez-Garrido, M.A.; Leon, S.; Dieguez, C.; Cordido, F.; Matagne, V.; Dissen, G.A.; et al. Changes in hypothalamic expression of the Lin28/let-7 system and related MicroRNAs during postnatal maturation and after experimental manipulations of puberty. Endocrinology 2013, 154, 942–955. [Google Scholar] [CrossRef]
- Tognini, P.; Putignano, E.; Coatti, A.; Pizzorusso, T. Experience-dependent expression of miR-132 regulates ocular dominance plasticity. Nat. Neurosci. 2011, 14, 1237–1239. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Mao, S.; Wang, H.; Zen, K.; Zhang, C.; Li, L. MicroRNA-29a modulates axon branching by targeting doublecortin in primary neurons. Protein Cell 2014, 5, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazziotti, R.; Baroncelli, L.; Ceglia, N.; Chelini, G.; Sala, G.D.; Magnan, C.; Napoli, D.; Putignano, E.; Silingardi, D.; Tola, J.; et al. Mir-132/212 is required for maturation of binocular matching of orientation preference and depth perception. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xiao, J.; Fan, Y.; Yang, K.; Li, K.; Wang, X.; Lu, Y.; Zhou, Y. MiR-29 family regulates the puberty onset mediated by a novel Gnrh1 transcription factor TBX21. J. Endocrinol. 2019, 242, 185–197. [Google Scholar] [CrossRef]
- Napoli, D.; Lupori, L.; Mazziotti, R.; Sagona, G.; Bagnoli, S.; Samad, M.; Sacramento, E.K.; Kirkpartick, J.; Putignano, E.; Chen, S.; et al. MiR-29 coordinates age-dependent plasticity brakes in the adult visual cortex. EMBO Rep. 2020, 21, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Fando, J.L.; Salinas, M.; Wasterlain, C.G. Age-dependent changes in brain protein synthesis in the rat. Neurochem. Res. 1980, 5, 373–383. [Google Scholar] [CrossRef]
- Hovda, D.A.; Villablanca, J.R.; Chugani, H.T.; Barrio, J.R. Metabolic maturation of the brain: A study of local cerebral protein synthesis in the developing cat. Brain Res. 2006, 1113, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Deibler, G.E.; Jehle, J.; Macedonia, J.; Dumont, I.; Dang, T.; Smith, C.B. Rates of local cerebral protein synthesis in the rat during normal postnatal development. Am. J. Physiol. 1995, 268, R549–R561. [Google Scholar] [CrossRef]
- Vargas, R.; Castañeda, M. Age-dependent decrease in the activity of protein-synthesis initiation factors in rat brain. Mech. Ageing Dev. 1983, 21, 183–191. [Google Scholar] [CrossRef]
- Qin, M.; Kang, J.; Burlin, T.V.; Jiang, C.; Smith, C.B. Postadolescent changes in regional cerebral protein synthesis: An in vivo study in the FMR1 null mouse. J. Neurosci. 2005, 25, 5087–5095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayola, R.; Condorelli, D.F.; Ragusa, N.; Renis, M.; Alberghina, M.; Stella, A.M.G.; Lajtha, A. Protein synthesis rates in rat brain regions and subcellular fractions during aging. Neurochem. Res. 1988, 13, 337–342. [Google Scholar] [CrossRef] [PubMed]
- English, J.A.; Fan, Y.; Föcking, M.; Lopez, L.M.; Hryniewiecka, M.; Wynne, K.; Dicker, P.; Matigian, N.; Cagney, G.; Mackay-Sim, A.; et al. Reduced protein synthesis in schizophrenia patient-derived olfactory cells. Transl. Psychiatry 2015, 5, e663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arioka, Y.; Shishido, E.; Kushima, I.; Suzuki, T.; Saito, R.; Aiba, A.; Mori, D.; Ozaki, N. Chromosome 22q11.2 deletion causes PERK-dependent vulnerability in dopaminergic neurons. EBioMedicine 2021, 63, 103138. [Google Scholar] [CrossRef]
- Wei, Y.N.; Hu, H.Y.; Xie, G.C.; Fu, N.; Ning, Z.B.; Zeng, R.; Khaitovich, P. Transcript and protein expression decoupling reveals RNA binding proteins and miRNAs as potential modulators of human aging. Genome Biol. 2015, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Breen, M.S.; Ozcan, S.; Ramsey, J.M.; Wang, Z.; Ma’ayan, A.; Rustogi, N.; Gottschalk, M.G.; Webster, M.J.; Weickert, C.S.; Buxbaum, J.D.; et al. Temporal proteomic profiling of postnatal human cortical development. Transl. Psychiatry 2018, 8, 267. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Messina, A.; Langlet, F.; Chachlaki, K.; Roa, J.; Rasika, S.; Jouy, N.; Gallet, S.; Gaytan, F.; Parkash, J.; Tena-Sempere, M.; et al. A microRNA switch regulates the rise in hypothalamic GnRH production before puberty. Nat. Neurosci. 2016, 19, 835–844. [Google Scholar] [CrossRef]
- Herting, M.M.; Sowell, E.R. Puberty and structural brain development in humans. Front. Neuroendocrinol. 2017, 44, 122–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juraska, J.M.; Sisk, C.L.; DonCarlos, L.L. Sexual differentiation of the adolescent rodent brain: Hormonal influences and developmental mechanisms. Horm. Behav. 2013, 64, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L.; Thompson, A.P.; Krenzel, E.; Teicher, M.H. Pubertal changes in gonadal hormones do not underlie adolescent dopamine receptor overproduction. Psychoneuroendocrinology 2002, 27, 683–691. [Google Scholar] [CrossRef]
- Vetter-O’Hagen, C.S.; Spear, L.P. Hormonal and physical markers of puberty and their relationship to adolescent-typical novelty-directed behavior. Dev. Psychobiol. 2012, 54, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.J.; Probst, C.K.; Brown, L.M.; de Vries, G.J. Dissociation of Puberty and Adolescent Social Development in a Seasonally Breeding Species. Curr. Biol. 2018, 28, 1116–1123.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef] [Green Version]
- Bédard, A.; Parent, A. Evidence of newly generated neurons in the human olfactory bulb. Brain Res. Dev. Brain Res. 2004, 151, 159–168. [Google Scholar] [CrossRef]
- Wang, C.; Liu, F.; Liu, Y.-Y.; Zhao, C.-H.; You, Y.; Wang, L.; Zhang, J.; Wei, B.; Ma, T.; Zhang, Q.; et al. Identification and characterization of neuroblasts in the subventricular zone and rostral migratory stream of the adult human brain. Cell Res. 2011, 21, 1534–1550. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Liebl, J.; Bernard, S.; Alkass, K.; Yeung, M.S.Y.; Steier, P.; Kutschera, W.; Johnson, L.; Landén, M.; Druid, H.; et al. The age of olfactory bulb neurons in humans. Neuron 2012, 74, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Ernst, A.; Alkass, K.; Bernard, S.; Salehpour, M.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; Frisén, J. Neurogenesis in the striatum of the adult human brain. Cell 2014, 156, 1072–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, O.; Spalding, K.L.; Frisén, J. Adult neurogenesis in humans. Cold Spring Harb. Perspect. Med. 2015, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scolding, N.; Franklin, R.; Stevens, S.; Heldin, C.H.; Compston, A.; Newcombe, J. Oligodendrocyte progenitors are present in the normal adult human CNS and in the lesions of multiple sclerosis. Brain 1998, 121, 2221–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, M.S.Y.; Zdunek, S.; Bergmann, O.; Bernard, S.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Brundin, L.; et al. Dynamics of Oligodendrocyte Generation and Myelination in the Human Brain. Cell 2014, 159, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, R.D.; Curtis, M.A.; Spalding, K.L.; Buchholz, B.A.; Fink, D.; Björk-Eriksson, T.; Nordborg, C.; Gage, F.H.; Druid, H.; Eriksson, P.S.; et al. Neocortical neurogenesis in humans is restricted to development. Proc. Natl. Acad. Sci. USA 2006, 103, 12564–12568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, M.C.; Roy, N.S.; Keyoung, H.M.; Goodman, R.R.; McKhann, G.; Jiang, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003, 9, 439–447. [Google Scholar] [CrossRef]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21 st century. J. Psychopharmacol. 2015, 29, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Nord, M.; Farde, L. Antipsychotic Occupancy of Dopamine Receptors in Schizophrenia. CNS Neurosci. Ther. 2011, 17, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Tseng, K.Y.; O’Donnell, P. Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb. Cortex 2007, 17, 1235–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, K.-Y.; Lewis, B.L.; Lipska, B.K.; O’Donnell, P. Post-pubertal disruption of medial prefrontal cortical dopamine-glutamate interactions in a developmental animal model of schizophrenia. Biol. Psychiatry 2007, 62, 730–738. [Google Scholar] [CrossRef] [Green Version]
- Goldman-Rakic, P.S.; Brown, R.M. Postnatal development of monoamine content and synthesis in the cerebral cortex of rhesus monkeys. Dev. Brain Res. 1982, 4, 339–349. [Google Scholar] [CrossRef]
- Rosenberg, D.R.; Lewis, D.A. Changes in the dopaminergic innervation of monkey prefrontal cortex during late postnatal development: A tyrosine hydroxylase immunohistochemical study. Biol. Psychiatry 1994, 36, 272–277. [Google Scholar] [CrossRef]
- Reynolds, L.M.; Pokinko, M.; Torres-Berrío, A.; Cuesta, S.; Lambert, L.C.; Del Cid Pellitero, E.; Wodzinski, M.; Manitt, C.; Krimpenfort, P.; Kolb, B.; et al. DCC Receptors Drive Prefrontal Cortex Maturation by Determining Dopamine Axon Targeting in Adolescence. Biol. Psychiatry 2018, 83, 181–192. [Google Scholar] [CrossRef]
- Torres-Berrío, A.; Lopez, J.P.; Bagot, R.C.; Nouel, D.; Dal Bo, G.; Cuesta, S.; Zhu, L.; Manitt, C.; Eng, C.; Cooper, H.M.; et al. DCC Confers Susceptibility to Depression-like Behaviors in Humans and Mice and Is Regulated by miR-218. Biol. Psychiatry 2017, 81, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Chun, S.; Du, F.; Westmoreland, J.J.; Han, S.B.; Wang, Y.-D.; Eddins, D.; Bayazitov, I.T.; Devaraju, P.; Yu, J.; Mellado Lagarde, M.M.; et al. Thalamic miR-338-3p mediates auditory thalamocortical disruption and its late onset in 22q11.2 microdeletion models. Nat. Med. 2016, 23, 1–39. [Google Scholar] [CrossRef]
- Eom, T.Y.; Han, S.B.; Kim, J.; Blundon, J.A.; Wang, Y.D.; Yu, J.; Anderson, K.; Kaminski, D.B.; Sakurada, S.M.; Pruett-Miller, S.M.; et al. Schizophrenia-related microdeletion causes defective ciliary motility and brain ventricle enlargement via microRNA-dependent mechanisms in mice. Nat. Commun. 2020, 11, 912. [Google Scholar] [CrossRef]
- Jia, X.; Wang, F.; Han, Y.; Geng, X.; Li, M.; Shi, Y.; Lu, L.; Chen, Y. miR-137 and miR-491 Negatively Regulate Dopamine Transporter Expression and Function in Neural Cells. Neurosci. Bull. 2016, 32, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Harris, L.W.; Lockstone, H.E.; Khaitovich, P.; Weickert, C.S.; Webster, M.J.; Bahn, S. Gene expression in the prefrontal cortex during adolescence: Implications for the onset of schizophrenia. BMC Med. Genomics 2009, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, N.; Das, S.; Kar Mahapatra, S.; Chakraborty, S.P.; Kundu, P.K.; Roy, S. Age associated oxidative damage in lymphocytes. Oxid. Med. Cell. Longev. 2010, 3, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef]
- De-Souza-Ferreira, E.; Rios-Neto, I.M.; Martins, E.L.; Galina, A. Mitochondria-coupled glucose phosphorylation develops after birth to modulate H2O2 release and calcium handling in rat brain. J. Neurochem. 2019, 149, 624–640. [Google Scholar] [CrossRef]
- Thorburne, S.K.; Juurlink, B.H. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J. Neurochem. 1996, 67, 1014–1022. [Google Scholar] [CrossRef]
- Back, S.A.; Gan, X.; Li, Y.; Rosenberg, P.A.; Volpe, J.J. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J. Neurosci. 1998, 18, 6241–6253. [Google Scholar] [CrossRef] [PubMed]
- Monin, A.; Baumann, P.S.; Griffa, A.; Xin, L.; Mekle, R.; Fournier, M.; Butticaz, C.; Klaey, M.; Cabungcal, J.H.; Steullet, P.; et al. Glutathione deficit impairs myelin maturation: Relevance for white matter integrity in schizophrenia patients. Mol. Psychiatry 2015, 20, 827–838. [Google Scholar] [CrossRef] [Green Version]
- Kann, O. The interneuron energy hypothesis: Implications for brain disease. Neurobiol. Dis. 2016, 90, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Papageorgiou, I.E.; Draguhn, A. Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J. Cereb. Blood Flow Metab. 2014, 34, 1270–1282. [Google Scholar] [CrossRef] [Green Version]
- Sowell, E.R.; Thompson, P.M.; Holmes, C.J.; Jernigan, T.L.; Toga, A.W. In vivo evidence for post-adolescent brain maturation in frontal and striatal regions. Nat. Neurosci. 1999, 2, 859–861. [Google Scholar] [CrossRef] [PubMed]
- Lebel, C.; Walker, L.; Leemans, A.; Phillips, L.; Beaulieu, C. Microstructural maturation of the human brain from childhood to adulthood. Neuroimage 2008, 40, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev PL, L.A. The myelogenetic cycles of regional maturation of the brain. In Regional Development of the Brain in Early Life; Minkowski, A., Ed.; Blackwell Scientific: Oxford, UK, 1967; pp. 3–70. [Google Scholar]
- Benes, F.M. Myelination of cortical-hippocampal relays during late adolescence. Schizophr. Bull. 1989, 15, 585–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, D.A.; Moore, C.S. MiRNAs as emerging regulators of oligodendrocyte development and differentiation. Front. Cell Dev. Biol. 2016, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Silveri, M.M.; Sneider, J.T.; Crowley, D.J.; Covell, M.J.; Acharya, D.; Rosso, I.M.; Jensen, J.E. Frontal lobe γ-aminobutyric acid levels during adolescence: Associations with impulsivity and response inhibition. Biol. Psychiatry 2013, 74, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoftman, G.D.; Volk, D.W.; Bazmi, H.H.; Li, S.; Sampson, A.R.; Lewis, D.A. Altered cortical expression of GABA-related genes in schizophrenia: Illness progression vs developmental disturbance. Schizophr. Bull. 2015, 41, 180–191. [Google Scholar] [CrossRef]
- Cruz, D.A.; Eggan, S.M.; Lewis, D.A. Postnatal development of pre- and postsynaptic GABA markers at chandelier cell connections with pyramidal neurons in monkey prefrontal cortex. J. Comp. Neurol. 2003, 465, 385–400. [Google Scholar] [CrossRef]
- Datta, D.; Arion, D.; Lewis, D.A. Developmental Expression Patterns of GABAA Receptor Subunits in Layer 3 and 5 Pyramidal Cells of Monkey Prefrontal Cortex. Cereb. Cortex 2015, 25, 2295–2305. [Google Scholar] [CrossRef] [Green Version]
- Fung, S.J.; Webster, M.J.; Sivagnanasundaram, S.; Duncan, C.; Elashoff, M.; Weickert, C.S. Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am. J. Psychiatry 2010, 167, 1479–1488. [Google Scholar] [CrossRef]
- Hoftman, G.D.; Lewis, D.A. Postnatal developmental trajectories of neural circuits in the primate prefrontal cortex: Identifying sensitive periods for vulnerability to schizophrenia. Schizophr. Bull. 2011, 37, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, S.J.; Lewis, D.A. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol. Dis. 2019, 131, 104208. [Google Scholar] [CrossRef] [PubMed]
- Schür, R.R.; Draisma, L.W.R.; Wijnen, J.P.; Boks, M.P.; Koevoets, M.G.J.C.; Joëls, M.; Klomp, D.W.; Kahn, R.S.; Vinkers, C.H. Brain GABA levels across psychiatric disorders: A systematic literature review and meta-analysis of 1H-MRS studies. Hum. Brain Mapp. 2016, 37, 3337–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egerton, A.; Modinos, G.; Ferrera, D.; McGuire, P. Neuroimaging studies of GABA in schizophrenia: A systematic review with meta-analysis. Transl. Psychiatry 2017, 7, e1147-10. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Vajawat, B.; Rao, N.P. Frontal GABA in schizophrenia: A meta-analysis of 1H-MRS studies. World J. Biol. Psychiatry 2021, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Fernandes, C.C.; Ewell, L.A.; John, D.; Romoli, B.; Curia, G.; Taylor, S.R.; Frady, E.P.; Jensen, A.B.; Liu, J.C.; et al. MicroRNA-101 Regulates Multiple Developmental Programs to Constrain Excitation in Adult Neural Networks. Neuron 2016, 92, 1337–1351. [Google Scholar] [CrossRef] [Green Version]
- Huttenlocher, P.R.; Dabholkar, A.S. Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 1997, 387, 167–178. [Google Scholar] [CrossRef]
- Huttenlocher, P.R. Synaptic density in human frontal cortex—Developmental changes and effects of aging. Brain Res. 1979, 163, 195–205. [Google Scholar] [CrossRef]
- Petanjek, Z.; Judaš, M.; Šimić, G.; Rašin, M.R.; Uylings, H.B.M.; Rakic, P.; Kostović, I. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc. Natl. Acad. Sci. USA 2011, 108, 13281–13286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallya, A.P.; Wang, H.D.; Lee, H.N.R.; Deutch, A.Y. Microglial Pruning of Synapses in the Prefrontal Cortex during Adolescence. Cereb. Cortex 2019, 29, 1634–1643. [Google Scholar] [CrossRef]
- Koss, W.A.; Belden, C.E.; Hristov, A.D.; Juraska, J.M. Dendritic remodeling in the adolescent medial prefrontal cortex and the basolateral amygdala of male and female rats. Synapse 2014, 68, 61–72. [Google Scholar] [CrossRef]
- Johnson, C.M.; Loucks, F.A.; Peckler, H.; Thomas, A.W.; Janak, P.H.; Wilbrecht, L. Long-range orbitofrontal and amygdala axons show divergent patterns of maturation in the frontal cortex across adolescence. Dev. Cogn. Neurosci. 2016, 18, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Zheutlin, A.B.; Jeffries, C.D.; Perkins, D.O.; Chung, Y.; Chekroud, A.M.; Addington, J.; Bearden, C.E.; Cadenhead, K.S.; Cornblatt, B.A.; Mathalon, D.H.; et al. The Role of microRNA Expression in Cortical Development during Conversion to Psychosis. Neuropsychopharmacology 2017, 42, 2188–2195. [Google Scholar] [CrossRef] [Green Version]
- Drzewiecki, C.M.; Willing, J.; Juraska, J.M. Synaptic number changes in the medial prefrontal cortex across adolescence in male and female rats: A role for pubertal onset. Synapse 2016, 70, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, I. Schizophrenia: Caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 1982, 17, 319–334. [Google Scholar] [CrossRef]
- Valiathan, R.; Ashman, M.; Asthana, D. Effects of Ageing on the Immune System: Infants to Elderly. Scand. J. Immunol. 2016, 83, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Tollerud, D.J.; Ildstad, S.T.; Brown, L.M.; Clark, J.W.; Blattner, W.A.; Mann, D.L.; Neuland, C.Y.; Pankiw-Trost, L.; Hoover, R.N. T-cell subsets in healthy teenagers: Transition to the adult phenotype. Clin. Immunol. Immunopathol. 1990, 56, 88–96. [Google Scholar] [CrossRef]
- Aldrimer, M.; Ridefelt, P.; Rödöö, P.; Niklasson, F.; Gustafsson, J.; Hellberg, D. Population-based pediatric reference intervals for hematology, iron and transferrin. Scand. J. Clin. Lab. Invest. 2013, 73, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Dalferes, E.R.J.; Srinivasan, S.R.; Webber, L.S.; Berenson, G.S. Normative distribution of complete blood count from early childhood through adolescence: The Bogalusa Heart Study. Prev. Med. 1993, 22, 825–837. [Google Scholar] [CrossRef]
- Simon, A.K.; Hollander, G.A.; McMichael, A. Evolution of the immune system in humans from infancy to old age. Proc. Biol. Sci. 2015, 282, 20143085. [Google Scholar] [CrossRef]
- Brenhouse, H.C.; Schwarz, J.M. Immunoadolescence: Neuroimmune development and adolescent behavior. Neurosci. Biobehav. Rev. 2016, 70, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Levinson, D.F.; Duan, J.; Sanders, A.R.; Zheng, Y.; Pe’er, I.; Dudbridge, F.; Holmans, P.A.; Whittemore, A.S.; Mowry, B.J.; et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009, 460, 753–757. [Google Scholar] [CrossRef]
- Stefansson, H.; Ophoff, R.A.; Steinberg, S.; Andreassen, O.A.; Cichon, S.; Rujescu, D.; Werge, T.; Pietiläinen, O.P.H.; Mors, O.; Mortensen, P.B.; et al. Common variants conferring risk of schizophrenia. Nature 2009, 460, 744–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, A.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Li, R.; McGeer, E.G.; McGeer, P.L. Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res. 1997, 769, 391–395. [Google Scholar] [CrossRef]
- Veerhuis, R.; Nielsen, H.M.; Tenner, A.J. Complement in the brain. Mol. Immunol. 2011, 48, 1592–1603. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.J.; Goldsmith, D.R. Inflammatory biomarkers in schizophrenia: Implications for heterogeneity and neurobiology. Biomark. Neuropsychiatry 2019, 1, 100006. [Google Scholar] [CrossRef]
- Howes, O.D.; McCutcheon, R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: A reconceptualization. Transl. Psychiatry 2017, 7, e1024-11. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, S.M.; Krüger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An unconventional role for miRNA: Let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Barak, B.; Feldman, N.; Okun, E. Toll-like receptors as developmental tools that regulate neurogenesis during development: An update. Front. Neurosci. 2014, 8, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolls, A.; Shechter, R.; London, A.; Ziv, Y.; Ronen, A.; Levy, R.; Schwartz, M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat. Cell Biol. 2007, 9, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Okun, E.; Griffioen, K.; Barak, B.; Roberts, N.J.; Castro, K.; Pita, M.A.; Cheng, A.; Mughal, M.R.; Wan, R.; Ashery, U.; et al. Toll-like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15625–15630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Y.; Lin, C.W.; Chang, C.Y.; Jian, S.T.; Hsueh, Y.P. Sarm1, a negative regulator of innate immunity, interacts with syndecan-2 and regulates neuronal morphology. J. Cell Biol. 2011, 193, 769–784. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lambris, J.D. More than complementing Tolls: Complement-Toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol. Rev. 2016, 274, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Woods, S.W.; Addington, J.; Cadenhead, K.S.; Cannon, T.D.; Cornblatt, B.A.; Heinssen, R.; Perkins, D.O.; Seidman, L.J.; Tsuang, M.T.; Walker, E.F.; et al. Validity of the Prodromal Risk Syndrome for First Psychosis: Findings From the North American Prodrome Longitudinal Study. Schizophr. Bull. 2009, 35, 894–908. [Google Scholar] [CrossRef] [Green Version]
- Cannon, T.D.; Cadenhead, K.; Cornblatt, B.; Woods, S.W.; Addington, J.; Walker, E.; Seidman, L.J.; Perkins, D.; Tsuang, M.; McGlashan, T.; et al. Prediction of psychosis in youth at high clinical risk: A multisite longitudinal study in North America. Arch. Gen. Psychiatry 2008, 65, 28–37. [Google Scholar] [CrossRef]
- Fusar-Poli, P.; Bonoldi, I.; Yung, A.R.; Borgwardt, S.; Kempton, M.J.; Valmaggia, L.; Barale, F.; Caverzasi, E.; McGuire, P. Predicting psychosis: Meta-analysis of transition outcomes in individuals at high clinical risk. Arch. Gen. Psychiatry 2012, 69, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Yung, A.R.; Nelson, B.; Stanford, C.; Simmons, M.B.; Cosgrave, E.M.; Killackey, E.; Phillips, L.J.; Bechdolf, A.; Buckby, J.; McGorry, P.D. Validation of “prodromal” criteria to detect individuals at ultra high risk of psychosis: 2 year follow-up. Schizophr. Res. 2008, 105, 10–17. [Google Scholar] [CrossRef]
- Ziermans, T.B.; Schothorst, P.F.; Sprong, M.; van Engeland, H. Transition and remission in adolescents at ultra-high risk for psychosis. Schizophr. Res. 2011, 126, 58–64. [Google Scholar] [CrossRef]
- Fusar-Poli, P.; Borgwardt, S.; Bechdolf, A.; Addington, J.; Riecher-Rössler, A.; Schultze-Lutter, F.; Keshavan, M.; Wood, S.; Ruhrmann, S.; Seidman, L.J.; et al. The psychosis high-risk state: A comprehensive state-of-the-art review. Arch. Gen. Psychiatry 2013, 70, 107–120. [Google Scholar] [CrossRef]
- Lee, T.Y.; Lee, J.; Kim, M.; Choe, E.; Kwon, J.S. Can we predict psychosis outside the clinical high-risk state? A systematic review of non-psychotic risk syndromes for mental disorders. Schizophr. Bull. 2018, 44, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Addington, J.; Cadenhead, K.S.; Cornblatt, B.A.; Mathalon, D.H.; McGlashan, T.H.; Perkins, D.O.; Seidman, L.J.; Tsuang, M.T.; Walker, E.F.; Woods, S.W.; et al. North American Prodrome Longitudinal Study (NAPLS 2): Overview and recruitment. Schizophr. Res. 2012, 142, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Addington, J.; Liu, L.; Buchy, L.; Cadenhead, K.S.; Cannon, T.D.; Cornblatt, B.A.; Perkins, D.O.; Seidman, L.J.; Tsuang, M.T.; Walker, E.F.; et al. North American Prodrome Longitudinal Study (NAPLS 2): The Prodromal Symptoms. J. Nerv. Ment. Dis. 2015, 203, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Jeffries, C.D.; Perkins, D.O.; Chandler, S.D.; Stark, T.; Yeo, E.; Addington, J.; Bearden, C.E.; Cadenhead, K.S.; Cannon, T.D.; Cornblatt, B.A.; et al. Insights into psychosis risk from leukocyte microRNA expression. Transl. Psychiatry 2016, 6, e981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, T.D.; Chung, Y.; He, G.; Sun, D.; Jacobson, A.; Van Erp, T.G.M.; McEwen, S.; Addington, J.; Bearden, C.E.; Cadenhead, K.; et al. Progressive reduction in cortical thickness as psychosis develops: A multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol. Psychiatry 2015, 77, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, W.H.; Kim, J.S.; Jang, J.H.; Choi, J.S.; Jung, M.H.; Park, J.Y.; Han, J.Y.; Choi, C.H.; Kang, D.H.; Chung, C.K.; et al. Cortical thickness reduction in individuals at ultra-high-risk for psychosis. Schizophr. Bull. 2011, 37, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Tamnes, C.K.; Herting, M.M.; Goddings, A.L.; Meuwese, R.; Blakemore, S.J.; Dahl, R.E.; Güroğlu, B.; Raznahan, A.; Sowell, E.R.; Crone, E.A.; et al. Development of the cerebral cortex across adolescence: A multisample study of inter-related longitudinal changes in cortical volume, surface area, and thickness. J. Neurosci. 2017, 37, 3402–3412. [Google Scholar] [CrossRef]
- Tamnes, C.K.; Østby, Y.; Fjell, A.M.; Westlye, L.T.; Due-Tønnessen, P.; Walhovd, K.B. Brain maturation in adolescence and young adulthood: Regional age-related changes in cortical thickness and white matter volume and microstructure. Cereb. Cortex 2010, 20, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, R.M.; Rybka, J.; Anderson, S.M.; Torrance, H.S.; Boxall, R.; Sussmann, J.E.; Porteous, D.J.; McIntosh, A.M.; Evans, K.L. Preliminary investigation of miRNA expression in individuals at high familial risk of bipolar disorder. J. Psychiatr. Res. 2015, 62, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Rasic, D.; Hajek, T.; Alda, M.; Uher, R. Risk of mental illness in offspring of parents with schizophrenia, bipolar disorder, and major depressive disorder: A meta-analysis of family high-risk studies. Schizophr. Bull. 2014, 40, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenstein, P.; Yip, B.H.; Björk, C.; Pawitan, Y.; Cannon, T.D.; Sullivan, P.F.; Hultman, C.M. Common genetic influences for schizophrenia and bipolar disorder: A population-based study of 2 million nuclear families. Lancet 2009, 373, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Jinde, S.; Koike, S.; Tada, M.; Satomura, Y.; Yoshikawa, A.; Nishimura, Y.; Takizawa, R.; Kinoshita, A.; Sakakibara, E.; et al. Altered expression of microRNA-223 in the plasma of patients with first-episode schizophrenia and its possible relation to neuronal migration-related genes. Transl. Psychiatry 2019, 9, 289. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, E.; Beveridge, N.J.; Wu, J.Q.; Carr, V.; Scott, R.J.; Tooney, P.A.; Cairns, M.J. Imprinted DLK1-DIO3 region of 14q32 defines a schizophrenia-associated miRNA signature in peripheral blood mononuclear cells. Mol. Psychiatry 2012, 17, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Lackinger, M.; Sungur, A.Ö.; Daswani, R.; Soutschek, M.; Bicker, S.; Stemmler, L.; Wüst, T.; Fiore, R.; Dieterich, C.; Schwarting, R.K.; et al. A placental mammal-specific micro RNA cluster acts as a natural brake for sociability in mice. EMBO Rep. 2019, 20, 1–11. [Google Scholar] [CrossRef]
- Fiore, R.; Khudayberdiev, S.; Christensen, M.; Siegel, G.; Flavell, S.W.; Kim, T.-K.; Greenberg, M.E.; Schratt, G.; Bagni, C.; Greenough, W.; et al. Mef2-mediated transcription of the miR379–410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 2009, 28, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.E.; Lee, P.R.; Chen, S.; Li, W.; Fields, R.D. MicroRNA regulation of homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 11650–11655. [Google Scholar] [CrossRef] [Green Version]
- Marty, V.; Labialle, S.; Bortolin-Cavaillé, M.L.; De Medeiros, G.F.; Moisan, M.P.; Florian, C.; Cavaillé, J. Deletion of the miR-379/miR-410 gene cluster at the imprinted Dlk1-Dio3 locus enhances anxiety-related behaviour. Hum. Mol. Genet. 2016, 25, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, F.; Wang, X.; Shugart, Y.Y.; Zhao, Y.; Li, X.; Liu, Z.; Sun, N.; Yang, C.; Zhang, K.; et al. Diagnostic value of blood-derived microRNAs for schizophrenia: Results of a meta-analysis and validation. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Chen, B.-Y.; Lin, J.-J.; Lu, M.-K.; Tan, H.-P.; Jang, F.-L.; Lin, S.-H. Neurodevelopment regulators miR-137 and miR-34 family as biomarkers for early and adult onset schizophrenia. npj Schizophr. 2021, 7, 35. [Google Scholar] [CrossRef]
- Gruzdev, S.K.; Yakovlev, A.A.; Druzhkova, T.A.; Guekht, A.B.; Gulyaeva, N.V. The Missing Link: How Exosomes and miRNAs can Help in Bridging Psychiatry and Molecular Biology in the Context of Depression, Bipolar Disorder and Schizophrenia. Cell. Mol. Neurobiol. 2019, 39, 729–750. [Google Scholar] [CrossRef] [PubMed]
- Khavari, B.; Cairns, M.J. Epigenomic Dysregulation in Schizophrenia: In Search of Disease Etiology and Biomarkers. Cells 2020, 9, 1837. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Baune, B.T.; Schubert, K.O.; Lavoie, S.; Smesny, S.; Rice, S.M.; Schäfer, M.R.; Benninger, F.; Feucht, M.; Klier, C.M.; et al. Prediction of transition from ultra-high risk to first-episode psychosis using a probabilistic model combining history, clinical assessment and fatty-acid biomarkers. Transl. Psychiatry 2016, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Perkins, D.O.; Gu, H.; Boteva, K.; Lieberman, J.A. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: A critical review and meta-analysis. Am. J. Psychiatry 2005, 162, 1785–1804. [Google Scholar] [CrossRef]
- Howes, O.D.; Whitehurst, T.; Shatalina, E.; Townsend, L.; Onwordi, E.C.; Mak, T.L.A.; Arumuham, A.; O’Brien, O.; Lobo, M.; Vano, L.; et al. The clinical significance of duration of untreated psychosis: An umbrella review and random-effects meta-analysis. World Psychiatry 2021, 20, 75–95. [Google Scholar] [CrossRef]
- Fusar-Poli, P.; Davies, C.; Solmi, M.; Brondino, N.; De Micheli, A.; Kotlicka-Antczak, M.; Shin, J.I.; Radua, J. Preventive Treatments for Psychosis: Umbrella Review (Just the Evidence). Front. Psychiatry 2019, 10, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitetti, A.; Mallory, A.C.; Golini, E.; Carrieri, C.; Carreño Gutiérrez, H.; Perlas, E.; Pérez-Rico, Y.A.; Tocchini-Valentini, G.P.; Enright, A.J.; Norton, W.H.J.; et al. MicroRNA degradation by a conserved target RNA regulates animal behavior. Nat. Struct. Mol. Biol. 2018, 25, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Qiao, H.; Lu, Z.; Hou, Y. miR-29 promotes the proliferation of cultured rat neural stem/progenitor cells via the PTEN/AKT signaling pathway. Mol. Med. Rep. 2019, 20, 2111–2118. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.Q.; Feng, J.G.; Li, M.; Wang, M.Z.; Liu, L.; Liu, X.; Duan, X.X.; Zhang, C.X.; Wang, X. Bin Prefrontal cortex miR-29b-3p plays a key role in the antidepressant-like effect of ketamine in rats. Exp. Mol. Med. 2018, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripa, R.; Dolfi, L.; Terrigno, M.; Pandolfini, L.; Savino, A.; Arcucci, V.; Groth, M.; Terzibasi Tozzini, E.; Baumgart, M.; Cellerino, A. MicroRNA miR-29 controls a compensatory response to limit neuronal iron accumulation during adult life and aging. BMC Biol. 2017, 15, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshan, R.; Shridhar, S.; Sarangdhar, M.A.; Banik, A.; Chawla, M.; Garg, M.; Singh, V.P.A.L.; Pillai, B. Brain-specific knockdown of miR-29 results in neuronal cell death and ataxia in mice. RNA 2014, 20, 1287–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellios, N.; Sugihara, H.; Castro, J.; Banerjee, A.; Le, C.; Kumar, A.; Crawford, B.; Strathmann, J.; Tropea, D.; Levine, S.S.; et al. miR-132, an experience-dependent microRNA, is essential for visual cortex plasticity. Nat. Neurosci. 2011, 14, 1240–1242. [Google Scholar] [CrossRef] [Green Version]
- Garey, L.J.; Ong, W.Y.; Patel, T.S.; Kanani, M.; Davis, A.; Mortimer, A.M.; Barnes, T.R.E.; Hirsch, S.R. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry 1998, 65, 446–453. [Google Scholar] [CrossRef]
- Glantz, L.A.; Lewis, D.A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 2000, 57, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Sweet, R.A.; Henteleff, R.A.; Zhang, W.; Sampson, A.R.; Lewis, D.A. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology 2009, 34, 374–389. [Google Scholar] [CrossRef] [Green Version]
- Konopaske, G.T.; Lange, N.; Coyle, J.T.; Benes, F.M. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 2014, 71, 1323–1331. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.F.; Sakamoto, K.; Aten, S.; Snider, K.H.; Loeser, J.; Hesse, A.M.; Page, C.E.; Pelz, C.; Simon, J.; Arthur, C.; et al. Targeted deletion of miR-132/-212 impairs memory and alters the hippocampal transcriptome. Learn. Mem. 2016, 23, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.Y.; Phang, R.Z.; Hsu, P.H.; Wang, W.H.; Huang, H.T.; Liu, I.Y. In vivo knockdown of hippocampal miR-132 expression impairs memory acquisition of trace fear conditioning. Hippocampus 2013, 23, 625–633. [Google Scholar] [CrossRef]
- Ronovsky, M.; Zambon, A.; Cicvaric, A.; Boehm, V.; Hoesel, B.; Moser, B.A.; Yang, J.; Schmid, J.A.; Haubensak, W.E.; Monje, F.J.; et al. A role for miR-132 in learned safety. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.J.; Clinton, J.M.; Taishi, P.; Bohnet, S.G.; Honn, K.A.; Krueger, J.M. MicroRNA 132 alters sleep and varies with time in brain. J. Appl. Physiol. 2011, 111, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.Y.M.; Papp, J.W.; Varlamova, O.; Dziema, H.; Russell, B.; Curfman, J.P.; Nakazawa, T.; Shimizu, K.; Okamura, H.; Impey, S.; et al. microRNA Modulation of Circadian-Clock Period and Entrainment. Neuron 2007, 54, 813–829. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Saavedra, M.; Antoun, G.; Yanagiya, A.; Oliva-Hernandez, R.; Cornejo-Palma, D.; Perez-Iratxeta, C.; Sonenberg, N.; Cheng, H.Y.M. miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum. Mol. Genet. 2011, 20, 731–751. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.Y.; Britton, J.C.; Nelson, E.E.; Angold, A.; Ernst, M.; Goldwin, M.; Grillon, C.; Leibenluft, E.; Lissek, S.; Norcross, M.; et al. Distinct neural signatures of threat learning in adolescents and adults. Proc. Natl. Acad. Sci. USA 2011, 108, 4500–4505. [Google Scholar] [CrossRef] [Green Version]
- Pattwell, S.S.; Duhoux, S.; Hartley, C.A.; Johnson, D.C.; Jing, D.; Elliott, M.D.; Ruberry, E.J.; Powers, A.; Mehta, N.; Yang, R.R.; et al. Altered fear learning across development in both mouse and human. Proc. Natl. Acad. Sci. USA 2012, 109, 16318–16323. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.; Theresiana, C.; Neumann, D.; Craske, M. Developmental differences in aversive conditioning, extinction, and reinstatement: A study with children, adolescents, and adults. J. Exp. Child Psychol. 2017, 159, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Ganella, D.E.; Drummond, K.D.; Ganella, E.P.; Whittle, S.; Kim, J.H. Extinction of conditioned fear in adolescents and adults: A human fmri study. Front. Hum. Neurosci. 2018, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagenauer, M.H.; Perryman, J.I.; Lee, T.M.; Carskadon, M.A. Adolescent changes in the homeostatic and circadian regulation of sleep. Dev. Neurosci. 2009, 31, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Crowley, S.J.; Acebo, C.; Carskadon, M.A. Sleep, circadian rhythms, and delayed phase in adolescence. Sleep Med. 2007, 8, 602–612. [Google Scholar] [CrossRef]
- Holt, D.J.; Lebron-Milad, K.; Milad, M.R.; Rauch, S.L.; Pitman, R.K.; Orr, S.P.; Cassidy, B.S.; Walsh, J.P.; Goff, D.C. Extinction Memory Is Impaired in Schizophrenia. Biol. Psychiatry 2009, 65, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Holt, D.J.; Coombs, G.; Zeidan, M.A.; Goff, D.C.; Milad, M.R. Failure of neural responses to safety cues in schizophrenia. Arch. Gen. Psychiatry 2012, 69, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, D.L.; Pan, H.; Weisholtz, D.S.; Root, J.C.; Tuescher, O.; Fischer, D.B.; Butler, T.; Vago, D.R.; Isenberg, N.; Epstein, J.; et al. Altered threat and safety neural processing linked to persecutory delusions in schizophrenia: A two-task fMRI study. Psychiatry Res. Neuroimag. 2015, 233, 352–366. [Google Scholar] [CrossRef] [Green Version]
- Wulff, K.; Dijk, D.J.; Middleton, B.; Foster, R.G.; Joyce, E.M. Sleep and circadian rhythm disruption in schizophrenia. Br. J. Psychiatry 2012, 200, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Hubel, D.H.; Wiesel, T.N. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J. Physiol. 1970, 206, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.C.; Hocking, D.R. Timing of the critical period for plasticity of ocular dominance columns in macaque striate cortex. J. Neurosci. 1997, 17, 3684–3709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, K.; Löwel, S. Age-dependent ocular dominance plasticity in adult mice. PLoS ONE 2008, 3, e3120. [Google Scholar] [CrossRef]
- Issa, N.P.; Trachtenberg, J.T.; Chapman, B.; Zahs, K.R.; Stryker, M.P. The critical period for ocular dominance plasticity in the Ferret’s visual cortex. J. Neurosci. 1999, 19, 6965–6978. [Google Scholar] [CrossRef]
- Pizzorusso, T.; Medini, P.; Berardi, N.; Chierzi, S.; Fawcett, J.W.; Maffei, L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science 2002, 298, 1248–1251. [Google Scholar] [CrossRef] [Green Version]
- Cabungcal, J.H.; Steullet, P.; Morishita, H.; Kraftsik, R.; Cuenod, M.; Hensch, T.K.; Do, K.Q. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 9130–9135. [Google Scholar] [CrossRef] [Green Version]
- Flatow, J.; Buckley, P.; Miller, B.J. Meta-analysis of oxidative stress in schizophrenia. Biol. Psychiatry 2013, 74, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraguas, D.; Díaz-Caneja, C.M.; Rodríguez-Quiroga, A.; Arango, C. Oxidative Stress and Inflammation in Early Onset First Episode Psychosis: A Systematic Review and Meta-Analysis. Int. J. Neuropsychopharmacol. 2017, 20, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Fraguas, D.; Díaz-Caneja, C.M.; Ayora, M.; Hernández-Álvarez, F.; Rodríguez-Quiroga, A.; Recio, S.; Leza, J.C.; Arango, C. Oxidative stress and inflammation in first-episode psychosis: A Systematic Review and Meta-analysis. Schizophr. Bull. 2019, 45, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Javadzadeh, A.; Dey, A.; Sabesan, P.; Théberge, J.; Radua, J.; Palaniyappan, L. Antioxidant defense in schizophrenia and bipolar disorder: A meta-analysis of MRS studies of anterior cingulate glutathione. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 91, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, S.; Noda, Y.; Tarumi, R.; Mimura, Y.; Yoshida, K.; Iwata, Y.; Elsalhy, M.; Kuromiya, M.; Kurose, S.; Masuda, F.; et al. Glutathione levels and activities of glutathione metabolism enzymes in patients with schizophrenia: A systematic review and meta-analysis. J. Psychopharmacol. 2019, 33, 1199–1214. [Google Scholar] [CrossRef]
- Steullet, P.; Cabungcal, J.H.; Coyle, J.; Didriksen, M.; Gill, K.; Grace, A.A.; Hensch, T.K.; Lamantia, A.S.; Lindemann, L.; Maynard, T.M.; et al. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol. Psychiatry 2017, 22, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Chang, H.; Li, E.; Fan, G. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res. 2005, 79, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [Green Version]
- Tognini, P.; Napoli, D.; Tola, J.; Silingardi, D.; Della Ragione, F.; D’esposito, M.; Pizzorusso, T. Experience-dependent DNA methylation regulates plasticity in the developing visual cortex. Nat. Neurosci. 2015, 18, 956–958. [Google Scholar] [CrossRef]
- Zhubi, A.; Veldic, M.; Puri, N.V.; Kadriu, B.; Caruncho, H.; Loza, I.; Sershen, H.; Lajtha, A.; Smith, R.C.; Guidotti, A.; et al. An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophr. Res. 2009, 111, 115–122. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, K.S.; McGregor, N.W.; Emsley, R.; Seedat, S.; Warnich, L. The potential role of regulatory genes (DNMT3A, HDAC5, and HDAC9) in antipsychotic treatment response in South African schizophrenia patients. Front. Genet. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Saradalekshmi, K.R.; Neetha, N.V.; Sathyan, S.; Nair, I.V.; Nair, C.M.; Banerjee, M. DNA methyl transferase (DNMT) gene polymorphisms could be a primary event in epigenetic susceptibility to schizophrenia. PLoS ONE 2014, 9, e98182. [Google Scholar] [CrossRef] [Green Version]
- Tenorio, J.; Alarcón, P.; Arias, P.; Dapía, I.; García-Miñaur, S.; Palomares Bralo, M.; Campistol, J.; Climent, S.; Valenzuela, I.; Ramos, S.; et al. Further delineation of neuropsychiatric findings in Tatton-Brown-Rahman syndrome due to disease-causing variants in DNMT3A: Seven new patients. Eur. J. Hum. Genet. 2020, 28, 469–479. [Google Scholar] [CrossRef]
- Christian, D.L.; Wu, D.Y.; Martin, J.R.; Moore, J.R.; Liu, Y.R.; Clemens, A.W.; Nettles, S.A.; Kirkland, N.M.; Papouin, T.; Hill, C.A.; et al. DNMT3A Haploinsufficiency Results in Behavioral Deficits and Global Epigenomic Dysregulation Shared across Neurodevelopmental Disorders. Cell Rep. 2020, 33, 108416. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Na, E.S.; Autry, A.E.; Monteggia, L.M. Impact of DNMT1 and DNMT3a forebrain knockout on depressive- and anxiety like behavior in mice. Neurobiol. Learn. Mem. 2016, 135, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Lavery, L.A.; Ure, K.; Wan, Y.W.; Luo, C.; Trostle, A.J.; Wang, W.; Jin, H.; Lopez, J.; Lucero, J.; Durham, M.A.; et al. Losing dnmt3a dependent methylation in inhibitory neurons impairs neural function by a mechanism impacting rett syndrome. Elife 2020, 9, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Hwang, V.J.; Dandekar, R.; Durbin-Johnson, B.; Charlet-Berguerand, N.; Ander, B.P.; Sharp, F.R.; Angkustsiri, K.; Simon, T.J.; Tassone, F. Decreased DGCR8 expression and miRNA dysregulation in individuals with 22q11.2 deletion syndrome. PLoS ONE 2014, 9, e103884. [Google Scholar] [CrossRef]
- De la Morena, M.T.; Eitson, J.L.; Dozmorov, I.M.; Belkaya, S.; Hoover, A.R.; Anguiano, E.; Pascual, M.V.; van Oers, N.S.C. Signature MicroRNA expression patterns identified in humans with 22q11.2 deletion/DiGeorge syndrome. Clin. Immunol. 2013, 147, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Bassett, A.S.; Chow, E.W.C.; AbdelMalik, P.; Gheorghiu, M.; Husted, J.; Weksberg, R. The schizophrenia phenotype in 22q11 deletion syndrome. Am. J. Psychiatry 2003, 160, 1580–1586. [Google Scholar] [CrossRef] [Green Version]
- Heckers, S.; Rauch, S.L.; Goff, D.; Savage, C.R.; Schacter, D.L.; Fischman, A.J.; Alpert, N.M. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Neuroimage 1998, 7, 827–836. [Google Scholar] [CrossRef]
- Jessen, F.; Scheef, L.; Germeshausen, L.; Tawo, Y.; Kockler, M.; Kuhn, K.U.; Maier, W.; Schild, H.H.; Heun, R. Reduced hippocampal activation during encoding and recognition of words in schizophrenia patients. Am. J. Psychiatry 2003, 160, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Ragland, J.D.; Gur, R.C.; Valdez, J.; Turetsky, B.I.; Elliott, M.; Kohler, C.; Siegel, S.; Kanes, S.; Gur, R.E. Event-related fMRI of frontotemporal activity during word encoding and recognition in schizophrenia. Am. J. Psychiatry 2004, 161, 1004–1015. [Google Scholar] [CrossRef] [Green Version]
- Pirnia, T.; Woods, R.P.; Hamilton, L.S.; Lyden, H.; Joshi, S.H.; Asarnow, R.F.; Nuechterlein, K.H.; Narr, K.L. Hippocampal dysfunction during declarative memory encoding in schizophrenia and effects of genetic liability. Schizophr. Res. 2015, 161, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragland, J.D.; Ranganath, C.; Harms, M.P.; Barch, D.M.; Gold, J.M.; Layher, E.; Lesh, T.A.; MacDonald, A.W.; Niendam, T.A.; Phillips, J.; et al. Functional and Neuroanatomic Specificity of Episodic Memory Dysfunction in Schizophrenia. JAMA Psychiatry 2015, 72, 909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragland, J.D.; Layher, E.; Hannula, D.E.; Niendam, T.A.; Lesh, T.A.; Solomon, M.; Carter, C.S.; Ranganath, C. Impact of schizophrenia on anterior and posterior hippocampus during memory for complex scenes. NeuroImage Clin. 2017, 13, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Earls, L.R.; Bayazitov, I.T.; Fricke, R.G.; Berry, R.B.; Illingworth, E.; Mittleman, G.; Zakharenko, S.S. Dysregulation of presynaptic calcium and synaptic plasticity in a mouse model of 22q11 deletion syndrome. J. Neurosci. 2010, 30, 15843–15855. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Mathur, P.; Gottesman, I.I.; Nagpal, R.; Nimgaonkar, V.L.; Deshpande, S.N. Correlates of hallucinations in schizophrenia: A cross-cultural evaluation. Schizophr. Res. 2007, 92, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.M.; Schanda, H.; Karakula, H.; Olajossy-Hilkesberger, L.; Rudaleviciene, P.; Okribelashvili, N.; Chaudhry, H.R.; Idemudia, S.E.; Gscheider, S.; Ritter, K.; et al. Culture and the prevalence of hallucinations in schizophrenia. Compr. Psychiatry 2011, 52, 319–325. [Google Scholar] [CrossRef]
- Chun, S.; Westmoreland, J.J.; Bayazitov, I.T.; Eddins, D.; Pani, A.K.; Smeyne, R.J.; Yu, J.; Blundon, J.A.; Zakharenko, S.S. Specific disruption of thalamic inputs to the auditory cortex in schizophrenia models. Science 2014, 344, 1178–1182. [Google Scholar] [CrossRef] [Green Version]
- Wright, I.C.; Rabe-Hesketh, S.; Woodruff, P.W.R.; David, A.S.; Murray, R.M.; Bullmore, E.T. Meta-analysis of regional brain volumes in schizophrenia. Am. J. Psychiatry 2000, 157, 16–25. [Google Scholar] [CrossRef]
- Van Erp, T.G.M.; Hibar, D.P.; Rasmussen, J.M.; Glahn, D.C.; Pearlson, G.D.; Andreassen, O.A.; Agartz, I.; Westlye, L.T.; Haukvik, U.K.; Dale, A.M.; et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol. Psychiatry 2016, 21, 547–553. [Google Scholar] [CrossRef]
- Lawrie, S.M.; Abukmeil, S.S. Brain abnormality in schizophrenia. A systematic and quantitative review of volumetric magnetic resonance imaging studies. Br. J. Psychiatry 1998, 172, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Cahn, W.; Pol, H.E.H.; Lems, E.B.T.E.; van Haren, N.E.M.; Schnack, H.G.; van der Linden, J.A.; Schothorst, P.F.; van Engeland, H.; Kahn, R.S. Brain Volume Changes in First-Episode Schizophrenia. Arch. Gen. Psychiatry 2002, 59, 1002. [Google Scholar] [CrossRef] [Green Version]
- Steen, R.G.; Mull, C.; McClure, R.; Hamer, R.M.; Lieberman, J.A. Brain volume in first-episode schizophrenia: Systematic review and meta-analysis of magnetic resonance imaging studies. Br. J. Psychiatry 2006, 188, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Vita, A.; De Peri, L.; Silenzi, C.; Dieci, M. Brain morphology in first-episode schizophrenia: A meta-analysis of quantitative magnetic resonance imaging studies. Schizophr. Res. 2006, 82, 75–88. [Google Scholar] [CrossRef]
- Olabi, B.; Ellison-Wright, I.; McIntosh, A.M.; Wood, S.J.; Bullmore, E.; Lawrie, S.M. Are there progressive brain changes in schizophrenia? a meta-analysis of structural magnetic resonance imaging studies. Biol. Psychiatry 2011, 70, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempton, M.J.; Stahl, D.; Williams, S.C.R.; DeLisi, L.E. Progressive lateral ventricular enlargement in schizophrenia: A meta-analysis of longitudinal MRI studies. Schizophr. Res. 2010, 120, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Arcila, M.L.; Betizeau, M.; Cambronne, X.A.; Guzman, E.; Doerflinger, N.; Bouhallier, F.; Zhou, H.; Wu, B.; Rani, N.; Bassett, D.S.; et al. Novel primate miRNAs coevolved with ancient target genes in germinal zone-specific expression patterns. Neuron 2014, 81, 1255–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodromidou, K.; Vlachos, I.S.; Gaitanou, M.; Kouroupi, G.; Hatzigeorgiou, A.G.; Matsas, R. MicroRNA-934 is a novel primate-specific small non-coding RNA with neurogenic function during early development. eLife 2020, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R.; Lugli, G.; Zhang, H.; Rizavi, H.; Cook, E.H.; Dwivedi, Y. Expression of micrornas and other small RNAs in prefrontal cortex in schizophrenia, bipolar disorder and depressed subjects. PLoS ONE 2014, 9, e86469. [Google Scholar] [CrossRef] [PubMed]
- Lett, T.; Chakravarty, M.; Chakavarty, M.; Felsky, D.; Brandl, E.; Tiwari, A.; Gonçalves, V.; Rajji, T.; Daskalakis, Z.; Meltzer, H.; et al. The genome-wide supported microRNA-137 variant predicts phenotypic heterogeneity within schizophrenia. Mol. Psychiatry 2013, 18, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.S.; Kelly, S.; Wright, C.; Gupta, C.N.; Arias-Vasquez, A.; Perrone-Bizzozero, N.; Ehrlich, S.; Wang, L.; Bustillo, J.R.; Morris, D.; et al. MIR137HG risk variant rs1625579 genotype is related to corpus callosum volume in schizophrenia. Neurosci. Lett. 2015, 602, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuswanto, C.N.; Sum, M.Y.; Qiu, A.; Sitoh, Y.Y.; Liu, J.; Sim, K. The impact of genome wide supported microRNA-137 (MIR137) risk variants on frontal and striatal white matter integrity, neurocognitive functioning, and negative symptoms in schizophrenia. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2015, 168, 317–326. [Google Scholar] [CrossRef]
- Green, M.J.; Cairns, M.J.; Wu, J.; Dragovic, M.; Jablensky, A.; Tooney, P.A.; Scott, R.J.; Carr, V.J. Genome-wide supported variant MIR137 and severe negative symptoms predict membership of an impaired cognitive subtype of schizophrenia. Mol. Psychiatry 2013, 18, 774–780. [Google Scholar] [CrossRef]
- Siegert, S.; Seo, J.; Kwon, E.J.; Rudenko, A.; Cho, S.; Wang, W.; Flood, Z.; Martorell, A.J.; Ericsson, M.; Mungenast, A.E.; et al. The schizophrenia risk gene product miR-137 alters presynaptic plasticity. Nat. Neurosci. 2015, 18, 1008–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, M.P.; Zhang, H.; Moy, W.; McGowan, H.; Leites, C.; Dionisio, L.E.; Xu, Z.; Shi, J.; Sanders, A.R.; Greenleaf, W.J.; et al. Open Chromatin Profiling in hiPSC-Derived Neurons Prioritizes Functional Noncoding Psychiatric Risk Variants and Highlights Neurodevelopmental Loci. Cell Stem Cell 2017, 21, 305–318.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guella, I.; Sequeira, A.; Rollins, B.; Morgan, L.; Torri, F.; van Erp, T.G.M.; Myers, R.M.; Barchas, J.D.; Schatzberg, A.F.; Watson, S.J.; et al. Analysis of miR-137 expression and rs1625579 in dorsolateral prefrontal cortex. J. Psychiatr. Res. 2013, 47, 1215–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Wang, Z.-M.; Tan, W.; Wang, X.; Li, Y.; Bai, B.; Li, Y.; Zhang, S.; Yan, H.; Chen, Z.-L.; et al. Partial loss of psychiatric risk gene Mir137 in mice causes repetitive behavior and impairs sociability and learning via increased Pde10a. Nat. Neurosci. 2018, 21, 1689–1703. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, M.H.; Vallès, A.; Kirkels, L.A.M.H.; Mastebroek, M.; Olde Loohuis, N.; Kos, A.; Wissink-Lindhout, W.M.; de Brouwer, A.P.M.; Nillesen, W.M.; Pfundt, R.; et al. Chromosome 1p21.3 microdeletions comprising DPYD and MIR137 are associated with intellectual disability. J. Med. Genet. 2011, 48, 810–818. [Google Scholar] [CrossRef]
- Carter, M.; Nikkel, S.; Fernandez, B.; Marshall, C.; Noor, A.; Lionel, A.; Prasad, A.; Pinto, D.; Joseph-George, A.; Noakes, C.; et al. Hemizygous deletions on chromosome 1p21.3 involving the DPYD gene in individuals with autism spectrum disorder. Clin. Genet. 2011, 80, 435–443. [Google Scholar] [CrossRef]
- D’Angelo, C.S.; Moller dos Santos, M.F.; Alonso, L.G.; Koiffmann, C.P. Two New Cases of 1p21.3 Deletions and an Unbalanced Translocation t(8;12) among Individuals with Syndromic Obesity. Mol. Syndromol. 2015, 6, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of Genes and Cellular Pathways Dysregulated in Autism Spectrum Disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [Green Version]
- Tucci, A.; Ciaccio, C.; Scuvera, G.; Esposito, S.; Milani, D. MIR137 is the key gene mediator of the syndromic obesity phenotype of patients with 1p21.3 microdeletions. Mol. Cytogenet. 2016, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Smrt, R.D.; Szulwach, K.E.; Pfeiffer, R.L.; Li, X.; Guo, W.; Pathania, M.; Teng, Z.Q.; Luo, Y.; Peng, J.; Bordey, A.; et al. MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells 2010, 28, 1060–1070. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, Y.; Yokoyama, K.; Tasaki, S.; Kato, J.; Nakashima, K.; Takeyama, M.; Nakatani, A.; Suzuki, M. Transgenic mice overexpressing miR-137 in the brain show schizophrenia-associated behavioral deficits and transcriptome profiles. PLoS ONE 2019, 14, e0220389. [Google Scholar] [CrossRef] [Green Version]
- Thomas, K.T.; Anderson, B.R.; Shah, N.; Zimmer, S.E.; Hawkins, D.; Valdez, A.N.; Gu, Q.; Bassell, G.J. Inhibition of the Schizophrenia-Associated MicroRNA miR-137 Disrupts Nrg1α Neurodevelopmental Signal Transduction. Cell Rep. 2017, 20, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Vallès, A.; Martens, G.J.M.; De Weerd, P.; Poelmans, G.; Aschrafi, A. MicroRNA-137 regulates a glucocorticoid receptor-dependent signalling network: Implications for the etiology of schizophrenia. J. Psychiatry Neurosci. 2014, 39, 312–320. [Google Scholar] [CrossRef] [Green Version]
- He, E.; Lozano, M.A.G.; Stringer, S.; Watanabe, K.; Sakamoto, K.; den Oudsten, F.; Koopmans, F.; Giamberardino, S.N.; Hammerschlag, A.; Cornelisse, L.N.; et al. MIR137 schizophrenia-associated locus controls synaptic function by regulating synaptogenesis, synapse maturation and synaptic transmission. Hum. Mol. Genet. 2018, 27, 1879–1891. [Google Scholar] [CrossRef]
- Olde Loohuis, N.F.M.; Ba, W.; Stoerchel, P.H.; Kos, A.; Jager, A.; Schratt, G.; Martens, G.J.M.; van Bokhoven, H.; Nadif Kasri, N.; Aschrafi, A. MicroRNA-137 Controls AMPA-Receptor-Mediated Transmission and mGluR-Dependent LTD. Cell Rep. 2015, 11, 1876–1884. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Li, H.; Guo, R.; Ma, T.; Hou, R.; Ma, X.; Du, Y. miR-137, a new target for post-stroke depression? Neural Regen. Res. 2013, 8, 2441–2448. [Google Scholar] [CrossRef]
- Luby, E.D.; Cohen, B.D.; Rosenbaum, G.; Gottlieb, J.S.; Kelley, R. Study of a new schizophrenomimetic drug; sernyl. AMA. Arch. Neurol. Psychiatry 1959, 81, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Itil, T.; Keskiner, A.; Kiremitci, N.; Holden, J.M. Effect of phencyclidine in chronic schizophrenics. Can. Psychiatr. Assoc. J. 1967, 12, 209–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Dai, J.; Xu, L.J.; Han, G.D.; Sun, H.L.; Zhu, G.T.; Jiang, H.T.; Yu, G.Y.; Tang, X.M. MiR-137 attenuates spinal cord injury by modulating NEUROD4 through reducing inflammation and oxidative stress. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1884–1890. [Google Scholar] [CrossRef]
- Tang, Y.; Li, Y.; Yu, G.; Ling, Z.; Zhong, K.; Zilundu, P.L.M.; Li, W.; Fu, R.; Zhou, L.H. MicroRNA-137-3p Protects PC12 Cells Against Oxidative Stress by Downregulation of Calpain-2 and nNOS. Cell. Mol. Neurobiol. 2021, 41, 1373–1387. [Google Scholar] [CrossRef]
- Niwa, M.; Jaaro-Peled, H.; Tankou, S.; Seshadri, S.; Hikida, T.; Matsumoto, Y.; Cascella, N.G.; Kano, S.; Ozaki, N.; Nabeshima, T.; et al. Adolescent stress-induced epigenetic control of dopaminergic neurons via glucocorticoids. Science 2013, 339, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Maynard, T.M.; Sikich, L.; Lieberman, J.A.; LaMantia, A.S. Neural development, cell-cell signaling, and the “two-hit” hypothesis of schizophrenia. Schizophr. Bull. 2001, 27, 457–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoops, D.; Flores, C. Making Dopamine Connections in Adolescence. Trends Neurosci. 2017, 40, 709–719. [Google Scholar] [CrossRef]
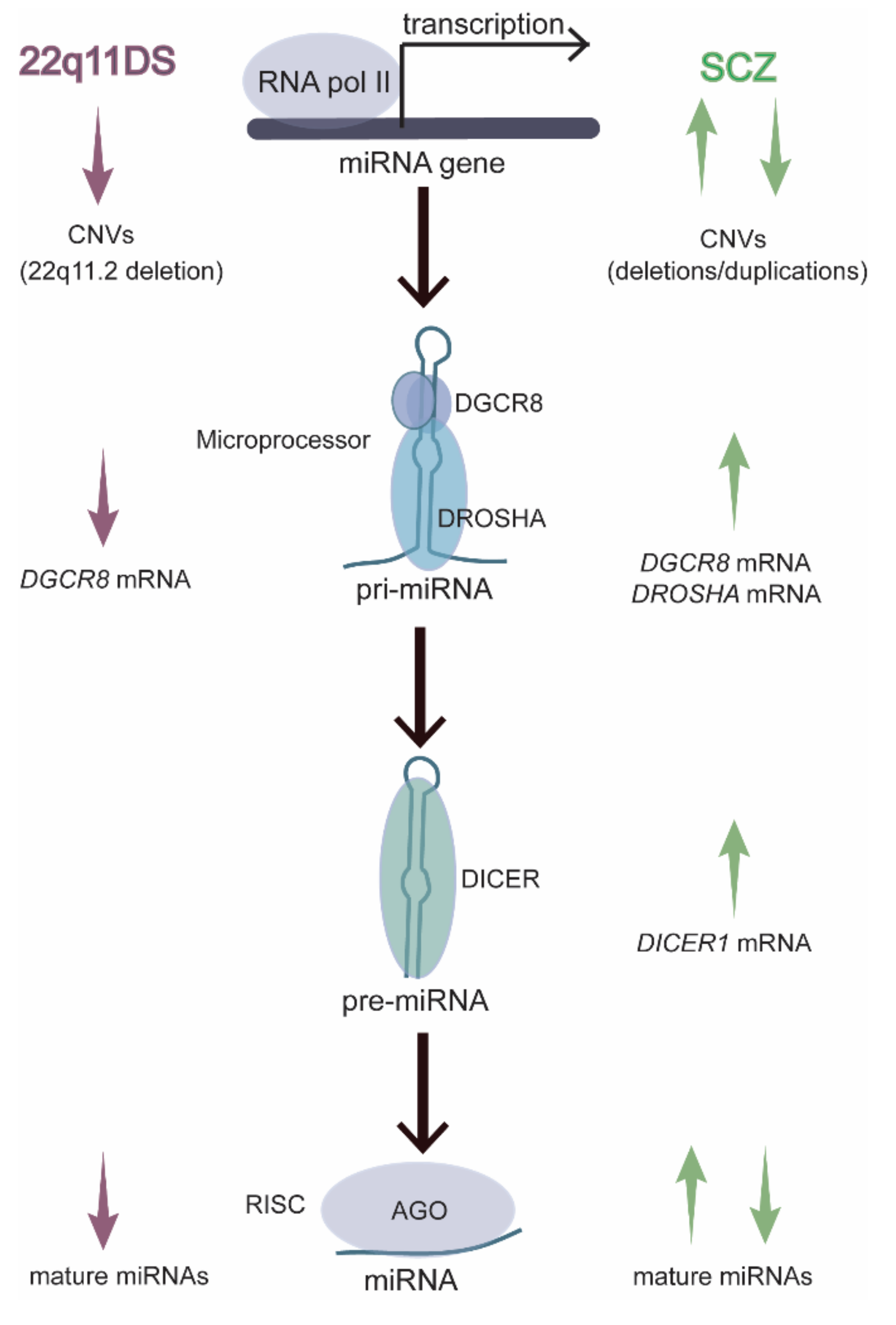
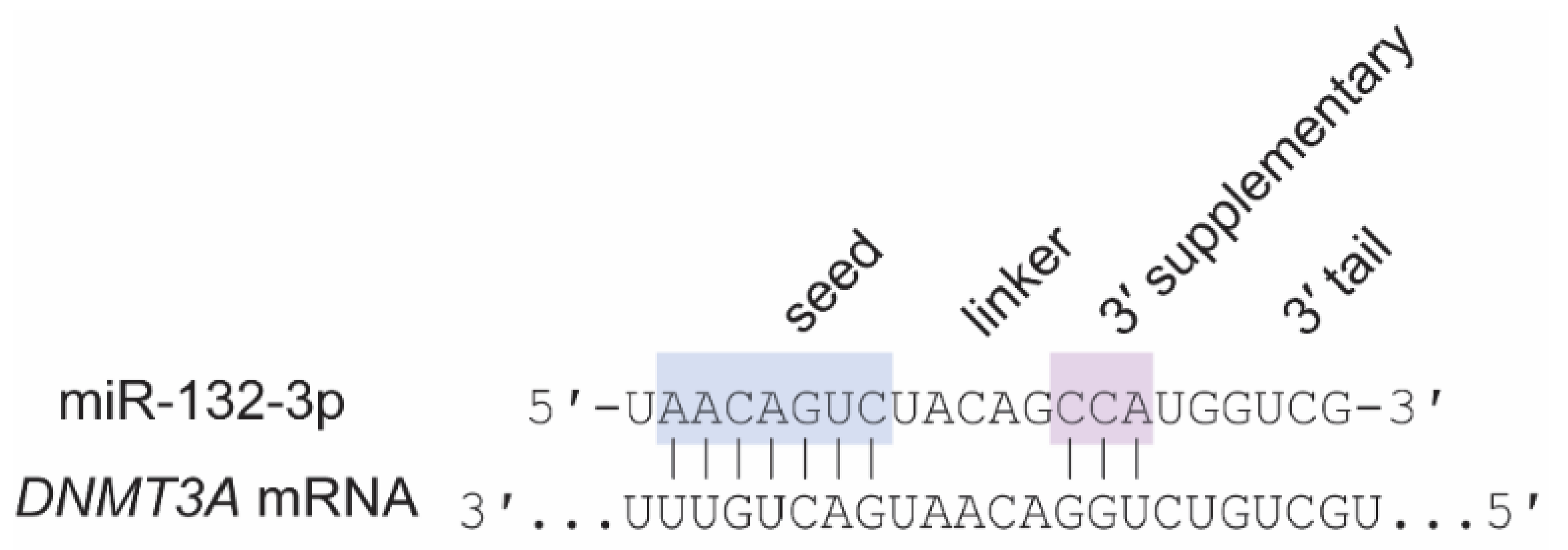
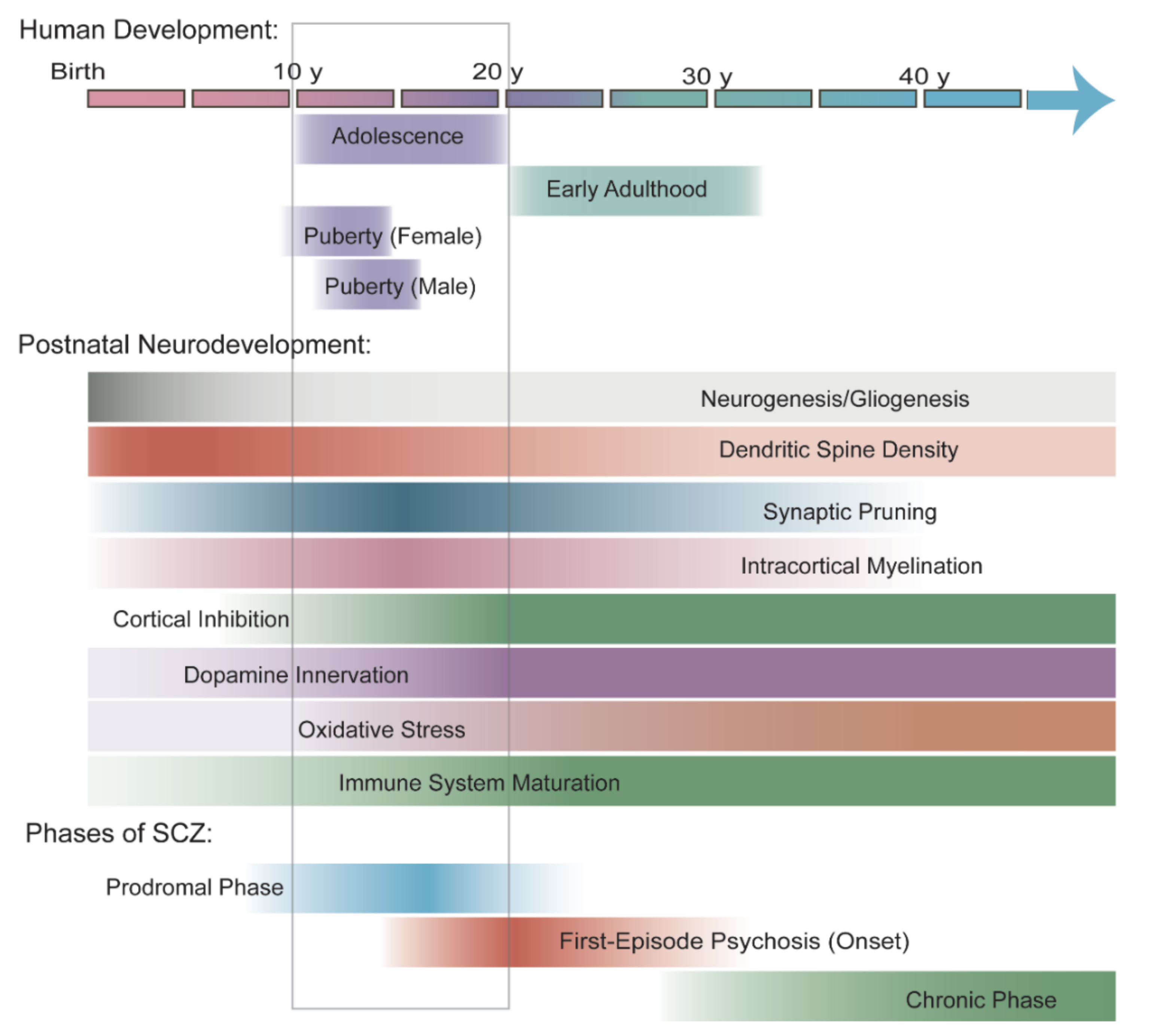
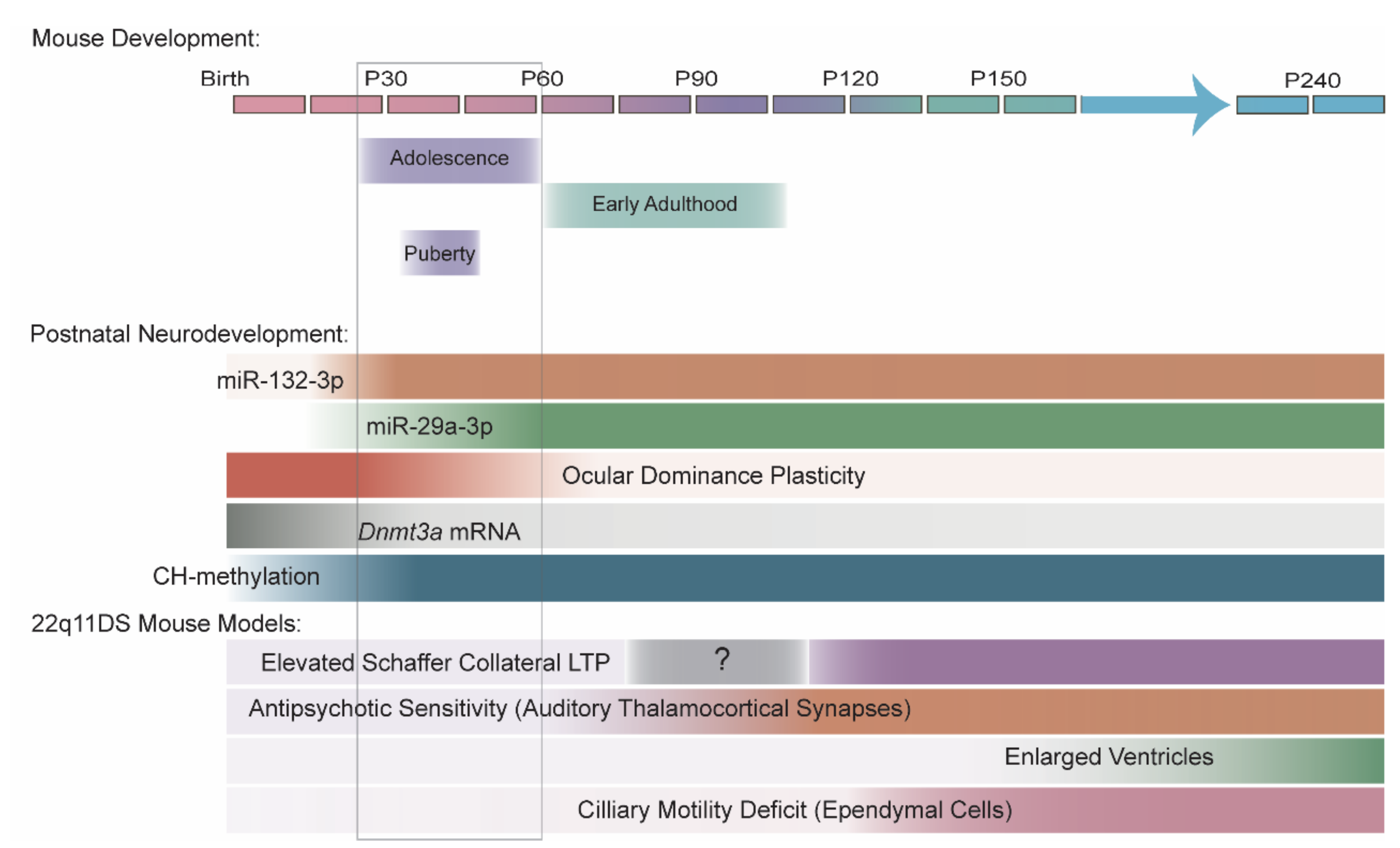

{kind=link}
{kind=link}
{kind=link}
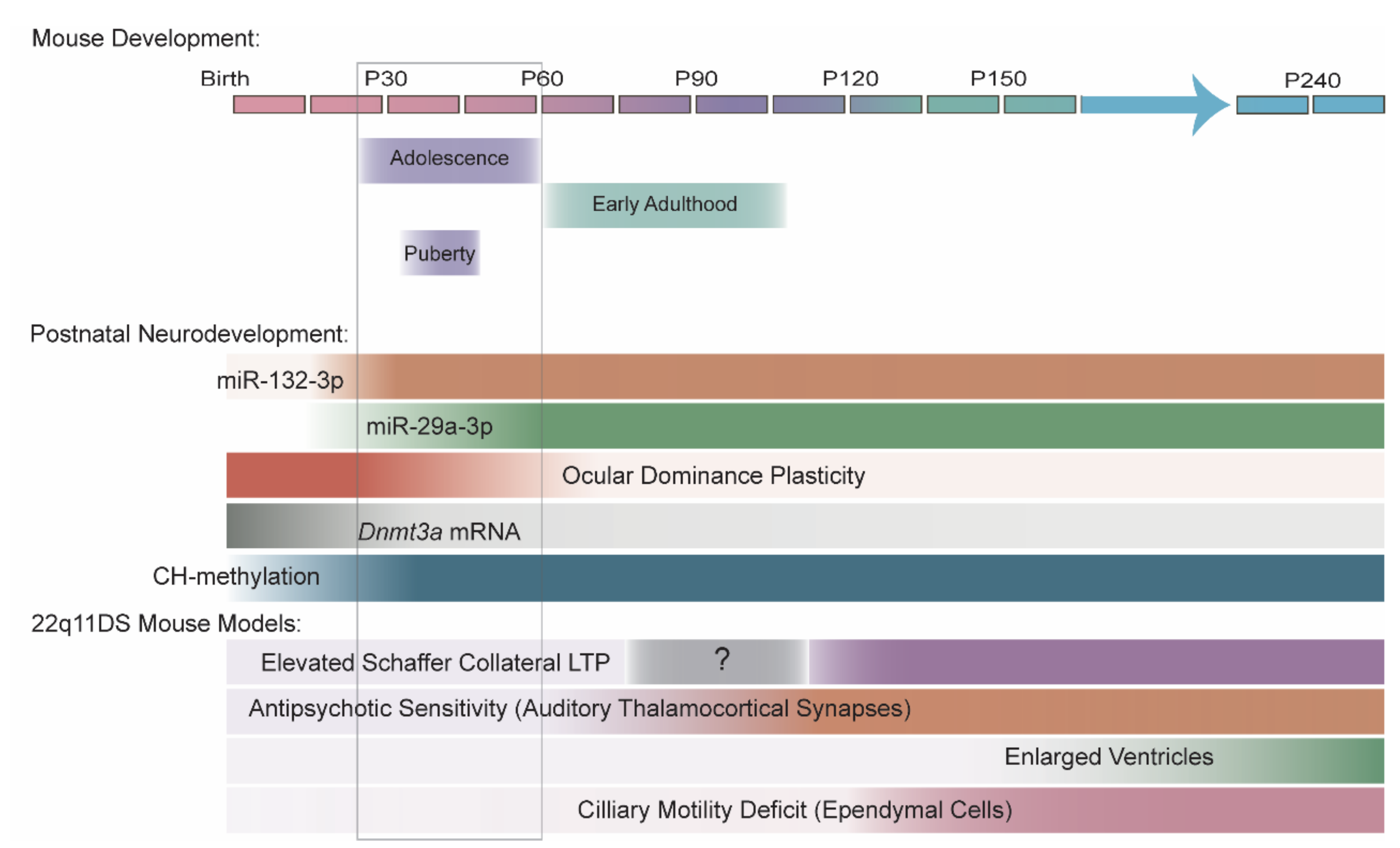{kind=link}
| Species | Reference | Region | Method | Age Range | Adolescent Time Points? | Global Patterns in miRNA Levels |
|---|---|---|---|---|---|---|
| Human | Somel et al., 2010 [45] | Superior frontal gyrus of PFC | Small RNA-seq | 0–98 y | Yes (one 13 y) | Highly dynamic with age, with inflection points at ~4 y and ~20 y |
| Somel et al., 2011 [46] | PFC and cerebellum | Microarray | 0–98 y | No | None noted | |
| Moreau et al., 2013 [47] | Cerebrum | Microarray | GW14–adult a | No | Increase with age | |
| Beveridge et al., 2014 [48] | DLPFC (BA 46) | Microarray | 2 mo–78 y | Yes | Decrease with age, with inflection point at late adolescence | |
| Ziats and Rennert 2014 [44] | OPFC, DLPFC, MPFC, VLPFC, hippocampus and cerebellum | Small RNA-seq | 4 mo–19 y | Yes | Most dynamic during transition from infancy to early childhood | |
| Hu et al., 2019 [11] | DLPFC (BA 46) | Small RNA-seq | Second trimester–74 y | Yes | Most dynamic/peak expression before puberty | |
| Pig | Podolska et al., 2011 [49] | Cortex and cerebellum | Microarray | F50–3 mo | Yes (3 mo) | None noted |
| Rat | Krichevsky et al., 2003 [50] | Forebrain | Array b | E12–adult | No | Highly dynamic with age |
| Yao et al., 2012 [51] | Dorsolateral cortex | Small RNA-seq | E17–P28 | Yes (P28) | Highly dynamic across most time points | |
| Mouse | Miska et al., 2004 [52] | Whole brain | Microarray c | E12.5–4 mo | No | Increase with age |
| Eda et al., 2011 [53] | Cerebrum, cerebellum, hippocampus | Microarray | E16.5–19 mo | Yes (1 mo) | Most dynamic in early postnatal brain (P6–1 mo) | |
| Fertuzinhos et al., 2014 [54] | Primary somatosensory cortex | Small RNA-seq | P4–P180 | No | None noted | |
| Swahari et al., 2021 [55] | Cerebellum | Small RNA-seq | P18 and P250 | No | None noted |
| Study Information | miR-29a/b/c-3p | miR-132-3p | miR-137-3p | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | Reference | Method | p-Value | Age Range | Region | Direction | Differential Age Range | Direction | Differential Age Range | Direction | Differential Age Range |
| Human | Somel et al., 2010 [45] | Small RNA-seq | Yes | 2 d–98 y | Superior frontal gyrus (BA9) | Increased (a) NR (b/c) | 2 d–98 y (a) | NR | -- | NR | -- |
| Moreau et al., 2013 [47] | Microarray | No | fetal (GW14–GW24), early postnatal (5 d–4 y), adult * | Cerebrum | Increased (a) Increased (b) NR (c) | Postnatal-adult (a) postnatal (5 d–4 y) (b) | Increased | Fetal-postnatal | Increased | Fetal (GW14–GW20) | |
| Beveridge et al., 2014 [48] | Microarray | Yes | 2 mo–78 y | DLPFC (BA46) | Decreased (a/c) NR (b) | 2 mo–78 y | Decreased | 2 mo–78 y | Decreased | 2 mo–78 y | |
| Ziats and Rennert 2014 [44] | Small RNA-seq | Yes | 4 mo–19 y | DLPFC (BA9, 46) | Increased (b) NS (a/c) | Infancy-early childhood (b) | NS | -- | NS | -- | |
| cerebellum (CBC) | NS | -- | NS | -- | Decreased | Infancy-early childhood | |||||
| OPFC (BA11), MPFC (BA32-34), VLPFC (BA44-45), hippocampus | NS | -- | NS | -- | NS | -- | |||||
| Macaque | Somel et al., 2010 [45] | Small RNA-seq | Yes | 16 d–28 y | Superior frontal gyrus (cortex) | Increased (a) NR (b/c) | 16 d–28 y (a) | NR | -- | NR | -- |
| Pig | Podolska et al., 2011 [49] | Microarray | No | F50, F100, 3 mo | Cortex | Increased (a/b/c) | F100–3 mo | NR | -- | NR | -- |
| Cerebellum | Increased (a/b/c) | F100–3 mo | NR | -- | NR | -- | |||||
| Study Information | miR-29a/b/c-3p | miR-132-3p | miR-137-3p | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | Reference | Method | p-Value | Age Range | Region | Direction | Differential Age Range | Direction | Differential Age Range | Direction | Differential Age Range |
| Rat | Yao et al., 2012 [51] | Small RNA-seq | No | E17–P28 | Whole cortex (E10 and E13) dorsolateral cortex (E17–P28) | Increased (a/b/c) | P3–P28 | Increased | P3–P28 | Peak at P0 | Increased (E10–P0) Decreased (P0–P28) |
| Sangiao-Alvarellos et al., 2013 [78] | RT-qPCR | Yes | P1–adult (>P75) | Hypothalamus | NE | — | Increased | Neonatal-juvenile | NE | — | |
| Mouse | Eda et al., 2011 [53] | Microarray | No | E16.5–19 mo | Cerebrum | Increased (a/b/c) | E16.5–3 mo | Increased | E16.5–1 mo | Increased | E16.5–P6 |
| P2–19 mo | Hippocampus | Increased (a/b/c) | P2–6 mo | Increased | P2–6 mo | ND | — | ||||
| E16.5–19 mo | Cerebellum | ND | — | Increased | E16.5–1 mo | Two peaks | Increased (E16.5–P2) Decreased (P2–6 mo), Increased (6 mo–19 mo) | ||||
| Tognini et al., 2011 [79] | RT-qPCR | Yes | P7–P35 | Visual cortex | NE | — | Increased | P7–P30 | NE | — | |
| Miller et al., 2012 [10] | RT-qPCR | Yes | E15–P60 | PFC | NE | — | Increased | P7–P28 | NE | — | |
| Fertuzinhos et al., 2014 [54] | Small RNA-seq | Yes | P4–P180 | S1 cortex | Increased (a) ND (b/c) | P4–P180 | Increased | P4–P180 | ND | — | |
| Li et al., 2014 [80] | Microarray | No | E12.5–E18.5, P60 | Cortex | Increased (a) ND (b/c) | E18.5–P60 | Increased | E12.5–P60 | NR | — | |
| RT-qPCR | Yes | E12.5–P60 | Cortex | Increased (a) NE (b/c) | P1–P60 (a) | NE | — | NE | — | ||
| E18.5–P60 | Hippocampus | Increased (a) NE (b/c) | P1–P60 (a) | NE | — | NE | — | ||||
| Mazziotti et al., 2017 [81] | Small RNA-seq | Yes | P10, P28 | V1 cortex | Increased (a/c), NS (b) | P10–P28 | Increased | P10–P28 | NS | — | |
| Li et al., 2019 [82] | RT-qPCR | Yes | P10–P120 | Hypothalamus | Increased (a/b/c) | P10–P120 | NE | — | NE | — | |
| Swahari et al., 2021 [55] | RT-qPCR | No | P0–P60 | Cortex | Increased (a/b/c) | P0–P40 (a/c), P0–P60 (b) | NE | — | NE | — | |
| Cerebellum | Increased (b) NE (a/c) | P0–P60 (b) | NE | — | NE | — | |||||
| Small RNA-seq | No | P18, P250 | Cerebellum | Increased (a/b/c) | P18–P250 | NR | — | Increased | P18–P250 | ||
| Napoli et al., 2020 [83] | RT-qPCR | Yes | P10–P200 | Visual cortex | Increased (a) NE (b/c) | P10–P60 | NE | — | NE | — | |
| MiRNA | Sequence (Human/hsa-) | Sequence (Mouse/mmu-) | Match a | Targets of Interest |
|---|---|---|---|---|
| miR-25-3p | CAUUGCACUUGUCUCGGUCUGA | CAUUGCACUUGUCUCGGUCUGA | Yes | ATP2A2 (Serca2)b |
| miR-29a-3p | UAGCACCAUCUGAAAUCGGUUA | UAGCACCAUCUGAAAUCGGUUA | Yes | DNMT3A |
| miR-29b-3p | UAGCACCAUUUGAAAUCAGUGUU | UAGCACCAUUUGAAAUCAGUGUU | Yes | DNMT3A |
| miR-29c-3p | UAGCACCAUUUGAAAUCGGUUA | UAGCACCAUUUGAAAUCGGUUA | Yes | DNMT3A |
| miR-132-3p | UAACAGUCUACAGCCAUGGUCG | UAACAGUCUACAGCCAUGGUCG | Yes | DNMT3A |
| miR-137-3pb | UUAUUGCUUAAGAAUACGCGUAG | UUAUUGCUUAAGAAUACGCGUAG | Yes | GRIA1b, GRIN2Ab |
| miR-185-5p | UGGAGAGAAAGGCAGUUCCUGA | UGGAGAGAAAGGCAGUUCCUGA | Yes | ATP2A2 (Serca2)b |
| miR-338-3p | UCCAGCAUCAGUGAUUUUGUUG | UCCAGCAUCAGUGAUUUUGUUG | Yes | DRD2b |
| miR-382-3p | AAUCAUUCACGGACAACACUU | UCAUUCACGGACAACACUUUUU | No | DRD1 |
| miR-674-3p | no human ortholog | CACAGCUCCCAUCUCAGAACAA | No | DRD1 |
| Brain Region | Reference | Method | miR-29a/b/c-3p | miR-132-3p | miR-137-3p |
|---|---|---|---|---|---|
| DLPFC (BA9) | Perkins et al., 2007 [6] | Microarray, RT-qPCR (miR-29b) | Decreased (a/b/c) | NS | NS |
| Beveridge et al., 2010 [7] | Microarray | Increased (c) NS (a/b) | NS | NR | |
| Moreau et al., 2011 [8] | RT-qPCR | NS | NS | NE | |
| DLPFC (BA46) | Kim et al., 2010 [9] | RT-qPCR | NS | Increased | NS |
| Miller et al., 2012 [10] | Microarray RT-qPCR (miR-132-3p) | NS | Decreased | NS | |
| Hu et al., 2019 [11] | Small RNA-seq | NS | NS | NS | |
| STG (BA22) | Beveridge et al., 2008 [12] | Microarray | NS | NS | NE |
| Beveridge et al., 2010 [7] | Microarray | NS | NS | NR | |
| Amygdala | Liu et al., 2018 [13] | Small RNA-seq | NS | Decreased | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, K.T.; Zakharenko, S.S. MicroRNAs in the Onset of Schizophrenia. Cells 2021, 10, 2679. https://doi.org/10.3390/cells10102679
Thomas KT, Zakharenko SS. MicroRNAs in the Onset of Schizophrenia. Cells. 2021; 10(10):2679. https://doi.org/10.3390/cells10102679
Chicago/Turabian StyleThomas, Kristen T., and Stanislav S. Zakharenko. 2021. "MicroRNAs in the Onset of Schizophrenia" Cells 10, no. 10: 2679. https://doi.org/10.3390/cells10102679
APA StyleThomas, K. T., & Zakharenko, S. S. (2021). MicroRNAs in the Onset of Schizophrenia. Cells, 10(10), 2679. https://doi.org/10.3390/cells10102679