
MicroRNAs in the Onset of Schizophrenia


Department of Developmental Neurobiology, St. Jude Children’s Research Hospital, Memphis, TN 38105, USA
*
Authors to whom correspondence should be addressed.
Cells 2021, 10(10), 2679; https://doi.org/10.3390/cells10102679
Submission received: 30 August 2021
/
Revised: 30 September 2021
/
Accepted: 2 October 2021
/
Published: 6 October 2021
(This article belongs to the Special Issue MicroRNA-Mediated Silencing Pathways in the Nervous System and Neurological Diseases)
Abstract
:Mounting evidence implicates microRNAs (miRNAs) in the pathology of schizophrenia. These small noncoding RNAs bind to mRNAs containing complementary sequences and promote their degradation and/or inhibit protein synthesis. A single miRNA may have hundreds of targets, and miRNA targets are overrepresented among schizophrenia-risk genes. Although schizophrenia is a neurodevelopmental disorder, symptoms usually do not appear until adolescence, and most patients do not receive a schizophrenia diagnosis until late adolescence or early adulthood. However, few studies have examined miRNAs during this critical period. First, we examine evidence that the miRNA pathway is dynamic throughout adolescence and adulthood and that miRNAs regulate processes critical to late neurodevelopment that are aberrant in patients with schizophrenia. Next, we examine evidence implicating miRNAs in the conversion to psychosis, including a schizophrenia-associated single nucleotide polymorphism in MIR137HG that is among the strongest known predictors of age of onset in patients with schizophrenia. Finally, we examine how hemizygosity for DGCR8, which encodes an obligate component of the complex that synthesizes miRNA precursors, may contribute to the onset of psychosis in patients with 22q11.2 microdeletions and how animal models of this disorder can help us understand the many roles of miRNAs in the onset of schizophrenia.
1. Introduction
MicroRNAs (miRNAs) are strongly implicated in neuronal function. These small noncoding RNAs first bind to mRNA targets containing complementary sequences, and then, they inhibit protein synthesis from the mRNA target or promote target degradation. A single miRNA may regulate hundreds of mRNA targets, and many mRNAs are dynamically regulated by multiple miRNAs. The mammalian brain expresses most of the hundreds of miRNAs that have been identified to date. MiRNAs are intimately intertwined with neurodevelopmental signaling, synaptic plasticity, and adult neuronal activity. Disruptions in the miRNA biogenesis pathway or those in a single miRNA can produce neurologic deficits in human subjects and rodent models. Significant dysregulation of miRNAs and the miRNA biogenesis machinery has been reported in almost every neurodevelopmental, neurodegenerative, or psychiatric disorder in which they have been examined. However, few disorders have received as much attention from miRNA research as schizophrenia (SCZ).
SCZ is a severe psychiatric disorder with a lifetime prevalence of 0.7% in populations worldwide [1]. SCZ symptoms include hallucinations, delusions, disorganized thoughts and speech, diminished or inappropriate emotional expression, apathy, reduced social drive, and deficits in attention and working memory; however, the specific symptoms and their severity vary greatly across patients [2]. SCZ is highly heritable [3], and hundreds of gene variants have been linked to SCZ risk. The disease is also associated with a wide array of environmental factors (e.g., prenatal viral infection, maternal and early life stress, adverse child rearing, and adolescent drug abuse) (reviewed in [4]). SCZ is incurable, chronic, devastating to patients and their families, and one of the most costly mental disorders for national health care systems worldwide [5].
Multiple lines of evidence suggest that the miRNA pathway is central to SCZ. First, postmortem brain studies suggest multiple miRNAs are dysregulated in patients with SCZ [6,7,8,9,10,11,12,13,14]. Second, when compared to other protein-coding genes, SCZ-risk genes contain significantly more predicted miRNA-binding sites [15]. Third, several single nucleotide polymorphisms (SNPs) and variable number tandem repeats near or within MIR137HG, the gene that encodes the miRNA miR-137, are highly associated with SCZ risk [16,17,18,19,20,21]. Fourth, brain miRNA levels are sensitive to environmental factors associated with increased SCZ risk. For example, maternal immune activation and adolescent cannabis exposure alter the levels of several miRNAs from the MIR379/410 gene cluster in the entorhinal cortex of adult rats [22], suggesting that early life events can have long-term effects on brain miRNAs. Finally, the genomes of patients with SCZ are more likely to contain rare copy number variants (CNVs) that overlap with miRNA-encoding genes [23].
Additional lines of evidence suggest that the disruption of protein components in the miRNA biogenesis pathway contributes to SCZ risk. The miRNA is synthesized in a multistep process (Figure 1), beginning with the transcription of a miRNA gene to form a primary miRNA transcript (or pri-miRNA). The pri-miRNA contains one or more hairpin structures that are recognized by the RNA-binding protein DGCR8 and cleaved by the ribonuclease DROSHA to form miRNA precursors (pre-miRNAs) that are exported from the nucleus [24,25,26,27,28,29,30]. Within the cytoplasm, DICER cleaves the pre-miRNA loop and selectively loads one strand into an Argonaute (AGO1-4) protein-containing complex, which mediates the miRNA’s effects on mRNA degradation and translation inhibition [31,32,33,34,35,36].
Dysregulation of miRNA synthesis may increase SCZ risk. Both increases and decreases in the levels of specific miRNAs have been reported in postmortem SCZ brain samples. Hemizygous microdeletions in the 22q11.2 region, CNVs which include the DGCR8 gene, lead to an ~20- to 25-fold increase in SCZ risk [37,38], and studies in animal models of 22q11.2 deletion syndrome (22q11DS) report widespread deficits in brain miRNA levels [39,40]. By contrast, several reports suggest that DGCR8, DROSHA, and/or DICER1 mRNA levels are increased in patients with SCZ but lacking 22q11.2 mutations [7,14,41]. MiRNA levels are tightly controlled in the mammalian brain. Studies in animal models suggest that overexpression or inhibition of miRNAs disrupts neuronal function, so it is quite possible that elevated or reduced activity in the miRNA pathway increases the risk of SCZ.
The roles of miRNAs in SCZ onset are poorly understood. MiRNAs are critical for fetal, infant, and childhood brain development. Other environmental and genetic risk factors associated with SCZ also appear to disrupt early development. However, SCZ onset before puberty is relatively rare, with an estimated prevalence of 1 in 40,000 children younger than 13 years [42]. The rates of psychiatric symptoms increase during puberty and post-pubertal adolescence, including the prodromal symptoms that precede psychosis and the eventual onset of SCZ in some subjects [2]. First-episode psychosis and a SCZ diagnosis typically occur between the ages of 15 (adolescence) and 25 (early adulthood), though some subjects, particularly females, experience onset after age 30 [43]. These studies strongly implicate disruptions in adolescent neurodevelopment in SCZ etiology. However, despite the substantial evidence supporting a role for miRNAs in SCZ etiology, the functions of miRNAs in the adolescent brain remain poorly understood.
In this review, we will examine evidence of miRNAs functioning in the typical maturation of adolescent and early adult brains and in the disrupted developmental trajectories that may underlie SCZ onset. Few studies have directly examined miRNA functions during typical adolescence, and fewer have examined their dysregulation in SCZ model systems. To determine the roles of miRNAs in SCZ onset, we will examine each component of this argument individually. First, the brain undergoes widespread changes during adolescence, and miRNA levels and activity also appear to be dynamic during that time. Next, although we cannot examine miRNA dynamics during SCZ onset in the brains of living patients, evidence from peripheral tissues suggests that miRNA dysregulation occurs during the prodromal phase of disease, as well as after disease onset. Then, we will present a case study of two miRNAs that are highly dynamic during adolescent brain development to explore how miRNA dysregulation during adolescence can interfere with typical development and contribute to disease onset. Finally, we will examine miRNAs and the miRNA pathway components that are strongly associated with SCZ risk but only poorly understood during adolescence and explore how they might contribute to disease onset.
2. Temporal Dynamics in the Levels of MiRNAs across the Lifespan
One finding is almost universal among studies examining how miRNA levels vary with age—miRNA levels are highly dynamic throughout the lifespan. A substantial portion of miRNAs are differentially expressed between two or more periods of development or aging (Table 1), and miRNA-profiles become more divergent across brain regions with age [44]. In this section, we will examine how age influences miRNA levels in the mammalian brain, with an emphasis on studies that contain either one or more adolescent time point or time points that span adolescence, thereby allowing changes in miRNA levels during adolescence or early adulthood to be inferred.
Because each miRNA can target hundreds of mRNAs, widespread changes in miRNA levels are predicted to have dramatic effects on cellular function. Several studies have reported global changes in brain miRNA levels associated with age, yet their results have been conflicting. The miRNA microarray studies conducted by Miska et al. [52] and Moreau et al. [47] found that miRNA levels increase with age in mouse and human brain, respectively. Beveridge et al. [48] used Illumina miRNA microarrays to examine miRNA levels in the human dorsolateral prefrontal cortex (DLPFC, Brodman’s area 46) of human subjects, ranging from 2 months to 78 years of age. They found that overall miRNA levels decline with age [48]. Intriguingly, the inflection point at which global miRNA levels switched from an upward to a downward trajectory occurred during late adolescence, at ~17 years of age. However, several of the miRNAs reported by Beveridge et al. to decrease with age (e.g., miR-29a-3p, miR-29c-3p, and miR-132-3p) were reported to increase with age in other studies (Table 2). Differences in study design and methods may explain the discrepancies in these findings. For example, Miska et al. used the first miRNA microarray, which detected far fewer unique miRNAs than later iterations of the method and did not adequately distinguish different miRNAs from the same family [52]. Furthermore, the samples analyzed by Moreau et al. were mostly derived from embryonic or early postnatal timepoints; the only adult-derived samples were commercially available samples, one from an elderly subject (81 years) and a second representing a pooled sample from 23 subjects with an average age of 68 years [47]. Moreau et al., therefore, had poor temporal resolution in childhood through elderly timepoints, and the adult timepoints may have been biased by differences in sample preparation, relative to the younger samples.
More recently, a larger study of 109 subjects used small RNA-seq to examine miRNA levels across the lifespan (second trimester to 78 years of age) in the human DLPFC (BA 46) [11]. Like Beveridge et al., Hu et al. [11] found that most miRNAs peaked in expression before adolescence. A smaller study conducted by Somel et al. [45] similarly found that most major transitions in miRNA expression occur during childhood (~3–4 years) or early adulthood (~20–25 years) [45]. However, that study included only one adolescent (~14 years) and one subject in early adulthood (25 years), so temporal resolution was poor in the indicated age ranges. Consistent with larger studies, Somel et al. found that a few miRNAs exhibit expression patterns that are specific to adult aging, and expression changes associated with aging tend to be extensions or reversals of patterns observed during brain development.
Overall, studies examining miRNA levels across the lifespan have been few in number, have come to sometimes conflicting conclusions, and have lacked consistency in their methods. Despite adolescence being critical in the onset of psychiatric disorders, those time points have been underrepresented in studies examining miRNAs in the brain. However, the following picture has emerged.
During brain development, from the prenatal stage to preadolescence, miRNAs are expressed in waves, with different combinations of miRNAs peaking at different stages. During adolescence and/or early adulthood, miRNA levels become less dynamic, as they transition from their developmental patterns. Most miRNAs are then placed on a trajectory that they will follow for decades during adult aging. This transition period might be particularly vulnerable because deviations from normal patterns might accumulate over time. However, further exploration is needed to understand miRNA dynamics in the human brain during this period, and further research in animal models is needed to understand the long-term impact of disruptions to miRNA levels during this time and whether those disruptions contribute to psychiatric disease.
3. Temporal Dynamics in the Activity of MiRNAs across the Lifespan
The activity of a miRNA can change with age in the absence of any change in its levels. Recent evidence demonstrates that miRNA activity also depends on sequence variation, the availability of mRNA target sites within a given cellular context, and the overall state of the protein-synthesis pathway within which the miRNA pathway functions.
MiRNA sequences in vivo often differ from the canonical form listed in miRBase [56,57,58,59,60,61] or other databases. MiRNA microarrays and RT-qPCR assays are typically designed to target the canonical form; thus, they often cannot reliably distinguish between miRNA sequence variants, or “isomiRs”, and they may fail to detect some functional variants entirely ([62], reviewed in [63]). Sequencing of miRNAs can detect isomiRs, but traditional analysis pipelines consider only the sum of a miRNA’s isomiRs rather than how individual isomiRs vary with age or other biological factors.
The loss or addition of nucleotides at the 5′ end of a miRNA shifts the seed sequence and can have a dramatic impact on miRNA targeting [64,65] (Figure 2). RNA editing in the seed sequence can also alter targeting [66]. Recent research suggests that sequence complementarity closer to the 3′ end of a miRNA (i.e., the 3′ supplementary region) can modulate targeting efficiency [67,68], suggesting that editing in this region also affects targeting, though to a lesser degree. However, most isomiR variation occurs at the terminal 3′ end (i.e., the 3′ tail), leaving the seed and 3′ supplementary regions largely unaffected [69]. In vitro studies suggest that nucleotide loss or addition at the 3′ tail can influence Argonaute protein 2 (AGO2) loading and dissociation [70] and may influence miRNA stability [71,72,73] (but also see [74]). Together, these studies suggest that age-associated variation in the sequences of individual miRNAs significantly influence miRNA activity independent of age-associated variation in miRNA levels.
To date, only one study has examined the effects of age on isomiR variation in the mammalian brain. Juvvuna et al. [75] found that overall miRNA length declines in mouse brain between embryonic day (E) 12.5 and postnatal day (P) 60 (early adulthood) due primarily to increased 3′ trimming of mature miRNAs [75]. Increased 3′ trimming was attributed to an increase in the ratio of AGO2 to AGO1 in the adult brain, though the enzyme responsible for the trimming was not identified. Further research is needed to elucidate the mechanisms underlying increased 3′ trimming; to determine its functional consequences; and to clarify whether it occurs during early development, adolescence, and/or adult aging.
The cellular function of a miRNA is determined almost entirely by the mRNAs that it targets. The mRNA targets available to a miRNA depend on the mRNAs co-expressed with the miRNA within a cell, the subcellular localization of those mRNAs relative to the miRNA, and the accessibility of miRNA-binding sites along the mRNA, all of which may vary with age in ways that are poorly understood. Notably, in most studies, miR-137 levels in the brain are relatively stable with age (Table 2 and Table 3). However, AGO HITS-CLIP (high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation) studies reveal that miR-137 targets largely nonoverlapping mRNAs in the developing human brain (gestation week (GW) 15–16 or GW19–20) [76], relative to adulthood (44–68 years) [77]. This suggests that miR-137 has different functions at different developmental stages.
To date, no HITS-CLIP studies have reported AGO-binding sites within the adolescent or early adult brain in any mammalian brain tissues. Furthermore, most miRNA–mRNA interactions have been validated in cell lines using in vitro reporter systems (e.g., luciferase assays); such systems cannot replicate the effects of age or brain-specific cellular contexts on miRNA targeting. Therefore, to identify miRNA–mRNA target interactions that contribute to adolescent brain maturation and may contribute to age of onset of psychiatric disease, it is crucial to identify the specific miRNA–mRNA target interactions that are present at these timepoints, during both typical development and disease onset.
Finally, several lines of evidence suggest that overall miRNA activity within the brain varies with age. Functionally, miRNAs primarily inhibit protein synthesis, either by directly inhibiting translation or by promoting the degradation of targeted mRNAs. Therefore, age-associated changes in miRNA activity are predicted to cause age-associated changes in protein synthesis. Studies in a variety of model systems suggest that the rates of protein synthesis peak during early postnatal development, before the onset of adolescence, and then decline with age throughout adolescence and adult aging, though the rate of this decline slows with age [84,85,86,87,88] (but also see [89]). This peak in protein synthesis rates also roughly coincides with the inflection point in miRNA levels noted in early childhood/postnatal development by some studies (Table 1), thereby suggesting that tight regulation of mRNA translation is critical to early development. The gradual age-associated decline in protein synthesis in the brain that occurs during adolescence and early adulthood may also implicate changes in miRNA regulation during that time. Theoretically, overall miRNA activity might increase with age, thereby contributing to a decline in protein synthesis. Alternatively, miRNA activity might decline with age due to a decreased need for miRNA regulation. Dysregulated protein synthesis is implicated in SCZ and other adult-onset psychiatric disorders. Olfactory neurosphere–derived cells from patients with SCZ exhibit reduced overall protein synthesis in vitro, relative to cells derived from control subjects [90]. No deficits were observed in fibroblasts, suggesting that the deficit is limited to specific cell types. Similarly, dopaminergic neurons derived from induced pluripotent stem cells from patients with 22q11DS exhibit an ~50% reduction in global protein synthesis, relative to cells from control subjects [91]. Thus, age-associated changes in brain protein synthesis may interact with disease-associated deficits in protein synthesis in patients with SCZ to alter brain function; however, this hypothesis has not been tested in patients or animal models.
The relative importance of miRNAs in regulating gene expression (relative to other methods) may change with age. During preadolescent development, mRNA and protein levels tend to be more highly correlated [92,93], suggesting that transcription drives most changes in gene expression. However, in the human cortex, the correlation between mRNA and protein levels weakens during postadolescent adult aging [92,93], suggesting that posttranscriptional regulation becomes more important with age. At least some of this uncoupling has been attributed to RNA-binding protein–binding sites within the mRNAs [92], thereby implicating age-associated changes in mRNA translation, mRNA stability, and protein synthesis in this uncoupling.
Age-dependent miRNA activity is particularly evident in a group of discordant genes in which the mRNA and protein levels both increase during childhood and adolescence but then diverge during adulthood, with mRNAs continuing to increase but protein levels declining during postadolescent aging [92]. This gene set exhibits evidence of an enrichment in AGO2-binding sites and predicted target sites for miRNAs that are dynamic during aging [45,92,94]. Together, these data suggest that miRNA targeting exerts greater influence over protein levels with increased age, and the brain becomes more sensitive to miRNA dysregulation during the transition from adolescence to adulthood than during earlier stages of development.
In summary, these studies indicate that the miRNA pathway undergoes strong temporal regulation throughout the lifespan, including during adolescence. The levels of at least a subset of miRNAs appear to be highly dynamic during adolescence, and miRNA activity may change independent of miRNA levels. Some of these changes may directly contribute to adolescent brain development, and their dysregulation may cause the onset of psychiatric disease including SCZ. However, further research is needed to fully elucidate the miRNA pathway during this critical period of brain development, during its typical trajectory and during disease onset.
4. Neurobiology during the Age of SCZ Onset
Adolescence marks the end of childhood, the onset of sexual maturity, and a transition into adulthood. Rapid changes occur in body growth, secondary sexual characteristics, stress response, cognition, and social development through the coordinated action of multiple signaling axes in the adolescent brain. Cognitively, adolescence is marked by increased emotionality and better capacity for executive function and abstract thinking, but also increased vulnerability to stress. Behaviorally, adolescence is marked by not only increased risk taking, exploration, sociability, and independence but also increased susceptibility to drugs of abuse and addiction. Widespread changes in the neural circuity underlie these cognitive and behavioral changes, and disruption of adolescent brain maturation may contribute to the onset of many psychiatric disorders. In SCZ, cognitive, negative, and subthreshold psychotic symptoms often first appear during adolescence (in the prodromal phase) and predict the onset of first-episode psychosis during late adolescence or early adulthood (Figure 3).
MiRNAs are implicated in the control of puberty (i.e., the physical changes during adolescence that lead to sexual and reproductive maturity). Gonadotropin-releasing hormone (GnRH) is essential for puberty onset and adult fertility. In mice, conditional knockout of the Dicer gene within hypothalamic neurons causes the progressive loss of GnRH at the onset of adolescence, an almost complete loss of GnRH in adulthood, and infertility [95]. Therefore, miRNAs are essential to the onset and successful completion of puberty. Puberty is also associated with its own neurodevelopmental changes independent of chronological age, both in humans (reviewed in [96]) and animal models (reviewed in [97]). Conversely, adolescent maturation of some neural circuits and behaviors occurs independent of puberty and in the absence of gonadal hormones [98,99,100]. For most adolescent neurodevelopmental processes, however, the relative contributions of puberty versus chronological age remain poorly understood.
Some neurodevelopmental processes typically associated with fetal, infant, or childhood development extend into adolescence and early adulthood. For example, the majority of neurogenesis occurs prior to birth, yet limited neurogenesis continues in the adolescent and adult striatum and hippocampus and may continue in olfactory bulb [101,102,103,104,105,106] (also reviewed in [107]). Similarly, although the majority of gliogenesis occurs prior to birth, new oligodendrocytes and microglia are generated throughout adulthood, and astrocytes continue to proliferate in response to environmental cues [108,109,110,111,112].
Dysregulation of dopamine (DA) signaling underlies psychotic symptoms and may contribute to other SCZ symptoms (reviewed in [113]). Antipsychotics that alleviate positive symptoms of SCZ primarily target D2 DA receptors (DRD2s) [114]. DA signaling within the PFC dramatically changes during adolescence and may directly contribute to SCZ onset. Prior to adolescence, the DRD2 agonist quinpirole has no effect on fast-spiking interneuron excitability. By P50 in rats, DRD2 agonists are strongly excitatory in this cell type [115] and have a net inhibitory effect on pyramidal neuron activity [116]. Overall, DA concentration and innervation peak in the PFC during adolescence in nonhuman primates [117,118]. In mice, DA axons originating in the ventral tegmental area extend from the nucleus accumbens to PFC during adolescence [119], i.e., the only known example of long-range axonal growth during adolescence. In the mouse PFC, miR-218 inhibits DCC [120], a receptor that is critical for correct DA axon targeting [119]. MiRNAs also limit D1 DA receptor, DRD2, and DA transporter levels [121,122,123]; some of this regulation is age-dependent, as we will discuss later in this review. Therefore, miRNAs are essential for shaping DA signaling during adolescence and may contribute to DA dysregulation during SCZ onset in patients through multiple mechanisms.
Markers of oxidative stress increase at the onset of adolescence and during adult aging, both in the brain and periphery [124,125,126]. In brain-derived mitochondria, the rate of aerobic respiration increases during early postnatal and adolescent development, reaching adult levels at ~P60 (end of adolescence/onset of adulthood) in rats [127]. Oxidative stress occurs when the reactive oxygen species formed as natural byproducts of aerobic respiration accumulate to deleterious levels and cause damage to DNA, lipids, and other cellular components susceptible to oxidation. Cells contain endogenous antioxidants (reducing agents), such as glutathione, to maintain redox balance, but reactive oxygen species that accumulate beyond a cell’s capacity to balance them can be particularly detrimental to cells with higher levels of aerobic respiration, such as oligodendrocytes [128,129,130] and fast-spiking parvalbumin-positive (PV+) interneurons (reviewed in [131,132]).
Oxidative stress damages oligodendrocytes and disrupts myelination in adolescent mice [130]. Although most long-range axonal projections complete myelination during infancy, many intracortical axons undergo myelination during adolescence or early adulthood [110,133,134,135,136]. MiRNAs are essential for oligodendrocyte maturation and myelination (reviewed in [137]), but research to date has largely focused on their functions in early development or in more severe disruptions observed in demyelinating diseases, e.g., multiple sclerosis.
Like the DA system, GABA signaling within the frontal lobe matures during adolescence in primates [138,139,140,141,142] (reviewed in [143]). Decreases in GABAergic interneuron markers are frequently observed in postmortem SCZ brain and may underlie some cognitive deficits in patients (reviewed in [144]). However, GABA deficiencies are less consistently observed in neuroimaging studies in patients with SCZ [145,146,147]. MiR-101 promotes the maturation of GABA signaling during early development [148] and may be elevated in the DLPFC of patients with SCZ [7]. Later in this review, we will examine a potential role for miR-29a-3p in regulating the maturation of perineuronal nets (PNNs) surrounding cortical PV+ interneurons during adolescence [83], which limits some forms of age-dependent plasticity in adulthood. Thus, miRNA dysregulation might disrupt GABA signaling in SCZ through several mechanisms.
Overall synaptic density peaks during early childhood, but synaptogenesis continues in the PFC until mid-adolescence [149]. Synaptic pruning begins in childhood, causing a net decline in synapse numbers that extends through the third decade of life in humans [150,151]. Studies in animal models suggest the rate of pruning may peak in adolescence [152,153,154,155,156]. According to the “Feinberg Hypothesis” [157], excessive synaptic pruning during adolescence may underlie gray matter deficits and reduced dendritic spine densities in patients with SCZ. Although the roles of miRNAs in synapse formation and morphology have been heavily studied, those in synaptic pruning have not been directly examined.
Immune signals influence most aspects of neuronal function. Both the innate and adaptive immune systems mature during adolescence [158,159,160,161] (reviewed in [162,163]). Variation in the major histocompatibility complex immune loci is strongly associated with SCZ [16,18,164,165] and mediated, in part, by the Complement Component 4 genes C4A and C4B [166]. Both neurons and microglia express components of the complement system [167], which is implicated in microglia-mediated synaptic pruning during adolescence and local inflammatory signaling within the brain [152,168,169,170]. Elevated inflammation has been observed in patients at all stages of SCZ (reviewed in [171]). During adolescence, excess inflammation may cause excess synaptic pruning that disrupts neuronal circuits that are critical for adult cognition and behaviors [172].
Extracellular miRNAs can stimulate inflammation by directly binding to toll-like receptors (TLRs) [173], rather than by binding to mRNA. The significance of this function in adolescence or earlier development remains unknown; however, TLRs and related adapter proteins are highly dynamic during postnatal neurodevelopment (reviewed in [174]) and directly contribute to neurogenesis and other aspects of brain maturation [175,176,177]. Whether miRNA binding to TLRs directly contributes to immune and/or neurodevelopmental dysfunction in SCZ remains unknown. However, extensive cross-communication occurs between TLRs and the complement system–signaling pathways [178], suggesting a novel mechanism by which miRNAs might influence synaptic pruning during adolescence.
In summary, the human brain undergoes widespread changes during adolescence. Some developmental processes (e.g., myelination, synaptic pruning, DA axon outgrowth) converge with other processes that mark a transition into adulthood and adult aging (e.g., increased oxidative stress, immune maturation, and increased inflammation). This convergence coincides with the onset of symptoms in SCZ (Figure 3) and underlies many proposed mechanisms of SCZ pathogenesis. Few studies have examined the roles of miRNAs in these processes during adolescent development or directly implicated miRNAs in their disruption at SCZ onset. However, miRNAs remain particularly attractive candidates for SCZ research because a single miRNA can have a wide variety of functions in adolescent brain development, suggesting that miRNAs influence many aspects of SCZ etiology.
5. Peripheral MiRNAs during Conversion to Psychosis
Ideally, miRNA levels could be tracked within brain regions of interest before, during, and after SCZ onset. Using family history and early onset symptoms, we can identify individuals at high risk of psychosis before SCZ onset; however, only 12–35% of individuals at high risk convert to psychosis within 2–2.5 years [179,180,181,182,183] (reviewed in [184]). Although this conversion rate is dramatically higher than that in the general population (~500-fold increase by some estimates [185]), current criteria cannot predict psychosis or SCZ onset with certainty. Consequently, we cannot use postmortem brain samples to identify miRNA changes in the brain that immediately precede symptom onset and are most likely to directly contribute to that onset. In contrast, blood samples can be collected from living subjects at high-risk of SCZ at all stages of disease. Here, we describe recent studies examining miRNAs in peripheral tissues in persons at high risk of psychosis (and SCZ).
Phase II of the North American Prodrome Longitudinal Study (NAPLS-2) assessed symptoms prospectively in participants at clinical high risk (CHR) and in controls over a 5-year period [186]. Approximately 25% of individuals at CHR progressed to psychosis within 2 years [187]. A subset of patients consented to blood collections, from which leukocytes were harvested for miRNA sequencing [188]. Subjects were divided into three groups based on baseline symptoms and diagnoses over the 2-year period: (1) control subjects, (2) subjects at CHR who did not progress to psychosis, and (3) subjects at CHR who progressed to psychosis. Although no single miRNA was identified as significantly different across the groups, subjects who progressed to psychosis differed significantly in the combined levels of five miRNAs: four downregulated miRNAs (miR-941, miR-199a-3p, miR-92a-3p, and miR-31-5p) and one upregulated miRNA (miR-103a-3p) [188]. Furthermore, miRNA levels within the controls and the group at CHR who did not progress were strongly correlated, whereas miRNA levels in those who progressed to psychosis were more weakly correlated [188]. Samples were collected only at baseline; therefore, longitudinal data on symptom-associated changes in miRNA levels are not available.
A later study utilizing the same NAPLS-2 miRNA-seq data set examined the relations between leukocyte miRNAs and gray matter reduction in the superior frontal cortex [155,189]. Although cortical thinning is a normal feature of adolescent brain development, the rate of cortical thinning is accelerated in subjects at CHR who progress to psychosis [189,190,191,192]. A set of nine miRNAs was used to develop a classifier function to predict the rate of cortical thinning in the NAPLS-2 data set: miR-103a-3p, miR-140-3p, miR-142-5p, miR-26b-5p, miR-27b-5p, miR-501-3p, miR-183-5p, miR-339-3p, and miR-193a-5p [155]. Notably, although the same miRNA-seq data were used in both studies, only miR-103a-3p appeared in both the miRNA classifier set associated with conversion to psychosis and the classifier set associated with cortical thinning [155,188]. Neither study found that peripheral miRNA levels are sufficient to predict conversion status, even among individuals at CHR, but together they suggest that miRNA data may be useful in predicting conversion to psychosis and structural phenotypes associated with that conversion.
Although the NAPLS-2 miRNA studies included individuals at CHR who did not progress to psychosis, the investigators did not examine the differences between those subjects and the other groups. Such comparisons might identify miRNAs that contribute to SCZ risk without directly contributing to disease conversion or miRNAs that protect individuals at CHR from disease conversion. To date, no studies have examined peripheral miRNAs specifically in individuals at CHR of SCZ but without disease. One study examined miRNAs in whole blood from individuals with high familial risk of bipolar disorder [193], who also have an elevated risk of SCZ [194,195]. Using RT-qPCR, the authors found that miR-15b, miR-132-3p, and miR-652 are elevated in the blood of subjects at high risk of bipolar disorder, relative to controls. Upregulation of all three miRNAs has also been reported in postmortem SCZ brains [7,9,14].
Finally, miRNA levels may change in the periphery in response to the onset of SCZ. One study examined plasma from patients with first-episode SCZ by using miRNA microarrays and found that miR-223-3p is significantly elevated in patients, relative to age-matched controls [196]. A validation experiment using RT-qPCR found that miR-223-3p is also elevated in plasma from patients with SCZ at later stages of disease. Therefore, miR-223-3p may be a useful peripheral biomarker for SCZ onset. Notably, a separate microarray study found that miR-223-3p is also elevated in postmortem DLPFC of SCZ brains [7], suggesting that miR-223-3p plays a role in disease etiology.
Many studies have examined peripheral miRNAs in individuals with advanced SCZ in hopes of identifying miRNA biomarkers. For example, a large, early study of peripheral blood mononuclear cells observed significant downregulation of 17 miRNAs from the MIR379/410 cluster in subjects with SCZ [197]. Studies in animal models suggest miRNAs in this cluster are critical for neuronal function [198,199,200], sensitive to environmental risk factors for SCZ [22], and required for typical sociability [198] and anxiety-like behaviors [201]. Other studies have observed peripheral upregulation of miR-137 [202,203], a miRNA with strong SCZ associations that we will discuss later in this review. These and other biomarker studies have been reviewed extensively elsewhere [202,204,205]. However, miRNA levels in subjects with advanced SCZ may also be altered in response to cellular damage during chronic disease or due to extended use of antipsychotics and/or other prescription drugs. These caveats limit the usefulness of these studies in predicting disease onset or understanding the underlying mechanisms by which miRNAs might contribute to its onset.
Recent work incorporating biomarker information with conventional CHR criteria substantially improved predictions of conversion to psychosis [206]. Additional research is needed to determine whether miRNA data can further improve these predictions. Ultimately, the earlier, more accurate identification of patients who will progress to psychosis will facilitate earlier treatment and interventions. Currently available early interventions improve prognosis for patients [207,208,209], and with continued improvement, future interventions might prevent disease manifestation entirely. This goal can be achieved only with greater understanding of the molecular mechanisms underlying disease onset.
Collectively, these studies suggest that miRNAs in peripheral tissues undergo changes in response to the various stages of SCZ in humans. However, peripheral miRNAs are unlikely to directly contribute to disease onset. Next, we will review work from animal models demonstrating that miRNA dynamics contribute to adolescent brain development.
6. MiR-29 and MiR-132-3p in Adolescent Neurodevelopment and Disease
Despite large discrepancies in the overall outcomes of studies examining miRNAs across the lifespan, several miRNAs robustly and consistently increase during postnatal brain maturation: the miR-29 family (primarily miR-29a/b/c-3p) and miR-132-3p (Table 2 and Table 3). We will use these miRNAs as a case study to illustrate how miRNAs are crucial for normal brain development, may be dysregulated in the brains of patients with SCZ, and regulate neurological processes implicated in SCZ etiology. These miRNAs were selected primarily because they are highly represented in the existing research literature, particularly in studies of adolescent brain. However, we acknowledge that future research may reveal other miRNAs that play a greater role in adolescent brain development and SCZ onset.
6.1. The MiR-29 Family and MiR-132-3p Levels Increase with Age and May Be Dysregulated in SCZ
The miR-29 family primarily consists of three miRNAs generated from two MIR29 gene clusters in mammals. These miRNAs are identical in their seed sequences (Table 4) (miRBase [56,57,58,59,60,61]), are enriched in mammalian brain relative to other body tissues, and share largely overlapping expression patterns within the brain [210]. The miR-29 family is implicated in a myriad of roles in brain development and function, including regulating neural progenitor cell proliferation [211], axon branching [80], calcium signaling [212], iron accumulation [213], and neuronal survival [214].
Most studies examining human cortex have found that the level of at least one member of the miR-29 family increases significantly during postnatal development [44,45,47] (but also see [48]). Studies in macaque, pig, rat, and mouse have demonstrated that postnatal miR-29 upregulation with age is highly conserved among mammals [45,49,51,53,54,55,80,81,82,83]. Notably, Swahari et al. [55] examined the levels of miR-29 in mouse cortex at P0–P60 and observed a robust increase in all three family members before the onset of adolescence (P0–P20) and during the transition from early to mid-adolescence (P20–P40). Napoli et al. [83] also observed a robust increase of miR-29a-3p in mouse visual cortex prior to adolescence (P10–P25) and throughout adolescence (P25–P60) [83]. Many other studies found an increase in one or more miR-29 family members between embryonic or early postnatal timepoints and adulthood but lacked adolescent time points; therefore, they could not demonstrate when exactly the increase occurred (Table 2 and Table 3).
The MIR132/212 gene cluster gives rise primarily to miR-132-3p, miR-212-5p, and miR-212-3p. MiR-132-3p is highly implicated in the regulation of dendritic spine morphology in primary neurons and in adolescent brain. Inhibition of miR-132-3p reduces dendritic spine density and inhibits dendritic spine maturation, without affecting overall dendritic morphology in pyramidal neurons in the mouse visual cortex [215]. In contrast, miR-132-3p mimic increases the proportion of spines with a mushroom (mature) morphology in mouse visual cortex [79]. Reduced dendritic spine density on cortical pyramidal neurons is also a well-documented phenomenon in the postmortem brains of patients with SCZ [216,217,218,219]. MiR-132-3p also regulates fear memory [220,221], learned safety [222], sleep [223], and circadian rhythms [224,225], all of which undergo changes during adolescent development [226,227,228,229,230,231] and are disrupted in SCZ [232,233,234,235]. Reduced levels of miR-132-3p during adolescence or adulthood might contribute to these pathologies in some patients with SCZ.
Human studies using microarrays have yielded conflicting results regarding the effects of age on miR-132-3p in the brain; Beveridge et al. [48] reported an overall decrease in miR-132-3p with age, and Moreau et al. [47] reported an increase between fetal and postnatal development [47,48] (Table 2). Notably, Beveridge et al. also reported a decrease in miR-29a/c-3p levels with age. Studies in rodent models have more consistently reported increased miR-132-3p levels during postnatal brain development: miR-132-3p increases during preadolescent development in the rat cortex and hypothalamus and in mouse PFC and visual cortex (Table 3). Tognini et al. [79] also observed upregulation of miR-132-3p in mouse visual cortex between P7 and P30, with strong upregulation in early adolescence (P20–P30). Whether miR-132-3p undergoes additional upregulation during later stages of adolescence or early adulthood is less clear.
Studies investigating miRNA levels in individuals with SCZ have produced conflicting results on the relations between the miR-29 family and miR-132-3p and disease (Table 5). In most studies, neither miR-29 nor miR-132-3p levels were significantly different in the postmortem brains of persons with SCZ [7,8,9,10,11,12,13]. The few studies that have found differences are not in agreement, with some demonstrating decreased levels and others demonstrating increased levels in persons with SCZ [6,7]. A possible explanation for these conflicting results is the strong relation between the miRNAs and age, as exhibited in Table 2 and noted by Beveridge et al. [7]. Any failure to properly age-match control subjects is likely to substantially affect the results of studies examining these miRNAs.
In summary, the evidence that miR-29 and miR-132-3p levels are higher in adolescent and adult brain than in childhood brain is robust, though evidence of miR-29 or miR-132-3p dysregulation in SCZ is inconsistent. Further research is needed to fully elucidate the trajectories of both families of miRNAs during typical neurodevelopment and in SCZ. In addition to their individual roles in adolescent brain development, miR-29 and miR-132-3p may also cooperate in regulating processes that are essential for adolescent brain function, specifically the closing of ocular dominance plasticity within the primary visual cortex and the downregulation of Dnmt3a mRNA to stabilize CH-methylation in cortical neuron genomes, which we discuss below (Figure 4).
6.2. Shared Roles of MiR-29 and MiR-132-3p: Cortical Ocular Dominance Plasticity and DNMT3A
Ocular dominance plasticity is an age-dependent form of cortical plasticity that occurs within the binocular portion of the visual cortex in response to sensory deprivation. If one eye is deprived of sensory input, the neuronal response to visual stimulation of that eye is weakened in the portion of the contralateral visual cortex that receives bilateral visual inputs, and the ocular dominance index (i.e., the relative strength of inputs) shifts toward the ipsilateral, nondeprived eye. Studies in animal models suggest the existence of early critical periods for this form of cortical plasticity: ocular dominance plasticity is strong before adolescence, undergoes an age-dependent decline during adolescence, and is virtually absent in adults [236,237,238,239].
Both miR-132-3p and miR-29a-3p are necessary for ocular dominance plasticity in the mouse primary visual cortex. MiR-29a-3p increases between P10 and P60 in an experience-independent manner [83]. In contrast, miR-132-3p upregulation coincides with the beginning of visual stimulation (i.e., eye opening at ~P13 in mice) [79]. This upregulation continues until early adolescence (~P30) in the presence of normal visual inputs but is substantially blunted if the mice are deprived of visual inputs via dark-rearing [79]. Typically, depriving mice of visual inputs to one eye (i.e., monocular deprivation) in early adolescence for 3–4 days produces a substantial shift in the ocular dominance index toward the nondeprived eye that is detectable at P28. Overexpression of either miR-29a-3p or miR-132-3p in the visual cortex in young mice prematurely blocks ocular dominance plasticity [79,83]. Furthermore, inhibition of miR-29a-3p in adult (~P120) mouse visual cortex restores ocular dominance plasticity that is normally absent by this age [83]. Together, these data suggest that the age-associated increases in miR-29a-3p and miR-132-3p underlie the closing of the critical period for ocular dominance plasticity in the adolescent cortex.
Both Mellios et al. [215] and Tognini et al. [79] attributed miR-132-3p’s role in ocular dominance plasticity to its role in regulating dendritic spine maturation and morphology. In contrast, miR-29a-3p’s role in ocular dominance plasticity has been attributed to its ability to promote maturation of PNNs around PV+ interneurons in the mouse visual cortex [83]. These specialized structures of the extracellular matrix limit synaptic plasticity but also protect neurons against oxidative stress [240,241]. Treatment with miR-29a-3p mimics increases PNN intensity during early adolescence, thereby mimicking the mature PNNs that surround neurons in adulthood and reducing plasticity to adult-like levels [83]. In contrast, inhibiting miR-29a-3p in adult mice weakens PNN intensity and reduces sulfonation of chondroitin sulfate position 4, a chemical modification that is typically increased in adult brains relative to adolescent brains. In other words, inhibiting miR-29a-3p in the adult mouse visual cortex partially reverts PNNs to their adolescent state, thereby enabling adolescent-like levels of plasticity.
However, PNNs also protect neurons against oxidative stress, to which fast-spiking PV+ interneurons are particularly susceptible. The mature PNNs that surround PV+ interneurons in adulthood offer better protection against oxidative stress than do the immature PNNs present in adolescence. Increased oxidative stress and deficits in PV+ interneurons and PNN labeling have been documented in patients with SCZ [242,243,244,245,246] and in most SCZ mouse models [247]. Dysregulation of miR-29a-3p, or other miRNAs that regulate PNN integrity, may render PV+ interneurons more susceptible to oxidative stress and contribute to PV+ interneuron deficits in patients with SCZ.
Both miR-29 and miR-132-3p target the DNA methyltransferase DNMT3A and may contribute to its downregulation during adolescence [10,55]. DNMT3A specifically catalyzes CH-methylation [248], where H indicates T, A, or C nucleotides. In the brain, DNMT3A is predominantly expressed in postmitotic neurons [249]. During early postnatal brain development, neuronal genomes undergo extensive CH-methylation, which ends at ~15 years of age in humans [250] and at ~P21 in mice [249], when DNMT3A levels dramatically decline and miR-29 and miR-132-3p are strongly upregulated. The loss of DNMT3A disrupts early brain development; however, failure to downregulate DNMT3A during adolescence is devastating to adult brain function. The mutation of miR-29–binding sites in Dnmt3a in mice leads to elevated DNMT3A protein levels, behavioral abnormalities (e.g., limb clasping), increased seizure susceptibility, and reduced lifespan [55]. Dnmt3a mRNA is also upregulated in response to monocular deprivation, and treatment with the DNA methyltransferase inhibitor RG108 blocks ocular dominance plasticity [251], suggesting that Dnmt3a downregulation by miR-29 and miR-132-3p contributes to these miRNAs’ roles in ocular dominance plasticity.
Many of the genes downregulated in response to excess CH-methylation are associated with SCZ or other neurologic disorders. Several reports have suggested that DNMT3A mRNA is upregulated in the brains of patients with SCZ, though upregulation may be limited to specific neuron subpopulations [252] and/or subjects with chronic (not first-episode) disease [253]. DNMT3A rs2304429 CC genotype may also predict better response to antipsychotics in patients with SCZ [253], and the DNMT3A rs2289195 locus is associated with SCZ risk [254]. SCZ-like symptoms (including auditory hallucinations) were also recently reported in three patients with novel, predicted deleterious variants in the DNMT3A-coding sequence [255]. Mice hemizygous for Dnmt3a also exhibit behavioral deficits, including reduced exploration and increased anxiety-like behaviors [256]. However, no deficits in pre-pulse inhibition (PPI), a common deficit in patients with SCZ, were observed in these mice. Mice with Dnmt3a knockout specifically in the forebrain excitatory neurons also exhibit normal PPI and anxiety-like behaviors [257]. However, mice with conditional Dnmt3a knockout in inhibitory neurons begin to show behavioral deficits around the time of weaning (~P21) or early adolescence; these deficits include repetitive behaviors, motor deficits, hippocampal learning and memory deficits, and enhanced acoustic startle [258].
In summary, upregulation of miR-132-3p and miR-29 during adolescence is central to the closing of ocular dominance plasticity, regulation of dendritic spine morphology, maturation of PNNs around PV+ interneurons, and the downregulation of Dnmt3a in adolescent and adult cortex. Dysregulation of either miRNA or their downstream effector DNMT3A disrupts neuronal function in the adolescent brain and may contribute to SCZ onset. However, further research is needed on these miRNAs and their targets in patients with SCZ and mouse models, particularly during adolescent brain maturation.
The cases of miR-29 and miR-132-3p highlight some of the obstacles and opportunities facing miRNA research, particularly as it pertains to adolescent brain maturation and SCZ onset. Research targeting miR-29 demonstrates that the effects of miRNA dysregulation vary with age and that up- or downregulation of the same miRNA at different time points can have very different impacts on neuronal function. Although miRNAs are highly dynamic with age, most studies examine only a few widely spaced time points to draw conclusions about the effects of age on miRNA levels. These studies often completely miss temporary fluxes in miRNA levels and may ignore critical developmental stages, including adolescence. MiRNAs also rarely act alone. Although most functional studies focus on only one miRNA or miRNA family, multiple miRNAs with similar expression patterns may cooperate or compete to regulate the same biological processes and, in some cases, the same mRNA targets. Dysregulation of these targets in patients with SCZ may reflect dysregulation of one or more of these miRNAs, with different miRNAs underlying the same deficit in different patients.
MiR-29 and miR-132-3p are strongly implicated in adolescent brain maturation, but their associations with SCZ remain unclear. In contrast, 22q11.2 microdeletions and genetic variants in MIR137HG are strongly associated with SCZ risk and provide more direct evidence that miRNA disruptions underlie this risk, yet how these variants disrupt adolescent brain maturation remains poorly understood. In the following section, we will review recent work suggesting that age-dependent phenotypes in 22q11DS mouse models mimic the age-dependent onset of symptoms in persons with SCZ. We will also review the evidence from human genomic studies suggesting that MIR137HG variants predict not only SCZ risk but also variance in age of onset, along with evidence from animal models suggesting how disrupted miR-137 function contributes to SCZ onset.
7. 22q11DS Disrupts the MiRNA Pathway in an Age-Dependent Manner
Hemizygous microdeletions in the 22q11.2 region lead to SCZ in approximately one in four patients, representing a 20- to 25-fold increase in risk compared to that in the general population [37,38]. The 22q11.2 region contains DGCR8, which encodes a protein component of the Microprocessor (Figure 1), along with several miRNAs, most notably miR-185-5p. The dysregulation of miRNA levels occurs in patients with 22q11DS [259,260], and Dgcr8 haploinsufficiency impairs miRNA biogenesis in mouse models of 22q11DS [39,40]. The age of onset of SCZ symptoms in patients with 22q11DS is also indistinguishable from that in patients with idiopathic SCZ [261]. Therefore, 22q11DS mouse models provide a valuable tool for investigating the roles of miRNAs in SCZ onset. Of the many phenotypes that have been identified in 22q11DS mouse models, several have a clearly documented age dependence that replicates the timeline of onset in patients with SCZ, and several of the age-dependent phenotypes have been linked to miRNA deficits resulting from Dgcr8 haploinsufficiency.
Abnormal hippocampal activity may contribute to cognitive symptoms in patients with SCZ [262,263,264,265,266,267]. Our lab previously reported that long-term potentiation (LTP) at hippocampal excitatory synapses between CA3 and CA1 pyramidal neurons (CA3-CA1 synapses) is elevated at ~P120 (mature adulthood) [268] but not at ~P60 (end of adolescence/early adulthood) in 22q11DS mouse models [40]. Dgcr8 haploinsufficiency is sufficient to replicate both the elevated LTP and its age-dependence. DGCR8 loss leads to reduced hippocampal levels of miR-25-3p and miR-185-5p, which both target Serca2 mRNA. DGCR8 and miRNA loss leads to an age-dependent increase in synaptic SERCA2, an enzyme that loads Ca2+ into the endoplasmic reticulum. Restoration of miR-25-3p or miR-185-5p is sufficient to reduce SERCA2 to wild-type levels and restore LTP in 22q11DS mouse models. Notably, elevated SERCA2 was observed in the PFC and hippocampus of patients with SCZ [40]. The ATP2A2 locus, the human homologue of Serca2, was also identified as one of 108 loci meeting genome-wide significance for association with SCZ by the Schizophrenia Working Group of the Psychiatric Genomics Consortium [18].
Auditory hallucinations are one of the most prevalent symptoms in SCZ, occurring in as many as 80% of patients (though prevalence varies by culture) [269,270]. The onset of hallucinations and other psychotic symptoms typically marks the end of the prodromal phase and the onset of early SCZ disease. Our lab has found that Dgcr8 haploinsufficiency underlies two age-dependent deficits in 22q11DS mouse models that may be related to auditory hallucinations in patients: behavioral deficits in PPI in an acoustic startle reflex and synaptic deficits in thalamocortical excitatory projections from the medial geniculate nucleus (i.e., auditory thalamus) to the primary auditory cortex [271]. Both deficits are absent at P60 but are present by P120 in mice, suggesting an early adult onset typical of hallucination onset in patients [121]. Both deficits are also sensitive to antipsychotics, the typical therapy for hallucinations and other positive symptoms of SCZ.
Additionally, our lab found that DGCR8 loss in mice leads to reduced levels of miR-338-3p in the auditory thalamus, which in turn leads to elevated levels of DRD2s (encoded by Drd2) [121]. Restoration of miR-338-3p or Drd2 to wild-type levels is sufficient to rescue behavioral and synaptic deficits in Dgcr8-deficient mice. Remarkably, Mir338 haploinsufficiency alone removes the age-dependence of some of these deficits; behavioral deficits and antipsychotic sensitivity are present by P45 (adolescence). This suggests that partial loss of a single miRNA underlies thalamocortical deficits in 22q11DS mouse models. Notably, our lab also observed reduced miR-338-3p levels in auditory thalamus samples from patients with SCZ, relative to age-matched control subjects, though the sample size for this experiment was fairly small (n = 7 per group). Finally, like ATP2A2, DRD2 is one of 108 loci that meets genome-wide significance for association with SCZ, per the Schizophrenia Working Group [18]. This finding suggests that the disruption of miR-338-3p targeting of DRD2 mRNA contributes to thalamocortical disruptions in patients with SCZ.
Together, these studies demonstrate that miRNA deficits underlie multiple synaptic or behavioral phenotypes with age-dependent, early adult onsets that replicate the onset of SCZ in patients with 22q11DS. However, DGCR8 deficiency is present throughout life, raising the question of why these deficits arise during late brain maturation rather than earlier in development. Our lab’s work with miR-338-3p revealed that miR-338-3p levels in the auditory thalamus decline between P60 and P120, independent of genotype [121]. This age-dependent decline, in combination with miR-338-3p loss due to DGCR8 deficiency, may cause miR-338-3p levels to drop below a critical level necessary for DRD2 regulation, allowing the emergence of synaptic and behavioral deficits.
The age-dependent decline in miR-338-3p is not dramatic (only ~20%). As previously discussed, the results of studies examining temporal regulation of miRNA levels are often poorly replicated, even within a single model system and for miRNAs that undergo several-fold increases or decreases in their levels. Furthermore, no mechanism has been proposed for this apparent decline in miR-338-3p levels, and no decline has been reported for miR-25-3p or miR-185-5p in the hippocampus to explain their age-dependent regulation of SERCA2 [40]. Further examination of miRNA levels and activity during adolescence and early adulthood in humans and mouse models is needed to elucidate how they contribute to SCZ onset.
The developmental trajectories observed in mice do not always model those observed in patients. Ventricular enlargement is one of the most replicated structural defects observed in the brains of patients with SCZ [272,273,274]. Our lab found that Dgcr8 haploinsufficiency underlies enlargement of the lateral and third ventricles in 22q11DS mouse models through a miRNA-dependent mechanism [122]. Although this phenotype is age-dependent, its onset occurs significantly later than that observed in patients. Specifically, ventricular enlargement is present in 8-month-old mice; at 4 months, the ventricles of 22q11DS mice are indistinguishable from those of wild-type mice. In contrast, ventricular enlargement has been observed in patients at SCZ onset, and it progresses with age [275,276,277,278,279].
Therefore, mouse models of 22q11DS imperfectly model the age dependence of SCZ pathologies, with some phenotypes mimicking onset in patients and others following a delayed trajectory. Ventricular enlargement in 22q11DS mice has been attributed to reduced ciliary motility in ependymal cells in the ventricle walls, which may lead to a slow, progressive accumulation of cerebrospinal fluid [122]. This defect in ciliary motility is present by ~P120 in mice, similar to other Dgcr8- and miRNA-dependent phenotypes. Therefore, the ventricular enlargement is developmentally delayed in mice relative to humans, but the underlying molecular mechanisms contributing to it may be initiated by developmental events that are more closely aligned across species.
Our lab’s work examining miRNAs in ventricular enlargement highlights an additional obstacle to using 22q11DS mouse models––species differences in the miRNA pathway. In 22q11DS mouse models, decreased levels of miR-382-3p and miR-674-3p lead to elevated D1 dopamine receptors, reduced ciliary motility, and enlarged ventricles [122]. However, the sequence of murine miR-382 listed in miRBase is shifted toward the 3ʹ end of pre–miR-382, relative to the human form, which would alter the seed sequence and could dramatically alter mRNA targeting (Table 4). Furthermore, mouse miR-674-3p appears to have no human ortholog, so this miRNA cannot contribute to ventricular enlargement in patients with SCZ. Conversely, mouse models cannot model the effects of human- or primate-specific miRNAs, some of which are crucial for human brain development [76,280,281] and/or dysregulated in postmortem SCZ brain [282]. Therefore, the specific miRNAs that underlie phenotypes in mouse models may play no role in the SCZ symptoms that they model. Nonetheless, in most cases, miRNAs critical for mouse brain function and dysregulated in SCZ mouse models are highly conserved between mice and humans (Table 4).
Despite these limitations, mouse models of 22q11DS provide a valuable tool for identifying novel miRNAs with potential relevance to SCZ onset, identifying novel miRNA-dependent pathways that underlie SCZ-associated phenotypes, and investigating basic mechanisms by which miRNAs contribute to adolescent and early adult brain maturation.
8. MIR137HG Variance Predicts Variance in the Age at SCZ Onset
The age of onset of psychosis in patients with SCZ is highly variable. Onset typically occurs between 15 and 35 years of age, and male patients typically have an earlier onset than females [43]. One of the best-known predictors of age of onset occurs at the rs1625579 SNP near MIR137HG. In subjects with SCZ, Lett et al. [283] found that the mean age of onset for those with the TT genotype at rs1625579 was 20.8 ± 5.7 years, whereas that for those carrying a protective G allele (GT/GG genotype) was 23.7 ± 9.1 years. In patients with SCZ, the T allele is also associated with larger left ventricle volume, smaller hippocampal volume, and reduced white matter integrity [283] (also see [284]) and may predict more severe negative symptoms and worse cognitive performance [285] (also see [286]).
The rs1625579 SNP was first identified in a SCZ genome-wide association study [16]. The T allele was identified as the major allele and the risk allele for SCZ, while the minor G allele appeared to be protective. Later studies identified additional SNPs in MIR137HG and in predicted and validated miR-137 targets that display significant enrichment in the genomes of patients with SCZ [17,18,19,20]. Furthermore, common genetic variants implicated in SCZ are enriched for predicted miR-137–binding sites [15]. Together, these studies strongly implicate miR-137 in genetic risk for SCZ.
Experiments in several cell culture systems suggest that the protective alleles at multiple MIR137HG loci are associated with elevated miR-137 levels [20,21,287,288], and some evidence suggests miR-137-3p levels are lower in the brains of neurotypical subjects homozygous for the risk allele (TT) at rs1625579 [289]. Together, these studies suggest that elevated miR-137 is protective against SCZ and delays its onset among patients. However, miR-137 levels may be higher in the peripheral blood of subjects with SCZ [203], and postmortem brain studies have mostly found no evidence of miR-137 dysregulation in SCZ when MIR137HG genetic variants are ignored (Table 5). Furthermore, the high frequency of the risk allele among most populations, the low prevalence of SCZ in most populations, and the lack of evidence of miR-137 dysregulation in most patients with SCZ suggest that miR-137 dysregulation alone is not sufficient to promote SCZ onset.
Experiments in mouse models suggest that overexpression and inhibition of miR-137 are disruptive to neuronal function. Mice hemizygous for neuronal Mir137 exhibit deficits in synaptic plasticity, social behavior, and learning, along with repetitive behaviors [290]. Similarly, hemizygous microdeletions in the 1p21.3 region, which contains MIR137HG, are associated with autism spectrum disorders and intellectual disability in human subjects [291,292,293,294,295]. In mice, overexpression of miR-137 in neurons impairs dendrite outgrowth, hippocampal learning, PPI, social behavior, and performance in the novel object-recognition task [287,296,297]. These studies demonstrate that both up- and downregulation of miR-137 disrupt neurodevelopment and suggest that either might contribute to neurodevelopmental disorders.
MiR-137 regulates many neuronal targets that are implicated in SCZ, including several of the 108 loci identified by the Schizophrenia Working Group [18]. Many miR-137 targets are involved in signaling pathways that are central to early neurodevelopment and/or synaptic plasticity, including: neuregulin signaling [298], PI3K–mTOR signaling [298], PKA signaling [290], and glucocorticoid signaling [299]. MiR-137 is also intimately involved in glutamatergic synaptic transmission, as it regulates many presynaptic targets involved in neurotransmitter release and multiple glutamatergic receptors [287,300], including AMPA and NMDA receptor subunits GluA1 and GluN2A, respectively [301,302]. NMDA receptor antagonists mimic positive and negative symptoms of SCZ [303,304,305], and altered DA signaling might arise as a result of dysregulated NMDA receptor activity [113]. Therefore, miR-137′s roles in regulating glutamate signaling may be particularly relevant to SCZ onset.
Although miR-137 regulates neurodevelopment and appears to contribute to SCZ onset, it is unclear how miR-137 contributes to adolescent brain maturation. Several studies suggest that miR-137 levels are highest and most dynamic during perinatal and early postnatal brain maturation and remain high but relatively stable during adolescence and adulthood (Table 2 and Table 3). However, HITS-CLIP studies suggest that miR-137 targeting varies with age [76,77], which suggests that its functions are also age dependent, but those functions during adolescence have yet to be examined.
MiR-137 dysregulation alone is highly unlikely to cause SCZ, but miR-137 may influence how the brain responds to other genetic and environmental risk factors for SCZ. A few studies suggest that miR-137 is protective against oxidative stress and inflammation [306,307], so lower levels of miR-137 might increase susceptibility to those stresses during adolescence. In mouse models, glucocorticoid signaling mediates interactions between genetic risk and adolescent stress that disrupt DA signaling and lead to deficits in PPI and other behaviors during adulthood [308]. MiR-137 targets many components of glucocorticoid signaling, suggesting miR-137 dysregulation could also increase susceptibility to adolescent stress. However, miR-137 function has not been examined within the context of SCZ mouse models, so it is unclear how miR-137 interacts with other genetic or environmental risk factors for SCZ in an adolescent brain.
9. Possible Mechanisms of MiRNA-Dependent Onset of SCZ
Although the miRNA pathway is strongly implicated in SCZ, the precise mechanisms by which miRNA dysregulation contributes to disease onset remain poorly understood, in part, because the mechanisms by which miRNAs contribute to typical brain maturation remain poorly understood, especially during adolescence. However, research demonstrates that miRNAs are highly dynamic during all stages of brain maturation and are essential for developmental processes implicated in SCZ onset.
SCZ is a highly heterogeneous disorder (and possibly an umbrella term for several disorders), with an array of genetic and environmental risk factors implicated in its etiology. Research in the 22q11DS field strongly implicates the miRNA pathway, but most studies of miRNA dysregulation in SCZ have failed to implicate the same set of miRNAs. Furthermore, as observed with miR-29 and miR-132-3p, studies often come to opposite conclusions about specific miRNAs. Perhaps the field has been too focused on specific miRNAs, when it is highly likely that no individual miRNA underlies SCZ onset or any aspect of its pathology in all patients.
Heterogeneity in miRNA dysregulation might contribute to heterogeneity of symptoms and their onset times in patients with SCZ. Because different miRNAs perform different functions, they might contribute to SCZ onset by different mechanisms. We propose the following mechanistic categories by which miRNAs might contribute to SCZ onset.
9.1. Direct Mechanism: Aberrations in MiRNAs during Adolescence Disrupt Adolescent Brain Maturation and Cause Disease Onset
We propose that the failure to correctly up- or downregulate miRNAs during adolescence will specifically disrupt adolescent brain development and directly contribute to SCZ onset. In this mechanism, early development may proceed as normal, when the miRNAs of interest are either nonessential or expressed at normal levels for early developmental stages. During adolescence, the miRNAs are incorrectly expressed, leading to dysregulation of their targets and disruption of neurodevelopment. For example, both miR-29 and miR-132-3p downregulate DNMT3A during adolescence and adulthood [10,55], but neither miRNA is essential for regulating DNMT3A levels during early postnatal development, when they are virtually undetectable. Genetic or environmental factors that inhibit miR-29 and miR-132-3p most likely have no effect on DNMT3A levels during early development because these miRNAs are not essential during that time. During adolescence, disruptions to miR-29 and miR-132-3p might cause an upregulation of DNMT3A; excess CH-methylation; and age-dependent, late-onset disruptions in neuronal function.
9.2. Delayed Mechanism: Aberrations in MiRNAs during Early Development Disrupt Brain Maturation during Adolescence
This mechanism is most closely related to the “two-hit” model of SCZ [309]. Here, dysregulation of miRNAs might provide the first hit to neurodevelopment, while the second occurs during adolescence and promotes SCZ onset. The second hit might be an additional disruption to neurodevelopment or a normal developmental process that reveals the effects of the first hit.
The Delayed Mechanism might underlie miR-137′s role in SCZ onset. MiR-137 levels are most dynamic in the brain during early postnatal development (Table 2 and Table 3), and miR-137 is implicated in many signaling pathways that are essential to early brain development and implicated in SCZ risk. Small disruptions in miR-137 early in development might not immediately affect neuronal function, but large disruptions might lead to disorders with earlier onset, e.g., autism spectrum disorders or intellectual disability [290,291]. However, small disruptions might render the brain more vulnerable to additional stress during adolescence. This mechanism is particularly attractive for explaining miR-137′s role, because this miRNA may regulate DA signaling [123], which matures during adolescence [310], and cellular responses to oxidative stress and inflammation [306,307]. Disruptions to miR-137 might, therefore, provide “hits” to both early and adolescent development. However, miR-137′s functions within the adolescent brain remain poorly understood.
In adulthood, the brain may become more dependent on posttranscriptional mechanisms of regulation, including miRNAs [92,93]. This mechanism might underlie several miRNA-associated deficits in 22q11DS mouse models [40,121,271]. As previously discussed, deficiencies in DGCR8 and miRNAs are present early in development, but several miRNA-dependent phenotypes do not emerge until early adulthood. Prior to adolescence, small disruptions to the miRNA pathway (the first “hit”) might be insufficient to disrupt the processes that they regulate, perhaps due to compensatory mechanisms that affect transcription. With age, cells may become more dependent on miRNAs, effectively providing a second “hit” to miRNA-dependent processes.
9.3. Progressive Mechanism: Aberrations in Individual MiRNAs Cause Small Deficits That Accumulate during Development and Become Disruptive during Adolescence
Some disruptions in brain function accumulate over time, before they cause detectable deficits. This is consistent with the observation that some SCZ phenotypes progress with age. However, few studies have directly examined how miRNA dysregulation contributes to this progression.
In patients with SCZ, ventricle enlargement is present during the prodromal phase [189] and first-episode psychosis [275,277], and it progresses with age through the chronic disease phase [278,279]. We previously described how deficiencies in DGCR8 and miRNAs contribute to ciliary deficits in ependymal cells and underlie enlarged ventricles in a 22q11DS mouse model [122]. DGCR8 deficiency is present throughout development. This ciliary deficit is detectable by ~P120, suggesting the miRNA deficit is also present by P120, though it is unclear whether the miRNAs underlying this deficit are regulated with age. By contrast, ventricular enlargement appears to be slow, progressive, and undetectable until ~P240. Thus, ventricular enlargement is developmentally delayed in these mice, suggesting that developmental stage may be less important than the duration of time following the onset of the miRNA deficit for this particular structural phenotype.
10. Conclusions
We speculate that the direct, delayed, and progressive mechanisms that we propose here contribute to SCZ onset, though different miRNAs may contribute to SCZ onset in different subsets of patients. Additional research is needed to fully elucidate miRNA trajectories during typical neurodevelopment and to determine normal levels of variance across neurotypical subjects. This will set the stage for comprehensive examination of miRNA deviations at different stages of development and their contribution to SCZ onset. We predict that a greater understanding of the molecular mechanisms underlying SCZ onset will facilitate the identification of at-risk subjects for earlier therapeutic intervention, provide novel targets for therapeutic interventions, and, ultimately, position us to delay or even prevent SCZ symptoms entirely.
Author Contributions
Conceptualization, K.T.T. and S.S.Z.; writing—original draft preparation, K.T.T.; writing—review and editing, S.S.Z.; visualization, K.T.T.; funding acquisition, S.S.Z. Both authors have read and agreed to the published version of the manuscript.
Funding
This work was supported, in part, by the National Institutes of Health (R01 MH097742, R01 DC012833), by the Stanford Maternal and Child Health Research Institute Uytengsu-Hamilton 22q11 Neuropsychiatry Research Program, and by the American Lebanese Syrian Associated Charities (ALSAC) to S.S.Z.
Acknowledgments
We thank the members of the Zakharenko lab for constructive comments and Angela McArthur for editing the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other granting agencies.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tandon, R.; Keshavan, M.S.; Nasrallah, H.A. Schizophrenia, “Just the Facts” What we know in 2008. 2. Epidemiology and etiology. Schizophr. Res. 2008, 102, 1–18. [Google Scholar] [CrossRef]
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr. Res. 2009, 110, 1–23. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Kendler, K.S.; Neale, M.C. Schizophrenia as a Complex Trait. Arch. Gen. Psychiatry 2003, 60, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Dean, K.; Murray, R.M. Environmental risk factors for psychosis. Dialogues Clin. Neurosci. 2005, 7, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.Y.; Teoh, S.L.; Wu, D.B.-C.; Kotirum, S.; Chiou, C.-F.; Chaiyakunapruk, N. Global economic burden of schizophrenia: A systematic review. Neuropsychiatr. Dis. Treat. 2016, 12, 357–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, D.O.; Jeffries, C.D.; Jarskog, L.F.; Thomson, J.M.; Woods, K.; Newman, M.A.; Parker, J.S.; Jin, J.; Hammond, S.M. microRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol. 2007, 8, R27. [Google Scholar] [CrossRef] [Green Version]
- Beveridge, N.J.; Gardiner, E.; Carroll, A.P.; Tooney, P.A.; Cairns, M.J. Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol. Psychiatry 2010, 15, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.P.; Bruse, S.E.; David-Rus, R.; Buyske, S.; Brzustowicz, L.M. Altered MicroRNA expression profiles in postmortem brain samples from individuals with schizophrenia and bipolar disorder. Biol. Psychiatry 2011, 69, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.H.; Reimers, M.; Maher, B.; Williamson, V.; McMichael, O.; McClay, J.L.; van den Oord, E.J.C.G.; Riley, B.P.; Kendler, K.S.; Vladimirov, V.I. MicroRNA expression profiling in the prefrontal cortex of individuals affected with schizophrenia and bipolar disorders. Schizophr. Res. 2010, 124, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.H.; Zeier, Z.; Xi, L.; Lanz, T.A.; Deng, S.; Strathmann, J.; Willoughby, D.; Kenny, P.J.; Elsworth, J.D.; Lawrence, M.S.; et al. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc. Natl. Acad. Sci. USA 2012, 109, 3125–3130. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Gao, S.; Lindberg, D.; Panja, D.; Wakabayashi, Y.; Li, K.; Kleinman, J.E.; Zhu, J.; Li, Z. Temporal dynamics of miRNAs in human DLPFC and its association with miRNA dysregulation in schizophrenia. Transl. Psychiatry 2019, 9, 196. [Google Scholar] [CrossRef]
- Beveridge, N.J.; Tooney, P.A.; Carroll, A.P.; Gardiner, E.; Bowden, N.; Scott, R.J.; Tran, N.; Dedova, I.; Cairns, M.J. Dysregulation of miRNA 181b in the temporal cortex in schizophrenia. Hum. Mol. Genet. 2008, 17, 1156–1168. [Google Scholar] [CrossRef]
- Liu, Y.; Chang, X.; Hahn, C.G.; Gur, R.E.; Sleiman, P.A.M.; Hakonarson, H. Non-coding RNA dysregulation in the amygdala region of schizophrenia patients contributes to the pathogenesis of the disease. Transl. Psychiatry 2018, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarelli, D.M.; Beveridge, N.J.; Tooney, P.A.; Cairns, M.J. Upregulation of dicer and MicroRNA expression in the dorsolateral prefrontal cortex Brodmann area 46 in schizophrenia. Biol. Psychiatry 2011, 69, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Hauberg, M.E.; Roussos, P.; Grove, J.; Børglum, A.D.; Mattheisen, M. Analyzing the Role of MicroRNAs in Schizophrenia in the Context of Common Genetic Risk Variants. JAMA Psychiatry 2016, 73, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011, 43, 969–976. [Google Scholar] [CrossRef]
- Ripke, S.; O’Dushlaine, C.; Chambert, K.; Moran, J.L.; Kähler, A.K.; Akterin, S.; Bergen, S.E.; Collins, A.L.; Crowley, J.J.; Fromer, M.; et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013, 45, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.R.; Farh, K.-H.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; Huang, H.; et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Goes, F.S.; McGrath, J.; Avramopoulos, D.; Wolyniec, P.; Pirooznia, M.; Ruczinski, I.; Nestadt, G.; Kenny, E.E.; Vacic, V.; Peters, I.; et al. Genome-wide association study of schizophrenia in Ashkenazi Jews. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2015, 168, 649–659. [Google Scholar] [CrossRef]
- Duan, J.; Shi, J.; Fiorentino, A.; Leites, C.; Chen, X.; Moy, W.; Chen, J.; Alexandrov, B.S.; Usheva, A.; He, D.; et al. A rare functional noncoding variant at the GWAS-Implicated MIR137/MIR2682 locus might confer risk to schizophrenia and bipolar disorder. Am. J. Hum. Genet. 2014, 95, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strazisar, M.; Cammaerts, S.; van der Ven, K.; Forero, D.A.; Lenaerts, A.; Nordin, A.; Almeida-Souza, L.; Genovese, G.; Timmerman, V.; Liekens, A.; et al. MIR137 variants identified in psychiatric patients affect synaptogenesis and neuronal transmission gene sets. Mol. Psychiatry 2014, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hollins, S.L.; Zavitsanou, K.; Walker, F.R.; Cairns, M.J. Alteration of imprinted Dlk1-Dio3 miRNA cluster expression in the entorhinal cortex induced by maternal immune activation and adolescent cannabinoid exposure. Transl. Psychiatry 2014, 4, e452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnica, W.; Merico, D.; Costain, G.; Alfred, S.E.; Wei, J.; Marshall, C.R.; Scherer, S.W.; Bassett, A.S. Copy number variable micrornas in schizophrenia and their neurodevelopmental gene targets. Biol. Psychiatry 2015, 77, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.-P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.; Kim, Y.; Jin, H.; Kim, V.N. The Drosha—DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 3016–3027. [Google Scholar] [CrossRef] [Green Version]
- Landthaler, M.; Yalcin, A.; Tuschl, T. The Human DiGeorge Syndrome Critical Region Gene 8 and Its D. melanogaster Homolog Are Required for miRNA Biogenesis. Curr. Biol. 2004, 14, 2162–2167. [Google Scholar] [CrossRef] [Green Version]
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [Green Version]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Hutvágner, G.; McLachlan, J.; Pasquinelli, A.E.; Bálint, E.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [Green Version]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Zhou, K.; Smith, A.M.; Noland, C.L.; Doudna, J.A. Differential roles of human Dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 2013, 41, 6568–6576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006, 20, 1885–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, M.R.; Mathonnet, G.; Sundermeier, T.; Mathys, H.; Zipprich, J.T.; Svitkin, Y.V.; Rivas, F.; Jinek, M.; Wohlschlegel, J.; Doudna, J.A.; et al. Mammalian miRNA RISC Recruits CAF1 and PABP to Affect PABP-Dependent Deadenylation. Mol. Cell 2009, 35, 868–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassett, A.S.; Chow, E.W.C. Schizophrenia and 22q11. 2 deletion syndrome. Curr. Psychiatry Rep. 2008, 10, 148–157. [Google Scholar] [CrossRef] [Green Version]
- McDonald-McGinn, D.M.; Sullivan, K.; Marino, B.; Swillen, A.; Vortsman, J.; Zackai, E.; Emanuel, B.; Vermeesch, J.; Morrow, B.; Scambler, P.; et al. 22q11.2 Deletion Syndrome. Nat. Rev. Dis. Prim. 2015, 1, 621–626.e1. [Google Scholar] [CrossRef] [Green Version]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef]
- Earls, L.R.; Fricke, R.G.; Yu, J.; Berry, R.B.; Baldwin, L.T.; Zakharenko, S.S. Age-Dependent MicroRNA Control of Synaptic Plasticity in 22q11 Deletion Syndrome and Schizophrenia. J. Neurosci. 2012, 32, 14132–14144. [Google Scholar] [CrossRef] [Green Version]
- Rey, R.; Suaud-Chagny, M.F.; Dorey, J.M.; Teyssier, J.R.; d’Amato, T. Widespread transcriptional disruption of the microRNA biogenesis machinery in brain and peripheral tissues of individuals with schizophrenia. Transl. Psychiatry 2020, 10, 376. [Google Scholar] [CrossRef]
- Gochman, P.; Miller, R.; Rapoport, J.L. Childhood-Onset Schizophrenia: The Challenge of Diagnosis. Curr. Psychiatry Rep. 2011, 13, 321–322. [Google Scholar] [CrossRef] [Green Version]
- Hafner, H.; Maurer, K.; Loffler, W.; Riecher-Rossler, A. The influence of age and sex on the onset of early course of schizophrenia. Br. J. Psychiatry 1993, 162, 80–86. [Google Scholar] [CrossRef]
- Ziats, M.N.; Rennert, O.M. Identification of differentially expressed microRNAs across the developing human brain. Mol. Psychiatry 2014, 19, 848–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somel, M.; Guo, S.; Fu, N.; Yan, Z.; Hu, H.Y.; Xu, Y.; Yuan, Y.; Ning, Z.; Hu, Y.; Menzel, C.; et al. MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain. Genome Res. 2010, 20, 1207–1218. [Google Scholar] [CrossRef] [Green Version]
- Somel, M.; Liu, X.; Tang, L.; Yan, Z.; Hu, H.; Guo, S.; Jiang, X.; Zhang, X.; Xu, G.; Xie, G.; et al. MicroRNA-driven developmental remodeling in the brain distinguishes humans from other primates. PLoS Biol. 2011, 9, e1001214. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.P.; Bruse, S.E.; Jornsten, R.; Liu, Y.; Brzustowicz, L.M. Chronological changes in microRNA expression in the developing human brain. PLoS ONE 2013, 8, e60480. [Google Scholar] [CrossRef] [Green Version]
- Beveridge, N.J.; Santarelli, D.M.; Wang, X.; Tooney, P.A.; Webster, M.J.; Weickert, C.S.; Cairns, M.J. Maturation of the human dorsolateral prefrontal cortex coincides with a dynamic shift in microRNA expression. Schizophr. Bull. 2014, 40, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Podolska, A.; Kaczkowski, B.; Kamp Busk, P.; Søkilde, R.; Litman, T.; Fredholm, M.; Cirera, S. MicroRNA expression profiling of the porcine developing brain. PLoS ONE 2011, 6, e14494. [Google Scholar] [CrossRef] [PubMed]
- Krichevsky, A.M.; King, K.S.; Donahue, C.P.; Khrapko, K.; Kosik, K.S. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 2003, 9, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Yao, M.J.; Chen, G.; Zhao, P.P.; Lu, M.H.; Jian, J.; Liu, M.F.; Yuan, X.B. Transcriptome analysis of microRNAs in developing cerebral cortex of rat. BMC Genom. 2012, 13, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miska, E.A.; Alvarez-Saavedra, E.; Townsend, M.; Yoshii, A.; Sestan, N.; Rakic, P.; Constantine-Paton, M.; Horvitz, H.R. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004, 5, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eda, A.; Takahashi, M.; Fukushima, T.; Hohjoh, H. Alteration of microRNA expression in the process of mouse brain growth. Gene 2011, 485, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Fertuzinhos, S.; Li, M.; Kawasawa, Y.I.; Ivic, V.; Franjic, D.; Singh, D.; Crair, M.; Sestan, N. Laminar and temporal expression dynamics of coding and noncoding RNAs in the mouse neocortex. Cell Rep. 2014, 6, 938–950. [Google Scholar] [CrossRef] [Green Version]
- Swahari, V.; Nakamura, A.; Hollville, E.; Stroud, H.; Simon, J.M.; Ptacek, T.S.; Beck, M.V.; Flowers, C.; Guo, J.; Plestant, C.; et al. MicroRNA-29 is an essential regulator of brain maturation through regulation of CH methylation. Cell Rep. 2021, 35, 108946. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S. The microRNA registry. Nucleic Acids Res. 2004, 32, 109–111. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, 140–144. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; Van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, 154–158. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, 152–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Magee, R.; Telonis, A.G.; Cherlin, T.; Rigoutsos, I.; Londin, E. Assessment of isomiR discrimination using commercial qPCR methods. Non-Coding RNA 2017, 3, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasello, L.; Distefano, R.; Nigita, G.; Croce, C.M. The MicroRNA Family Gets Wider: The IsomiRs Classification and Role. Front. Cell Dev. Biol. 2021, 9, 1–15. [Google Scholar] [CrossRef]
- Llorens, F.; Bañez-Coronel, M.; Pantano, L.; del Río, J.A.; Ferrer, I.; Estivill, X.; Martí, E. A highly expressed miR-101 isomiR is a functional silencing small RNA. BMC Genom. 2013, 14, 104. [Google Scholar] [CrossRef] [Green Version]
- Van der Kwast, R.V.C.T.; Woudenberg, T.; Quax, P.H.A.; Nossent, A.Y. MicroRNA-411 and Its 5′-IsomiR Have Distinct Targets and Functions and Are Differentially Regulated in the Vasculature under Ischemia. Mol. Ther. 2019, 28, 157–170. [Google Scholar] [CrossRef]
- Van der Kwast, R.V.C.T.; Parma, L.; van der Bent, M.L.; van Ingen, E.; Baganha, F.; Peters, H.A.B.; Goossens, E.A.C.; Simons, K.H.; Palmen, M.; de Vries, M.R.; et al. Adenosine-to-Inosine Editing of Vasoactive MicroRNAs Alters Their Targetome and Function in Ischemia. Mol. Ther. Nucleic Acids 2020, 21, 932–953. [Google Scholar] [CrossRef]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu-Gruttadauria, J.; Xiao, Y.; Gebert, L.F.; MacRae, I.J. Beyond the seed: Structural basis for supplementary micro RNA targeting by human Argonaute2. EMBO J. 2019, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Martí, E.; Pantano, L.; Bañez-Coronel, M.; Llorens, F.; Miñones-Moyano, E.; Porta, S.; Sumoy, L.; Ferrer, I.; Estivill, X. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010, 38, 7219–7235. [Google Scholar] [CrossRef] [PubMed]
- Wee, L.M.; Flores-Jasso, C.F.; Salomon, W.E.; Zamore, P.D. Argonaute divides Its RNA guide into domains with distinct functions and RNA-binding properties. Cell 2012, 151, 1055–1067. [Google Scholar] [CrossRef] [Green Version]
- Ameres, S.L.; Horwich, M.D.; Hung, J.-H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA-Directed Trimming and Tailing of Small Silencing RNAs. Science 2010, 328, 1534–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bail, S.; Swerdel, M.; Liu, H.; Jiao, X.; Goff, L.A.; Hart, R.P.; Kiledjian, M. Differential regulation of microRNA stability. Rna 2010, 16, 1032–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Mata, M.; Gaidatzis, D.; Vitanescu, M.; Stadler, M.B.; Wentzel, C.; Scheiffele, P.; Filipowicz, W.; Großhans, H. Potent degradation of neuronal miRNAs induced by highly complementary targets. EMBO Rep. 2015, 16, 500–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingston, E.R.; Bartel, D.P. Global analyses of the dynamics of mammalian microRNA metabolism. Genome Res. 2019, 29, 1777–1790. [Google Scholar] [CrossRef]
- Juvvuna, P.K.; Khandelia, P.; Lee, L.M.; Makeyev, E.V. Argonaute identity defines the length of mature mammalian microRNAs. Nucleic Acids Res. 2012, 40, 6808–6820. [Google Scholar] [CrossRef]
- Nowakowski, T.J.; Rani, N.; Golkaram, M.; Zhou, H.R.; Alvarado, B.; Huch, K.; West, J.A.; Leyrat, A.; Pollen, A.A.; Kriegstein, A.R.; et al. Regulation of cell-type-specific transcriptomes by microRNA networks during human brain development. Nat. Neurosci. 2018, 21, 1784–1792. [Google Scholar] [CrossRef]
- Boudreau, R.L.; Jiang, P.; Gilmore, B.L.; Spengler, R.M.; Tirabassi, R.; Nelson, J.A.; Ross, C.A.; Xing, Y.; Davidson, B.L. Transcriptome-wide discovery of microRNA binding sites in Human Brain. Neuron 2014, 81, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Sangiao-Alvarellos, S.; Manfredi-Lozano, M.; Ruiz-Pino, F.; Navarro, V.M.; Sánchez-Garrido, M.A.; Leon, S.; Dieguez, C.; Cordido, F.; Matagne, V.; Dissen, G.A.; et al. Changes in hypothalamic expression of the Lin28/let-7 system and related MicroRNAs during postnatal maturation and after experimental manipulations of puberty. Endocrinology 2013, 154, 942–955. [Google Scholar] [CrossRef]
- Tognini, P.; Putignano, E.; Coatti, A.; Pizzorusso, T. Experience-dependent expression of miR-132 regulates ocular dominance plasticity. Nat. Neurosci. 2011, 14, 1237–1239. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Mao, S.; Wang, H.; Zen, K.; Zhang, C.; Li, L. MicroRNA-29a modulates axon branching by targeting doublecortin in primary neurons. Protein Cell 2014, 5, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazziotti, R.; Baroncelli, L.; Ceglia, N.; Chelini, G.; Sala, G.D.; Magnan, C.; Napoli, D.; Putignano, E.; Silingardi, D.; Tola, J.; et al. Mir-132/212 is required for maturation of binocular matching of orientation preference and depth perception. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xiao, J.; Fan, Y.; Yang, K.; Li, K.; Wang, X.; Lu, Y.; Zhou, Y. MiR-29 family regulates the puberty onset mediated by a novel Gnrh1 transcription factor TBX21. J. Endocrinol. 2019, 242, 185–197. [Google Scholar] [CrossRef]
- Napoli, D.; Lupori, L.; Mazziotti, R.; Sagona, G.; Bagnoli, S.; Samad, M.; Sacramento, E.K.; Kirkpartick, J.; Putignano, E.; Chen, S.; et al. MiR-29 coordinates age-dependent plasticity brakes in the adult visual cortex. EMBO Rep. 2020, 21, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Fando, J.L.; Salinas, M.; Wasterlain, C.G. Age-dependent changes in brain protein synthesis in the rat. Neurochem. Res. 1980, 5, 373–383. [Google Scholar] [CrossRef]
- Hovda, D.A.; Villablanca, J.R.; Chugani, H.T.; Barrio, J.R. Metabolic maturation of the brain: A study of local cerebral protein synthesis in the developing cat. Brain Res. 2006, 1113, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Deibler, G.E.; Jehle, J.; Macedonia, J.; Dumont, I.; Dang, T.; Smith, C.B. Rates of local cerebral protein synthesis in the rat during normal postnatal development. Am. J. Physiol. 1995, 268, R549–R561. [Google Scholar] [CrossRef]
- Vargas, R.; Castañeda, M. Age-dependent decrease in the activity of protein-synthesis initiation factors in rat brain. Mech. Ageing Dev. 1983, 21, 183–191. [Google Scholar] [CrossRef]
- Qin, M.; Kang, J.; Burlin, T.V.; Jiang, C.; Smith, C.B. Postadolescent changes in regional cerebral protein synthesis: An in vivo study in the FMR1 null mouse. J. Neurosci. 2005, 25, 5087–5095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayola, R.; Condorelli, D.F.; Ragusa, N.; Renis, M.; Alberghina, M.; Stella, A.M.G.; Lajtha, A. Protein synthesis rates in rat brain regions and subcellular fractions during aging. Neurochem. Res. 1988, 13, 337–342. [Google Scholar] [CrossRef] [PubMed]
- English, J.A.; Fan, Y.; Föcking, M.; Lopez, L.M.; Hryniewiecka, M.; Wynne, K.; Dicker, P.; Matigian, N.; Cagney, G.; Mackay-Sim, A.; et al. Reduced protein synthesis in schizophrenia patient-derived olfactory cells. Transl. Psychiatry 2015, 5, e663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arioka, Y.; Shishido, E.; Kushima, I.; Suzuki, T.; Saito, R.; Aiba, A.; Mori, D.; Ozaki, N. Chromosome 22q11.2 deletion causes PERK-dependent vulnerability in dopaminergic neurons. EBioMedicine 2021, 63, 103138. [Google Scholar] [CrossRef]
- Wei, Y.N.; Hu, H.Y.; Xie, G.C.; Fu, N.; Ning, Z.B.; Zeng, R.; Khaitovich, P. Transcript and protein expression decoupling reveals RNA binding proteins and miRNAs as potential modulators of human aging. Genome Biol. 2015, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Breen, M.S.; Ozcan, S.; Ramsey, J.M.; Wang, Z.; Ma’ayan, A.; Rustogi, N.; Gottschalk, M.G.; Webster, M.J.; Weickert, C.S.; Buxbaum, J.D.; et al. Temporal proteomic profiling of postnatal human cortical development. Transl. Psychiatry 2018, 8, 267. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Messina, A.; Langlet, F.; Chachlaki, K.; Roa, J.; Rasika, S.; Jouy, N.; Gallet, S.; Gaytan, F.; Parkash, J.; Tena-Sempere, M.; et al. A microRNA switch regulates the rise in hypothalamic GnRH production before puberty. Nat. Neurosci. 2016, 19, 835–844. [Google Scholar] [CrossRef]
- Herting, M.M.; Sowell, E.R. Puberty and structural brain development in humans. Front. Neuroendocrinol. 2017, 44, 122–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juraska, J.M.; Sisk, C.L.; DonCarlos, L.L. Sexual differentiation of the adolescent rodent brain: Hormonal influences and developmental mechanisms. Horm. Behav. 2013, 64, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.L.; Thompson, A.P.; Krenzel, E.; Teicher, M.H. Pubertal changes in gonadal hormones do not underlie adolescent dopamine receptor overproduction. Psychoneuroendocrinology 2002, 27, 683–691. [Google Scholar] [CrossRef]
- Vetter-O’Hagen, C.S.; Spear, L.P. Hormonal and physical markers of puberty and their relationship to adolescent-typical novelty-directed behavior. Dev. Psychobiol. 2012, 54, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.J.; Probst, C.K.; Brown, L.M.; de Vries, G.J. Dissociation of Puberty and Adolescent Social Development in a Seasonally Breeding Species. Curr. Biol. 2018, 28, 1116–1123.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef] [Green Version]
- Bédard, A.; Parent, A. Evidence of newly generated neurons in the human olfactory bulb. Brain Res. Dev. Brain Res. 2004, 151, 159–168. [Google Scholar] [CrossRef]
- Wang, C.; Liu, F.; Liu, Y.-Y.; Zhao, C.-H.; You, Y.; Wang, L.; Zhang, J.; Wei, B.; Ma, T.; Zhang, Q.; et al. Identification and characterization of neuroblasts in the subventricular zone and rostral migratory stream of the adult human brain. Cell Res. 2011, 21, 1534–1550. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Liebl, J.; Bernard, S.; Alkass, K.; Yeung, M.S.Y.; Steier, P.; Kutschera, W.; Johnson, L.; Landén, M.; Druid, H.; et al. The age of olfactory bulb neurons in humans. Neuron 2012, 74, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Ernst, A.; Alkass, K.; Bernard, S.; Salehpour, M.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; Frisén, J. Neurogenesis in the striatum of the adult human brain. Cell 2014, 156, 1072–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, O.; Spalding, K.L.; Frisén, J. Adult neurogenesis in humans. Cold Spring Harb. Perspect. Med. 2015, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scolding, N.; Franklin, R.; Stevens, S.; Heldin, C.H.; Compston, A.; Newcombe, J. Oligodendrocyte progenitors are present in the normal adult human CNS and in the lesions of multiple sclerosis. Brain 1998, 121, 2221–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, M.S.Y.; Zdunek, S.; Bergmann, O.; Bernard, S.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Brundin, L.; et al. Dynamics of Oligodendrocyte Generation and Myelination in the Human Brain. Cell 2014, 159, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, R.D.; Curtis, M.A.; Spalding, K.L.; Buchholz, B.A.; Fink, D.; Björk-Eriksson, T.; Nordborg, C.; Gage, F.H.; Druid, H.; Eriksson, P.S.; et al. Neocortical neurogenesis in humans is restricted to development. Proc. Natl. Acad. Sci. USA 2006, 103, 12564–12568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, M.C.; Roy, N.S.; Keyoung, H.M.; Goodman, R.R.; McKhann, G.; Jiang, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003, 9, 439–447. [Google Scholar] [CrossRef]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21 st century. J. Psychopharmacol. 2015, 29, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Nord, M.; Farde, L. Antipsychotic Occupancy of Dopamine Receptors in Schizophrenia. CNS Neurosci. Ther. 2011, 17, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Tseng, K.Y.; O’Donnell, P. Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb. Cortex 2007, 17, 1235–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, K.-Y.; Lewis, B.L.; Lipska, B.K.; O’Donnell, P. Post-pubertal disruption of medial prefrontal cortical dopamine-glutamate interactions in a developmental animal model of schizophrenia. Biol. Psychiatry 2007, 62, 730–738. [Google Scholar] [CrossRef] [Green Version]
- Goldman-Rakic, P.S.; Brown, R.M. Postnatal development of monoamine content and synthesis in the cerebral cortex of rhesus monkeys. Dev. Brain Res. 1982, 4, 339–349. [Google Scholar] [CrossRef]
- Rosenberg, D.R.; Lewis, D.A. Changes in the dopaminergic innervation of monkey prefrontal cortex during late postnatal development: A tyrosine hydroxylase immunohistochemical study. Biol. Psychiatry 1994, 36, 272–277. [Google Scholar] [CrossRef]
- Reynolds, L.M.; Pokinko, M.; Torres-Berrío, A.; Cuesta, S.; Lambert, L.C.; Del Cid Pellitero, E.; Wodzinski, M.; Manitt, C.; Krimpenfort, P.; Kolb, B.; et al. DCC Receptors Drive Prefrontal Cortex Maturation by Determining Dopamine Axon Targeting in Adolescence. Biol. Psychiatry 2018, 83, 181–192. [Google Scholar] [CrossRef]
- Torres-Berrío, A.; Lopez, J.P.; Bagot, R.C.; Nouel, D.; Dal Bo, G.; Cuesta, S.; Zhu, L.; Manitt, C.; Eng, C.; Cooper, H.M.; et al. DCC Confers Susceptibility to Depression-like Behaviors in Humans and Mice and Is Regulated by miR-218. Biol. Psychiatry 2017, 81, 306–315. [Google Scholar] [CrossRef] [Green Version]
- Chun, S.; Du, F.; Westmoreland, J.J.; Han, S.B.; Wang, Y.-D.; Eddins, D.; Bayazitov, I.T.; Devaraju, P.; Yu, J.; Mellado Lagarde, M.M.; et al. Thalamic miR-338-3p mediates auditory thalamocortical disruption and its late onset in 22q11.2 microdeletion models. Nat. Med. 2016, 23, 1–39. [Google Scholar] [CrossRef]
- Eom, T.Y.; Han, S.B.; Kim, J.; Blundon, J.A.; Wang, Y.D.; Yu, J.; Anderson, K.; Kaminski, D.B.; Sakurada, S.M.; Pruett-Miller, S.M.; et al. Schizophrenia-related microdeletion causes defective ciliary motility and brain ventricle enlargement via microRNA-dependent mechanisms in mice. Nat. Commun. 2020, 11, 912. [Google Scholar] [CrossRef]
- Jia, X.; Wang, F.; Han, Y.; Geng, X.; Li, M.; Shi, Y.; Lu, L.; Chen, Y. miR-137 and miR-491 Negatively Regulate Dopamine Transporter Expression and Function in Neural Cells. Neurosci. Bull. 2016, 32, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Harris, L.W.; Lockstone, H.E.; Khaitovich, P.; Weickert, C.S.; Webster, M.J.; Bahn, S. Gene expression in the prefrontal cortex during adolescence: Implications for the onset of schizophrenia. BMC Med. Genomics 2009, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, N.; Das, S.; Kar Mahapatra, S.; Chakraborty, S.P.; Kundu, P.K.; Roy, S. Age associated oxidative damage in lymphocytes. Oxid. Med. Cell. Longev. 2010, 3, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef]
- De-Souza-Ferreira, E.; Rios-Neto, I.M.; Martins, E.L.; Galina, A. Mitochondria-coupled glucose phosphorylation develops after birth to modulate H2O2 release and calcium handling in rat brain. J. Neurochem. 2019, 149, 624–640. [Google Scholar] [CrossRef]
- Thorburne, S.K.; Juurlink, B.H. Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. J. Neurochem. 1996, 67, 1014–1022. [Google Scholar] [CrossRef]
- Back, S.A.; Gan, X.; Li, Y.; Rosenberg, P.A.; Volpe, J.J. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J. Neurosci. 1998, 18, 6241–6253. [Google Scholar] [CrossRef] [PubMed]
- Monin, A.; Baumann, P.S.; Griffa, A.; Xin, L.; Mekle, R.; Fournier, M.; Butticaz, C.; Klaey, M.; Cabungcal, J.H.; Steullet, P.; et al. Glutathione deficit impairs myelin maturation: Relevance for white matter integrity in schizophrenia patients. Mol. Psychiatry 2015, 20, 827–838. [Google Scholar] [CrossRef] [Green Version]
- Kann, O. The interneuron energy hypothesis: Implications for brain disease. Neurobiol. Dis. 2016, 90, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Papageorgiou, I.E.; Draguhn, A. Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J. Cereb. Blood Flow Metab. 2014, 34, 1270–1282. [Google Scholar] [CrossRef] [Green Version]
- Sowell, E.R.; Thompson, P.M.; Holmes, C.J.; Jernigan, T.L.; Toga, A.W. In vivo evidence for post-adolescent brain maturation in frontal and striatal regions. Nat. Neurosci. 1999, 2, 859–861. [Google Scholar] [CrossRef] [PubMed]
- Lebel, C.; Walker, L.; Leemans, A.; Phillips, L.; Beaulieu, C. Microstructural maturation of the human brain from childhood to adulthood. Neuroimage 2008, 40, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev PL, L.A. The myelogenetic cycles of regional maturation of the brain. In Regional Development of the Brain in Early Life; Minkowski, A., Ed.; Blackwell Scientific: Oxford, UK, 1967; pp. 3–70. [Google Scholar]
- Benes, F.M. Myelination of cortical-hippocampal relays during late adolescence. Schizophr. Bull. 1989, 15, 585–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, D.A.; Moore, C.S. MiRNAs as emerging regulators of oligodendrocyte development and differentiation. Front. Cell Dev. Biol. 2016, 4, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Silveri, M.M.; Sneider, J.T.; Crowley, D.J.; Covell, M.J.; Acharya, D.; Rosso, I.M.; Jensen, J.E. Frontal lobe γ-aminobutyric acid levels during adolescence: Associations with impulsivity and response inhibition. Biol. Psychiatry 2013, 74, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoftman, G.D.; Volk, D.W.; Bazmi, H.H.; Li, S.; Sampson, A.R.; Lewis, D.A. Altered cortical expression of GABA-related genes in schizophrenia: Illness progression vs developmental disturbance. Schizophr. Bull. 2015, 41, 180–191. [Google Scholar] [CrossRef]
- Cruz, D.A.; Eggan, S.M.; Lewis, D.A. Postnatal development of pre- and postsynaptic GABA markers at chandelier cell connections with pyramidal neurons in monkey prefrontal cortex. J. Comp. Neurol. 2003, 465, 385–400. [Google Scholar] [CrossRef]
- Datta, D.; Arion, D.; Lewis, D.A. Developmental Expression Patterns of GABAA Receptor Subunits in Layer 3 and 5 Pyramidal Cells of Monkey Prefrontal Cortex. Cereb. Cortex 2015, 25, 2295–2305. [Google Scholar] [CrossRef] [Green Version]
- Fung, S.J.; Webster, M.J.; Sivagnanasundaram, S.; Duncan, C.; Elashoff, M.; Weickert, C.S. Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am. J. Psychiatry 2010, 167, 1479–1488. [Google Scholar] [CrossRef]
- Hoftman, G.D.; Lewis, D.A. Postnatal developmental trajectories of neural circuits in the primate prefrontal cortex: Identifying sensitive periods for vulnerability to schizophrenia. Schizophr. Bull. 2011, 37, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, S.J.; Lewis, D.A. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol. Dis. 2019, 131, 104208. [Google Scholar] [CrossRef] [PubMed]
- Schür, R.R.; Draisma, L.W.R.; Wijnen, J.P.; Boks, M.P.; Koevoets, M.G.J.C.; Joëls, M.; Klomp, D.W.; Kahn, R.S.; Vinkers, C.H. Brain GABA levels across psychiatric disorders: A systematic literature review and meta-analysis of 1H-MRS studies. Hum. Brain Mapp. 2016, 37, 3337–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egerton, A.; Modinos, G.; Ferrera, D.; McGuire, P. Neuroimaging studies of GABA in schizophrenia: A systematic review with meta-analysis. Transl. Psychiatry 2017, 7, e1147-10. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Vajawat, B.; Rao, N.P. Frontal GABA in schizophrenia: A meta-analysis of 1H-MRS studies. World J. Biol. Psychiatry 2021, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Fernandes, C.C.; Ewell, L.A.; John, D.; Romoli, B.; Curia, G.; Taylor, S.R.; Frady, E.P.; Jensen, A.B.; Liu, J.C.; et al. MicroRNA-101 Regulates Multiple Developmental Programs to Constrain Excitation in Adult Neural Networks. Neuron 2016, 92, 1337–1351. [Google Scholar] [CrossRef] [Green Version]
- Huttenlocher, P.R.; Dabholkar, A.S. Regional differences in synaptogenesis in human cerebral cortex. J. Comp. Neurol. 1997, 387, 167–178. [Google Scholar] [CrossRef]
- Huttenlocher, P.R. Synaptic density in human frontal cortex—Developmental changes and effects of aging. Brain Res. 1979, 163, 195–205. [Google Scholar] [CrossRef]
- Petanjek, Z.; Judaš, M.; Šimić, G.; Rašin, M.R.; Uylings, H.B.M.; Rakic, P.; Kostović, I. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc. Natl. Acad. Sci. USA 2011, 108, 13281–13286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallya, A.P.; Wang, H.D.; Lee, H.N.R.; Deutch, A.Y. Microglial Pruning of Synapses in the Prefrontal Cortex during Adolescence. Cereb. Cortex 2019, 29, 1634–1643. [Google Scholar] [CrossRef]
- Koss, W.A.; Belden, C.E.; Hristov, A.D.; Juraska, J.M. Dendritic remodeling in the adolescent medial prefrontal cortex and the basolateral amygdala of male and female rats. Synapse 2014, 68, 61–72. [Google Scholar] [CrossRef]
- Johnson, C.M.; Loucks, F.A.; Peckler, H.; Thomas, A.W.; Janak, P.H.; Wilbrecht, L. Long-range orbitofrontal and amygdala axons show divergent patterns of maturation in the frontal cortex across adolescence. Dev. Cogn. Neurosci. 2016, 18, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Zheutlin, A.B.; Jeffries, C.D.; Perkins, D.O.; Chung, Y.; Chekroud, A.M.; Addington, J.; Bearden, C.E.; Cadenhead, K.S.; Cornblatt, B.A.; Mathalon, D.H.; et al. The Role of microRNA Expression in Cortical Development during Conversion to Psychosis. Neuropsychopharmacology 2017, 42, 2188–2195. [Google Scholar] [CrossRef] [Green Version]
- Drzewiecki, C.M.; Willing, J.; Juraska, J.M. Synaptic number changes in the medial prefrontal cortex across adolescence in male and female rats: A role for pubertal onset. Synapse 2016, 70, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, I. Schizophrenia: Caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 1982, 17, 319–334. [Google Scholar] [CrossRef]
- Valiathan, R.; Ashman, M.; Asthana, D. Effects of Ageing on the Immune System: Infants to Elderly. Scand. J. Immunol. 2016, 83, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Tollerud, D.J.; Ildstad, S.T.; Brown, L.M.; Clark, J.W.; Blattner, W.A.; Mann, D.L.; Neuland, C.Y.; Pankiw-Trost, L.; Hoover, R.N. T-cell subsets in healthy teenagers: Transition to the adult phenotype. Clin. Immunol. Immunopathol. 1990, 56, 88–96. [Google Scholar] [CrossRef]
- Aldrimer, M.; Ridefelt, P.; Rödöö, P.; Niklasson, F.; Gustafsson, J.; Hellberg, D. Population-based pediatric reference intervals for hematology, iron and transferrin. Scand. J. Clin. Lab. Invest. 2013, 73, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Dalferes, E.R.J.; Srinivasan, S.R.; Webber, L.S.; Berenson, G.S. Normative distribution of complete blood count from early childhood through adolescence: The Bogalusa Heart Study. Prev. Med. 1993, 22, 825–837. [Google Scholar] [CrossRef]
- Simon, A.K.; Hollander, G.A.; McMichael, A. Evolution of the immune system in humans from infancy to old age. Proc. Biol. Sci. 2015, 282, 20143085. [Google Scholar] [CrossRef]
- Brenhouse, H.C.; Schwarz, J.M. Immunoadolescence: Neuroimmune development and adolescent behavior. Neurosci. Biobehav. Rev. 2016, 70, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Levinson, D.F.; Duan, J.; Sanders, A.R.; Zheng, Y.; Pe’er, I.; Dudbridge, F.; Holmans, P.A.; Whittemore, A.S.; Mowry, B.J.; et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009, 460, 753–757. [Google Scholar] [CrossRef]
- Stefansson, H.; Ophoff, R.A.; Steinberg, S.; Andreassen, O.A.; Cichon, S.; Rujescu, D.; Werge, T.; Pietiläinen, O.P.H.; Mors, O.; Mortensen, P.B.; et al. Common variants conferring risk of schizophrenia. Nature 2009, 460, 744–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, A.; Bialas, A.R.; De Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Li, R.; McGeer, E.G.; McGeer, P.L. Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res. 1997, 769, 391–395. [Google Scholar] [CrossRef]
- Veerhuis, R.; Nielsen, H.M.; Tenner, A.J. Complement in the brain. Mol. Immunol. 2011, 48, 1592–1603. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.J.; Goldsmith, D.R. Inflammatory biomarkers in schizophrenia: Implications for heterogeneity and neurobiology. Biomark. Neuropsychiatry 2019, 1, 100006. [Google Scholar] [CrossRef]
- Howes, O.D.; McCutcheon, R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: A reconceptualization. Transl. Psychiatry 2017, 7, e1024-11. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, S.M.; Krüger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An unconventional role for miRNA: Let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Barak, B.; Feldman, N.; Okun, E. Toll-like receptors as developmental tools that regulate neurogenesis during development: An update. Front. Neurosci. 2014, 8, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolls, A.; Shechter, R.; London, A.; Ziv, Y.; Ronen, A.; Levy, R.; Schwartz, M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat. Cell Biol. 2007, 9, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Okun, E.; Griffioen, K.; Barak, B.; Roberts, N.J.; Castro, K.; Pita, M.A.; Cheng, A.; Mughal, M.R.; Wan, R.; Ashery, U.; et al. Toll-like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15625–15630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Y.; Lin, C.W.; Chang, C.Y.; Jian, S.T.; Hsueh, Y.P. Sarm1, a negative regulator of innate immunity, interacts with syndecan-2 and regulates neuronal morphology. J. Cell Biol. 2011, 193, 769–784. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lambris, J.D. More than complementing Tolls: Complement-Toll-like receptor synergy and crosstalk in innate immunity and inflammation. Immunol. Rev. 2016, 274, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Woods, S.W.; Addington, J.; Cadenhead, K.S.; Cannon, T.D.; Cornblatt, B.A.; Heinssen, R.; Perkins, D.O.; Seidman, L.J.; Tsuang, M.T.; Walker, E.F.; et al. Validity of the Prodromal Risk Syndrome for First Psychosis: Findings From the North American Prodrome Longitudinal Study. Schizophr. Bull. 2009, 35, 894–908. [Google Scholar] [CrossRef] [Green Version]
- Cannon, T.D.; Cadenhead, K.; Cornblatt, B.; Woods, S.W.; Addington, J.; Walker, E.; Seidman, L.J.; Perkins, D.; Tsuang, M.; McGlashan, T.; et al. Prediction of psychosis in youth at high clinical risk: A multisite longitudinal study in North America. Arch. Gen. Psychiatry 2008, 65, 28–37. [Google Scholar] [CrossRef]
- Fusar-Poli, P.; Bonoldi, I.; Yung, A.R.; Borgwardt, S.; Kempton, M.J.; Valmaggia, L.; Barale, F.; Caverzasi, E.; McGuire, P. Predicting psychosis: Meta-analysis of transition outcomes in individuals at high clinical risk. Arch. Gen. Psychiatry 2012, 69, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Yung, A.R.; Nelson, B.; Stanford, C.; Simmons, M.B.; Cosgrave, E.M.; Killackey, E.; Phillips, L.J.; Bechdolf, A.; Buckby, J.; McGorry, P.D. Validation of “prodromal” criteria to detect individuals at ultra high risk of psychosis: 2 year follow-up. Schizophr. Res. 2008, 105, 10–17. [Google Scholar] [CrossRef]
- Ziermans, T.B.; Schothorst, P.F.; Sprong, M.; van Engeland, H. Transition and remission in adolescents at ultra-high risk for psychosis. Schizophr. Res. 2011, 126, 58–64. [Google Scholar] [CrossRef]
- Fusar-Poli, P.; Borgwardt, S.; Bechdolf, A.; Addington, J.; Riecher-Rössler, A.; Schultze-Lutter, F.; Keshavan, M.; Wood, S.; Ruhrmann, S.; Seidman, L.J.; et al. The psychosis high-risk state: A comprehensive state-of-the-art review. Arch. Gen. Psychiatry 2013, 70, 107–120. [Google Scholar] [CrossRef]
- Lee, T.Y.; Lee, J.; Kim, M.; Choe, E.; Kwon, J.S. Can we predict psychosis outside the clinical high-risk state? A systematic review of non-psychotic risk syndromes for mental disorders. Schizophr. Bull. 2018, 44, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Addington, J.; Cadenhead, K.S.; Cornblatt, B.A.; Mathalon, D.H.; McGlashan, T.H.; Perkins, D.O.; Seidman, L.J.; Tsuang, M.T.; Walker, E.F.; Woods, S.W.; et al. North American Prodrome Longitudinal Study (NAPLS 2): Overview and recruitment. Schizophr. Res. 2012, 142, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Addington, J.; Liu, L.; Buchy, L.; Cadenhead, K.S.; Cannon, T.D.; Cornblatt, B.A.; Perkins, D.O.; Seidman, L.J.; Tsuang, M.T.; Walker, E.F.; et al. North American Prodrome Longitudinal Study (NAPLS 2): The Prodromal Symptoms. J. Nerv. Ment. Dis. 2015, 203, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Jeffries, C.D.; Perkins, D.O.; Chandler, S.D.; Stark, T.; Yeo, E.; Addington, J.; Bearden, C.E.; Cadenhead, K.S.; Cannon, T.D.; Cornblatt, B.A.; et al. Insights into psychosis risk from leukocyte microRNA expression. Transl. Psychiatry 2016, 6, e981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, T.D.; Chung, Y.; He, G.; Sun, D.; Jacobson, A.; Van Erp, T.G.M.; McEwen, S.; Addington, J.; Bearden, C.E.; Cadenhead, K.; et al. Progressive reduction in cortical thickness as psychosis develops: A multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol. Psychiatry 2015, 77, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, W.H.; Kim, J.S.; Jang, J.H.; Choi, J.S.; Jung, M.H.; Park, J.Y.; Han, J.Y.; Choi, C.H.; Kang, D.H.; Chung, C.K.; et al. Cortical thickness reduction in individuals at ultra-high-risk for psychosis. Schizophr. Bull. 2011, 37, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Tamnes, C.K.; Herting, M.M.; Goddings, A.L.; Meuwese, R.; Blakemore, S.J.; Dahl, R.E.; Güroğlu, B.; Raznahan, A.; Sowell, E.R.; Crone, E.A.; et al. Development of the cerebral cortex across adolescence: A multisample study of inter-related longitudinal changes in cortical volume, surface area, and thickness. J. Neurosci. 2017, 37, 3402–3412. [Google Scholar] [CrossRef]
- Tamnes, C.K.; Østby, Y.; Fjell, A.M.; Westlye, L.T.; Due-Tønnessen, P.; Walhovd, K.B. Brain maturation in adolescence and young adulthood: Regional age-related changes in cortical thickness and white matter volume and microstructure. Cereb. Cortex 2010, 20, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, R.M.; Rybka, J.; Anderson, S.M.; Torrance, H.S.; Boxall, R.; Sussmann, J.E.; Porteous, D.J.; McIntosh, A.M.; Evans, K.L. Preliminary investigation of miRNA expression in individuals at high familial risk of bipolar disorder. J. Psychiatr. Res. 2015, 62, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Rasic, D.; Hajek, T.; Alda, M.; Uher, R. Risk of mental illness in offspring of parents with schizophrenia, bipolar disorder, and major depressive disorder: A meta-analysis of family high-risk studies. Schizophr. Bull. 2014, 40, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenstein, P.; Yip, B.H.; Björk, C.; Pawitan, Y.; Cannon, T.D.; Sullivan, P.F.; Hultman, C.M. Common genetic influences for schizophrenia and bipolar disorder: A population-based study of 2 million nuclear families. Lancet 2009, 373, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Jinde, S.; Koike, S.; Tada, M.; Satomura, Y.; Yoshikawa, A.; Nishimura, Y.; Takizawa, R.; Kinoshita, A.; Sakakibara, E.; et al. Altered expression of microRNA-223 in the plasma of patients with first-episode schizophrenia and its possible relation to neuronal migration-related genes. Transl. Psychiatry 2019, 9, 289. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, E.; Beveridge, N.J.; Wu, J.Q.; Carr, V.; Scott, R.J.; Tooney, P.A.; Cairns, M.J. Imprinted DLK1-DIO3 region of 14q32 defines a schizophrenia-associated miRNA signature in peripheral blood mononuclear cells. Mol. Psychiatry 2012, 17, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Lackinger, M.; Sungur, A.Ö.; Daswani, R.; Soutschek, M.; Bicker, S.; Stemmler, L.; Wüst, T.; Fiore, R.; Dieterich, C.; Schwarting, R.K.; et al. A placental mammal-specific micro RNA cluster acts as a natural brake for sociability in mice. EMBO Rep. 2019, 20, 1–11. [Google Scholar] [CrossRef]
- Fiore, R.; Khudayberdiev, S.; Christensen, M.; Siegel, G.; Flavell, S.W.; Kim, T.-K.; Greenberg, M.E.; Schratt, G.; Bagni, C.; Greenough, W.; et al. Mef2-mediated transcription of the miR379–410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 2009, 28, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.E.; Lee, P.R.; Chen, S.; Li, W.; Fields, R.D. MicroRNA regulation of homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 11650–11655. [Google Scholar] [CrossRef] [Green Version]
- Marty, V.; Labialle, S.; Bortolin-Cavaillé, M.L.; De Medeiros, G.F.; Moisan, M.P.; Florian, C.; Cavaillé, J. Deletion of the miR-379/miR-410 gene cluster at the imprinted Dlk1-Dio3 locus enhances anxiety-related behaviour. Hum. Mol. Genet. 2016, 25, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, F.; Wang, X.; Shugart, Y.Y.; Zhao, Y.; Li, X.; Liu, Z.; Sun, N.; Yang, C.; Zhang, K.; et al. Diagnostic value of blood-derived microRNAs for schizophrenia: Results of a meta-analysis and validation. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Chen, B.-Y.; Lin, J.-J.; Lu, M.-K.; Tan, H.-P.; Jang, F.-L.; Lin, S.-H. Neurodevelopment regulators miR-137 and miR-34 family as biomarkers for early and adult onset schizophrenia. npj Schizophr. 2021, 7, 35. [Google Scholar] [CrossRef]
- Gruzdev, S.K.; Yakovlev, A.A.; Druzhkova, T.A.; Guekht, A.B.; Gulyaeva, N.V. The Missing Link: How Exosomes and miRNAs can Help in Bridging Psychiatry and Molecular Biology in the Context of Depression, Bipolar Disorder and Schizophrenia. Cell. Mol. Neurobiol. 2019, 39, 729–750. [Google Scholar] [CrossRef] [PubMed]
- Khavari, B.; Cairns, M.J. Epigenomic Dysregulation in Schizophrenia: In Search of Disease Etiology and Biomarkers. Cells 2020, 9, 1837. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Baune, B.T.; Schubert, K.O.; Lavoie, S.; Smesny, S.; Rice, S.M.; Schäfer, M.R.; Benninger, F.; Feucht, M.; Klier, C.M.; et al. Prediction of transition from ultra-high risk to first-episode psychosis using a probabilistic model combining history, clinical assessment and fatty-acid biomarkers. Transl. Psychiatry 2016, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Perkins, D.O.; Gu, H.; Boteva, K.; Lieberman, J.A. Relationship between duration of untreated psychosis and outcome in first-episode schizophrenia: A critical review and meta-analysis. Am. J. Psychiatry 2005, 162, 1785–1804. [Google Scholar] [CrossRef]
- Howes, O.D.; Whitehurst, T.; Shatalina, E.; Townsend, L.; Onwordi, E.C.; Mak, T.L.A.; Arumuham, A.; O’Brien, O.; Lobo, M.; Vano, L.; et al. The clinical significance of duration of untreated psychosis: An umbrella review and random-effects meta-analysis. World Psychiatry 2021, 20, 75–95. [Google Scholar] [CrossRef]
- Fusar-Poli, P.; Davies, C.; Solmi, M.; Brondino, N.; De Micheli, A.; Kotlicka-Antczak, M.; Shin, J.I.; Radua, J. Preventive Treatments for Psychosis: Umbrella Review (Just the Evidence). Front. Psychiatry 2019, 10, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitetti, A.; Mallory, A.C.; Golini, E.; Carrieri, C.; Carreño Gutiérrez, H.; Perlas, E.; Pérez-Rico, Y.A.; Tocchini-Valentini, G.P.; Enright, A.J.; Norton, W.H.J.; et al. MicroRNA degradation by a conserved target RNA regulates animal behavior. Nat. Struct. Mol. Biol. 2018, 25, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Qiao, H.; Lu, Z.; Hou, Y. miR-29 promotes the proliferation of cultured rat neural stem/progenitor cells via the PTEN/AKT signaling pathway. Mol. Med. Rep. 2019, 20, 2111–2118. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.Q.; Feng, J.G.; Li, M.; Wang, M.Z.; Liu, L.; Liu, X.; Duan, X.X.; Zhang, C.X.; Wang, X. Bin Prefrontal cortex miR-29b-3p plays a key role in the antidepressant-like effect of ketamine in rats. Exp. Mol. Med. 2018, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripa, R.; Dolfi, L.; Terrigno, M.; Pandolfini, L.; Savino, A.; Arcucci, V.; Groth, M.; Terzibasi Tozzini, E.; Baumgart, M.; Cellerino, A. MicroRNA miR-29 controls a compensatory response to limit neuronal iron accumulation during adult life and aging. BMC Biol. 2017, 15, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshan, R.; Shridhar, S.; Sarangdhar, M.A.; Banik, A.; Chawla, M.; Garg, M.; Singh, V.P.A.L.; Pillai, B. Brain-specific knockdown of miR-29 results in neuronal cell death and ataxia in mice. RNA 2014, 20, 1287–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellios, N.; Sugihara, H.; Castro, J.; Banerjee, A.; Le, C.; Kumar, A.; Crawford, B.; Strathmann, J.; Tropea, D.; Levine, S.S.; et al. miR-132, an experience-dependent microRNA, is essential for visual cortex plasticity. Nat. Neurosci. 2011, 14, 1240–1242. [Google Scholar] [CrossRef] [Green Version]
- Garey, L.J.; Ong, W.Y.; Patel, T.S.; Kanani, M.; Davis, A.; Mortimer, A.M.; Barnes, T.R.E.; Hirsch, S.R. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry 1998, 65, 446–453. [Google Scholar] [CrossRef]
- Glantz, L.A.; Lewis, D.A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 2000, 57, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Sweet, R.A.; Henteleff, R.A.; Zhang, W.; Sampson, A.R.; Lewis, D.A. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology 2009, 34, 374–389. [Google Scholar] [CrossRef] [Green Version]
- Konopaske, G.T.; Lange, N.; Coyle, J.T.; Benes, F.M. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 2014, 71, 1323–1331. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.F.; Sakamoto, K.; Aten, S.; Snider, K.H.; Loeser, J.; Hesse, A.M.; Page, C.E.; Pelz, C.; Simon, J.; Arthur, C.; et al. Targeted deletion of miR-132/-212 impairs memory and alters the hippocampal transcriptome. Learn. Mem. 2016, 23, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.Y.; Phang, R.Z.; Hsu, P.H.; Wang, W.H.; Huang, H.T.; Liu, I.Y. In vivo knockdown of hippocampal miR-132 expression impairs memory acquisition of trace fear conditioning. Hippocampus 2013, 23, 625–633. [Google Scholar] [CrossRef]
- Ronovsky, M.; Zambon, A.; Cicvaric, A.; Boehm, V.; Hoesel, B.; Moser, B.A.; Yang, J.; Schmid, J.A.; Haubensak, W.E.; Monje, F.J.; et al. A role for miR-132 in learned safety. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.J.; Clinton, J.M.; Taishi, P.; Bohnet, S.G.; Honn, K.A.; Krueger, J.M. MicroRNA 132 alters sleep and varies with time in brain. J. Appl. Physiol. 2011, 111, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.Y.M.; Papp, J.W.; Varlamova, O.; Dziema, H.; Russell, B.; Curfman, J.P.; Nakazawa, T.; Shimizu, K.; Okamura, H.; Impey, S.; et al. microRNA Modulation of Circadian-Clock Period and Entrainment. Neuron 2007, 54, 813–829. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Saavedra, M.; Antoun, G.; Yanagiya, A.; Oliva-Hernandez, R.; Cornejo-Palma, D.; Perez-Iratxeta, C.; Sonenberg, N.; Cheng, H.Y.M. miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum. Mol. Genet. 2011, 20, 731–751. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.Y.; Britton, J.C.; Nelson, E.E.; Angold, A.; Ernst, M.; Goldwin, M.; Grillon, C.; Leibenluft, E.; Lissek, S.; Norcross, M.; et al. Distinct neural signatures of threat learning in adolescents and adults. Proc. Natl. Acad. Sci. USA 2011, 108, 4500–4505. [Google Scholar] [CrossRef] [Green Version]
- Pattwell, S.S.; Duhoux, S.; Hartley, C.A.; Johnson, D.C.; Jing, D.; Elliott, M.D.; Ruberry, E.J.; Powers, A.; Mehta, N.; Yang, R.R.; et al. Altered fear learning across development in both mouse and human. Proc. Natl. Acad. Sci. USA 2012, 109, 16318–16323. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.; Theresiana, C.; Neumann, D.; Craske, M. Developmental differences in aversive conditioning, extinction, and reinstatement: A study with children, adolescents, and adults. J. Exp. Child Psychol. 2017, 159, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Ganella, D.E.; Drummond, K.D.; Ganella, E.P.; Whittle, S.; Kim, J.H. Extinction of conditioned fear in adolescents and adults: A human fmri study. Front. Hum. Neurosci. 2018, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagenauer, M.H.; Perryman, J.I.; Lee, T.M.; Carskadon, M.A. Adolescent changes in the homeostatic and circadian regulation of sleep. Dev. Neurosci. 2009, 31, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Crowley, S.J.; Acebo, C.; Carskadon, M.A. Sleep, circadian rhythms, and delayed phase in adolescence. Sleep Med. 2007, 8, 602–612. [Google Scholar] [CrossRef]
- Holt, D.J.; Lebron-Milad, K.; Milad, M.R.; Rauch, S.L.; Pitman, R.K.; Orr, S.P.; Cassidy, B.S.; Walsh, J.P.; Goff, D.C. Extinction Memory Is Impaired in Schizophrenia. Biol. Psychiatry 2009, 65, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Holt, D.J.; Coombs, G.; Zeidan, M.A.; Goff, D.C.; Milad, M.R. Failure of neural responses to safety cues in schizophrenia. Arch. Gen. Psychiatry 2012, 69, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, D.L.; Pan, H.; Weisholtz, D.S.; Root, J.C.; Tuescher, O.; Fischer, D.B.; Butler, T.; Vago, D.R.; Isenberg, N.; Epstein, J.; et al. Altered threat and safety neural processing linked to persecutory delusions in schizophrenia: A two-task fMRI study. Psychiatry Res. Neuroimag. 2015, 233, 352–366. [Google Scholar] [CrossRef] [Green Version]
- Wulff, K.; Dijk, D.J.; Middleton, B.; Foster, R.G.; Joyce, E.M. Sleep and circadian rhythm disruption in schizophrenia. Br. J. Psychiatry 2012, 200, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Hubel, D.H.; Wiesel, T.N. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J. Physiol. 1970, 206, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.C.; Hocking, D.R. Timing of the critical period for plasticity of ocular dominance columns in macaque striate cortex. J. Neurosci. 1997, 17, 3684–3709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, K.; Löwel, S. Age-dependent ocular dominance plasticity in adult mice. PLoS ONE 2008, 3, e3120. [Google Scholar] [CrossRef]
- Issa, N.P.; Trachtenberg, J.T.; Chapman, B.; Zahs, K.R.; Stryker, M.P. The critical period for ocular dominance plasticity in the Ferret’s visual cortex. J. Neurosci. 1999, 19, 6965–6978. [Google Scholar] [CrossRef]
- Pizzorusso, T.; Medini, P.; Berardi, N.; Chierzi, S.; Fawcett, J.W.; Maffei, L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science 2002, 298, 1248–1251. [Google Scholar] [CrossRef] [Green Version]
- Cabungcal, J.H.; Steullet, P.; Morishita, H.; Kraftsik, R.; Cuenod, M.; Hensch, T.K.; Do, K.Q. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 9130–9135. [Google Scholar] [CrossRef] [Green Version]
- Flatow, J.; Buckley, P.; Miller, B.J. Meta-analysis of oxidative stress in schizophrenia. Biol. Psychiatry 2013, 74, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraguas, D.; Díaz-Caneja, C.M.; Rodríguez-Quiroga, A.; Arango, C. Oxidative Stress and Inflammation in Early Onset First Episode Psychosis: A Systematic Review and Meta-Analysis. Int. J. Neuropsychopharmacol. 2017, 20, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Fraguas, D.; Díaz-Caneja, C.M.; Ayora, M.; Hernández-Álvarez, F.; Rodríguez-Quiroga, A.; Recio, S.; Leza, J.C.; Arango, C. Oxidative stress and inflammation in first-episode psychosis: A Systematic Review and Meta-analysis. Schizophr. Bull. 2019, 45, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Javadzadeh, A.; Dey, A.; Sabesan, P.; Théberge, J.; Radua, J.; Palaniyappan, L. Antioxidant defense in schizophrenia and bipolar disorder: A meta-analysis of MRS studies of anterior cingulate glutathione. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 91, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, S.; Noda, Y.; Tarumi, R.; Mimura, Y.; Yoshida, K.; Iwata, Y.; Elsalhy, M.; Kuromiya, M.; Kurose, S.; Masuda, F.; et al. Glutathione levels and activities of glutathione metabolism enzymes in patients with schizophrenia: A systematic review and meta-analysis. J. Psychopharmacol. 2019, 33, 1199–1214. [Google Scholar] [CrossRef]
- Steullet, P.; Cabungcal, J.H.; Coyle, J.; Didriksen, M.; Gill, K.; Grace, A.A.; Hensch, T.K.; Lamantia, A.S.; Lindemann, L.; Maynard, T.M.; et al. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol. Psychiatry 2017, 22, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Chang, H.; Li, E.; Fan, G. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res. 2005, 79, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [Green Version]
- Tognini, P.; Napoli, D.; Tola, J.; Silingardi, D.; Della Ragione, F.; D’esposito, M.; Pizzorusso, T. Experience-dependent DNA methylation regulates plasticity in the developing visual cortex. Nat. Neurosci. 2015, 18, 956–958. [Google Scholar] [CrossRef]
- Zhubi, A.; Veldic, M.; Puri, N.V.; Kadriu, B.; Caruncho, H.; Loza, I.; Sershen, H.; Lajtha, A.; Smith, R.C.; Guidotti, A.; et al. An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophr. Res. 2009, 111, 115–122. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, K.S.; McGregor, N.W.; Emsley, R.; Seedat, S.; Warnich, L. The potential role of regulatory genes (DNMT3A, HDAC5, and HDAC9) in antipsychotic treatment response in South African schizophrenia patients. Front. Genet. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Saradalekshmi, K.R.; Neetha, N.V.; Sathyan, S.; Nair, I.V.; Nair, C.M.; Banerjee, M. DNA methyl transferase (DNMT) gene polymorphisms could be a primary event in epigenetic susceptibility to schizophrenia. PLoS ONE 2014, 9, e98182. [Google Scholar] [CrossRef] [Green Version]
- Tenorio, J.; Alarcón, P.; Arias, P.; Dapía, I.; García-Miñaur, S.; Palomares Bralo, M.; Campistol, J.; Climent, S.; Valenzuela, I.; Ramos, S.; et al. Further delineation of neuropsychiatric findings in Tatton-Brown-Rahman syndrome due to disease-causing variants in DNMT3A: Seven new patients. Eur. J. Hum. Genet. 2020, 28, 469–479. [Google Scholar] [CrossRef]
- Christian, D.L.; Wu, D.Y.; Martin, J.R.; Moore, J.R.; Liu, Y.R.; Clemens, A.W.; Nettles, S.A.; Kirkland, N.M.; Papouin, T.; Hill, C.A.; et al. DNMT3A Haploinsufficiency Results in Behavioral Deficits and Global Epigenomic Dysregulation Shared across Neurodevelopmental Disorders. Cell Rep. 2020, 33, 108416. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Na, E.S.; Autry, A.E.; Monteggia, L.M. Impact of DNMT1 and DNMT3a forebrain knockout on depressive- and anxiety like behavior in mice. Neurobiol. Learn. Mem. 2016, 135, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Lavery, L.A.; Ure, K.; Wan, Y.W.; Luo, C.; Trostle, A.J.; Wang, W.; Jin, H.; Lopez, J.; Lucero, J.; Durham, M.A.; et al. Losing dnmt3a dependent methylation in inhibitory neurons impairs neural function by a mechanism impacting rett syndrome. Elife 2020, 9, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Hwang, V.J.; Dandekar, R.; Durbin-Johnson, B.; Charlet-Berguerand, N.; Ander, B.P.; Sharp, F.R.; Angkustsiri, K.; Simon, T.J.; Tassone, F. Decreased DGCR8 expression and miRNA dysregulation in individuals with 22q11.2 deletion syndrome. PLoS ONE 2014, 9, e103884. [Google Scholar] [CrossRef]
- De la Morena, M.T.; Eitson, J.L.; Dozmorov, I.M.; Belkaya, S.; Hoover, A.R.; Anguiano, E.; Pascual, M.V.; van Oers, N.S.C. Signature MicroRNA expression patterns identified in humans with 22q11.2 deletion/DiGeorge syndrome. Clin. Immunol. 2013, 147, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Bassett, A.S.; Chow, E.W.C.; AbdelMalik, P.; Gheorghiu, M.; Husted, J.; Weksberg, R. The schizophrenia phenotype in 22q11 deletion syndrome. Am. J. Psychiatry 2003, 160, 1580–1586. [Google Scholar] [CrossRef] [Green Version]
- Heckers, S.; Rauch, S.L.; Goff, D.; Savage, C.R.; Schacter, D.L.; Fischman, A.J.; Alpert, N.M. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Neuroimage 1998, 7, 827–836. [Google Scholar] [CrossRef]
- Jessen, F.; Scheef, L.; Germeshausen, L.; Tawo, Y.; Kockler, M.; Kuhn, K.U.; Maier, W.; Schild, H.H.; Heun, R. Reduced hippocampal activation during encoding and recognition of words in schizophrenia patients. Am. J. Psychiatry 2003, 160, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Ragland, J.D.; Gur, R.C.; Valdez, J.; Turetsky, B.I.; Elliott, M.; Kohler, C.; Siegel, S.; Kanes, S.; Gur, R.E. Event-related fMRI of frontotemporal activity during word encoding and recognition in schizophrenia. Am. J. Psychiatry 2004, 161, 1004–1015. [Google Scholar] [CrossRef] [Green Version]
- Pirnia, T.; Woods, R.P.; Hamilton, L.S.; Lyden, H.; Joshi, S.H.; Asarnow, R.F.; Nuechterlein, K.H.; Narr, K.L. Hippocampal dysfunction during declarative memory encoding in schizophrenia and effects of genetic liability. Schizophr. Res. 2015, 161, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragland, J.D.; Ranganath, C.; Harms, M.P.; Barch, D.M.; Gold, J.M.; Layher, E.; Lesh, T.A.; MacDonald, A.W.; Niendam, T.A.; Phillips, J.; et al. Functional and Neuroanatomic Specificity of Episodic Memory Dysfunction in Schizophrenia. JAMA Psychiatry 2015, 72, 909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragland, J.D.; Layher, E.; Hannula, D.E.; Niendam, T.A.; Lesh, T.A.; Solomon, M.; Carter, C.S.; Ranganath, C. Impact of schizophrenia on anterior and posterior hippocampus during memory for complex scenes. NeuroImage Clin. 2017, 13, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Earls, L.R.; Bayazitov, I.T.; Fricke, R.G.; Berry, R.B.; Illingworth, E.; Mittleman, G.; Zakharenko, S.S. Dysregulation of presynaptic calcium and synaptic plasticity in a mouse model of 22q11 deletion syndrome. J. Neurosci. 2010, 30, 15843–15855. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Mathur, P.; Gottesman, I.I.; Nagpal, R.; Nimgaonkar, V.L.; Deshpande, S.N. Correlates of hallucinations in schizophrenia: A cross-cultural evaluation. Schizophr. Res. 2007, 92, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.M.; Schanda, H.; Karakula, H.; Olajossy-Hilkesberger, L.; Rudaleviciene, P.; Okribelashvili, N.; Chaudhry, H.R.; Idemudia, S.E.; Gscheider, S.; Ritter, K.; et al. Culture and the prevalence of hallucinations in schizophrenia. Compr. Psychiatry 2011, 52, 319–325. [Google Scholar] [CrossRef]
- Chun, S.; Westmoreland, J.J.; Bayazitov, I.T.; Eddins, D.; Pani, A.K.; Smeyne, R.J.; Yu, J.; Blundon, J.A.; Zakharenko, S.S. Specific disruption of thalamic inputs to the auditory cortex in schizophrenia models. Science 2014, 344, 1178–1182. [Google Scholar] [CrossRef] [Green Version]
- Wright, I.C.; Rabe-Hesketh, S.; Woodruff, P.W.R.; David, A.S.; Murray, R.M.; Bullmore, E.T. Meta-analysis of regional brain volumes in schizophrenia. Am. J. Psychiatry 2000, 157, 16–25. [Google Scholar] [CrossRef]
- Van Erp, T.G.M.; Hibar, D.P.; Rasmussen, J.M.; Glahn, D.C.; Pearlson, G.D.; Andreassen, O.A.; Agartz, I.; Westlye, L.T.; Haukvik, U.K.; Dale, A.M.; et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol. Psychiatry 2016, 21, 547–553. [Google Scholar] [CrossRef]
- Lawrie, S.M.; Abukmeil, S.S. Brain abnormality in schizophrenia. A systematic and quantitative review of volumetric magnetic resonance imaging studies. Br. J. Psychiatry 1998, 172, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Cahn, W.; Pol, H.E.H.; Lems, E.B.T.E.; van Haren, N.E.M.; Schnack, H.G.; van der Linden, J.A.; Schothorst, P.F.; van Engeland, H.; Kahn, R.S. Brain Volume Changes in First-Episode Schizophrenia. Arch. Gen. Psychiatry 2002, 59, 1002. [Google Scholar] [CrossRef] [Green Version]
- Steen, R.G.; Mull, C.; McClure, R.; Hamer, R.M.; Lieberman, J.A. Brain volume in first-episode schizophrenia: Systematic review and meta-analysis of magnetic resonance imaging studies. Br. J. Psychiatry 2006, 188, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Vita, A.; De Peri, L.; Silenzi, C.; Dieci, M. Brain morphology in first-episode schizophrenia: A meta-analysis of quantitative magnetic resonance imaging studies. Schizophr. Res. 2006, 82, 75–88. [Google Scholar] [CrossRef]
- Olabi, B.; Ellison-Wright, I.; McIntosh, A.M.; Wood, S.J.; Bullmore, E.; Lawrie, S.M. Are there progressive brain changes in schizophrenia? a meta-analysis of structural magnetic resonance imaging studies. Biol. Psychiatry 2011, 70, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempton, M.J.; Stahl, D.; Williams, S.C.R.; DeLisi, L.E. Progressive lateral ventricular enlargement in schizophrenia: A meta-analysis of longitudinal MRI studies. Schizophr. Res. 2010, 120, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Arcila, M.L.; Betizeau, M.; Cambronne, X.A.; Guzman, E.; Doerflinger, N.; Bouhallier, F.; Zhou, H.; Wu, B.; Rani, N.; Bassett, D.S.; et al. Novel primate miRNAs coevolved with ancient target genes in germinal zone-specific expression patterns. Neuron 2014, 81, 1255–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prodromidou, K.; Vlachos, I.S.; Gaitanou, M.; Kouroupi, G.; Hatzigeorgiou, A.G.; Matsas, R. MicroRNA-934 is a novel primate-specific small non-coding RNA with neurogenic function during early development. eLife 2020, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R.; Lugli, G.; Zhang, H.; Rizavi, H.; Cook, E.H.; Dwivedi, Y. Expression of micrornas and other small RNAs in prefrontal cortex in schizophrenia, bipolar disorder and depressed subjects. PLoS ONE 2014, 9, e86469. [Google Scholar] [CrossRef] [PubMed]
- Lett, T.; Chakravarty, M.; Chakavarty, M.; Felsky, D.; Brandl, E.; Tiwari, A.; Gonçalves, V.; Rajji, T.; Daskalakis, Z.; Meltzer, H.; et al. The genome-wide supported microRNA-137 variant predicts phenotypic heterogeneity within schizophrenia. Mol. Psychiatry 2013, 18, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.S.; Kelly, S.; Wright, C.; Gupta, C.N.; Arias-Vasquez, A.; Perrone-Bizzozero, N.; Ehrlich, S.; Wang, L.; Bustillo, J.R.; Morris, D.; et al. MIR137HG risk variant rs1625579 genotype is related to corpus callosum volume in schizophrenia. Neurosci. Lett. 2015, 602, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuswanto, C.N.; Sum, M.Y.; Qiu, A.; Sitoh, Y.Y.; Liu, J.; Sim, K. The impact of genome wide supported microRNA-137 (MIR137) risk variants on frontal and striatal white matter integrity, neurocognitive functioning, and negative symptoms in schizophrenia. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2015, 168, 317–326. [Google Scholar] [CrossRef]
- Green, M.J.; Cairns, M.J.; Wu, J.; Dragovic, M.; Jablensky, A.; Tooney, P.A.; Scott, R.J.; Carr, V.J. Genome-wide supported variant MIR137 and severe negative symptoms predict membership of an impaired cognitive subtype of schizophrenia. Mol. Psychiatry 2013, 18, 774–780. [Google Scholar] [CrossRef]
- Siegert, S.; Seo, J.; Kwon, E.J.; Rudenko, A.; Cho, S.; Wang, W.; Flood, Z.; Martorell, A.J.; Ericsson, M.; Mungenast, A.E.; et al. The schizophrenia risk gene product miR-137 alters presynaptic plasticity. Nat. Neurosci. 2015, 18, 1008–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, M.P.; Zhang, H.; Moy, W.; McGowan, H.; Leites, C.; Dionisio, L.E.; Xu, Z.; Shi, J.; Sanders, A.R.; Greenleaf, W.J.; et al. Open Chromatin Profiling in hiPSC-Derived Neurons Prioritizes Functional Noncoding Psychiatric Risk Variants and Highlights Neurodevelopmental Loci. Cell Stem Cell 2017, 21, 305–318.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guella, I.; Sequeira, A.; Rollins, B.; Morgan, L.; Torri, F.; van Erp, T.G.M.; Myers, R.M.; Barchas, J.D.; Schatzberg, A.F.; Watson, S.J.; et al. Analysis of miR-137 expression and rs1625579 in dorsolateral prefrontal cortex. J. Psychiatr. Res. 2013, 47, 1215–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Wang, Z.-M.; Tan, W.; Wang, X.; Li, Y.; Bai, B.; Li, Y.; Zhang, S.; Yan, H.; Chen, Z.-L.; et al. Partial loss of psychiatric risk gene Mir137 in mice causes repetitive behavior and impairs sociability and learning via increased Pde10a. Nat. Neurosci. 2018, 21, 1689–1703. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, M.H.; Vallès, A.; Kirkels, L.A.M.H.; Mastebroek, M.; Olde Loohuis, N.; Kos, A.; Wissink-Lindhout, W.M.; de Brouwer, A.P.M.; Nillesen, W.M.; Pfundt, R.; et al. Chromosome 1p21.3 microdeletions comprising DPYD and MIR137 are associated with intellectual disability. J. Med. Genet. 2011, 48, 810–818. [Google Scholar] [CrossRef]
- Carter, M.; Nikkel, S.; Fernandez, B.; Marshall, C.; Noor, A.; Lionel, A.; Prasad, A.; Pinto, D.; Joseph-George, A.; Noakes, C.; et al. Hemizygous deletions on chromosome 1p21.3 involving the DPYD gene in individuals with autism spectrum disorder. Clin. Genet. 2011, 80, 435–443. [Google Scholar] [CrossRef]
- D’Angelo, C.S.; Moller dos Santos, M.F.; Alonso, L.G.; Koiffmann, C.P. Two New Cases of 1p21.3 Deletions and an Unbalanced Translocation t(8;12) among Individuals with Syndromic Obesity. Mol. Syndromol. 2015, 6, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of Genes and Cellular Pathways Dysregulated in Autism Spectrum Disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [Green Version]
- Tucci, A.; Ciaccio, C.; Scuvera, G.; Esposito, S.; Milani, D. MIR137 is the key gene mediator of the syndromic obesity phenotype of patients with 1p21.3 microdeletions. Mol. Cytogenet. 2016, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Smrt, R.D.; Szulwach, K.E.; Pfeiffer, R.L.; Li, X.; Guo, W.; Pathania, M.; Teng, Z.Q.; Luo, Y.; Peng, J.; Bordey, A.; et al. MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells 2010, 28, 1060–1070. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, Y.; Yokoyama, K.; Tasaki, S.; Kato, J.; Nakashima, K.; Takeyama, M.; Nakatani, A.; Suzuki, M. Transgenic mice overexpressing miR-137 in the brain show schizophrenia-associated behavioral deficits and transcriptome profiles. PLoS ONE 2019, 14, e0220389. [Google Scholar] [CrossRef] [Green Version]
- Thomas, K.T.; Anderson, B.R.; Shah, N.; Zimmer, S.E.; Hawkins, D.; Valdez, A.N.; Gu, Q.; Bassell, G.J. Inhibition of the Schizophrenia-Associated MicroRNA miR-137 Disrupts Nrg1α Neurodevelopmental Signal Transduction. Cell Rep. 2017, 20, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Vallès, A.; Martens, G.J.M.; De Weerd, P.; Poelmans, G.; Aschrafi, A. MicroRNA-137 regulates a glucocorticoid receptor-dependent signalling network: Implications for the etiology of schizophrenia. J. Psychiatry Neurosci. 2014, 39, 312–320. [Google Scholar] [CrossRef] [Green Version]
- He, E.; Lozano, M.A.G.; Stringer, S.; Watanabe, K.; Sakamoto, K.; den Oudsten, F.; Koopmans, F.; Giamberardino, S.N.; Hammerschlag, A.; Cornelisse, L.N.; et al. MIR137 schizophrenia-associated locus controls synaptic function by regulating synaptogenesis, synapse maturation and synaptic transmission. Hum. Mol. Genet. 2018, 27, 1879–1891. [Google Scholar] [CrossRef]
- Olde Loohuis, N.F.M.; Ba, W.; Stoerchel, P.H.; Kos, A.; Jager, A.; Schratt, G.; Martens, G.J.M.; van Bokhoven, H.; Nadif Kasri, N.; Aschrafi, A. MicroRNA-137 Controls AMPA-Receptor-Mediated Transmission and mGluR-Dependent LTD. Cell Rep. 2015, 11, 1876–1884. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Li, H.; Guo, R.; Ma, T.; Hou, R.; Ma, X.; Du, Y. miR-137, a new target for post-stroke depression? Neural Regen. Res. 2013, 8, 2441–2448. [Google Scholar] [CrossRef]
- Luby, E.D.; Cohen, B.D.; Rosenbaum, G.; Gottlieb, J.S.; Kelley, R. Study of a new schizophrenomimetic drug; sernyl. AMA. Arch. Neurol. Psychiatry 1959, 81, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Itil, T.; Keskiner, A.; Kiremitci, N.; Holden, J.M. Effect of phencyclidine in chronic schizophrenics. Can. Psychiatr. Assoc. J. 1967, 12, 209–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Dai, J.; Xu, L.J.; Han, G.D.; Sun, H.L.; Zhu, G.T.; Jiang, H.T.; Yu, G.Y.; Tang, X.M. MiR-137 attenuates spinal cord injury by modulating NEUROD4 through reducing inflammation and oxidative stress. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1884–1890. [Google Scholar] [CrossRef]
- Tang, Y.; Li, Y.; Yu, G.; Ling, Z.; Zhong, K.; Zilundu, P.L.M.; Li, W.; Fu, R.; Zhou, L.H. MicroRNA-137-3p Protects PC12 Cells Against Oxidative Stress by Downregulation of Calpain-2 and nNOS. Cell. Mol. Neurobiol. 2021, 41, 1373–1387. [Google Scholar] [CrossRef]
- Niwa, M.; Jaaro-Peled, H.; Tankou, S.; Seshadri, S.; Hikida, T.; Matsumoto, Y.; Cascella, N.G.; Kano, S.; Ozaki, N.; Nabeshima, T.; et al. Adolescent stress-induced epigenetic control of dopaminergic neurons via glucocorticoids. Science 2013, 339, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Maynard, T.M.; Sikich, L.; Lieberman, J.A.; LaMantia, A.S. Neural development, cell-cell signaling, and the “two-hit” hypothesis of schizophrenia. Schizophr. Bull. 2001, 27, 457–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoops, D.; Flores, C. Making Dopamine Connections in Adolescence. Trends Neurosci. 2017, 40, 709–719. [Google Scholar] [CrossRef]
Figure 1.
Overview of the miRNA pathway and its dysregulation in 22q11DS and idiopathic SCZ. The miRNA pathway begins when transcription produces a pri-miRNA, which undergoes sequential processing into a pre-miRNA by the Microprocessor and DICER to produce a mature miRNA. The mature miRNA is then loaded into an AGO protein complex to produce an active RNA-silencing complex (RISC), which binds to mRNAs containing complementary sequences and promotes mRNA degradation and/or inhibits protein synthesis. Current models of 22q11DS, which is associated with elevated SCZ risk, suggest that DGCR8 haploinsufficiency impairs Microprocessor activity, leading to widespread downregulation of miRNAs. In contrast, several studies suggest that several components of the miRNA pathway (i.e., DGCR8, DROSHA, and DICER1 mRNAs) are upregulated in idiopathic SCZ. Furthermore, in patients with idiopathic SCZ, overrepresentation of CNVs affecting miRNA genes (outside of the 22q11.2 region) and dysregulated miRNA levels in postmortem brain suggest that both up- and downregulation of miRNAs may contribute to SCZ risk. Abbreviations: AGO, Argonaute protein; CNV, copy number variant; RNA pol II, RNA polymerase II.
Figure 1.
Overview of the miRNA pathway and its dysregulation in 22q11DS and idiopathic SCZ. The miRNA pathway begins when transcription produces a pri-miRNA, which undergoes sequential processing into a pre-miRNA by the Microprocessor and DICER to produce a mature miRNA. The mature miRNA is then loaded into an AGO protein complex to produce an active RNA-silencing complex (RISC), which binds to mRNAs containing complementary sequences and promotes mRNA degradation and/or inhibits protein synthesis. Current models of 22q11DS, which is associated with elevated SCZ risk, suggest that DGCR8 haploinsufficiency impairs Microprocessor activity, leading to widespread downregulation of miRNAs. In contrast, several studies suggest that several components of the miRNA pathway (i.e., DGCR8, DROSHA, and DICER1 mRNAs) are upregulated in idiopathic SCZ. Furthermore, in patients with idiopathic SCZ, overrepresentation of CNVs affecting miRNA genes (outside of the 22q11.2 region) and dysregulated miRNA levels in postmortem brain suggest that both up- and downregulation of miRNAs may contribute to SCZ risk. Abbreviations: AGO, Argonaute protein; CNV, copy number variant; RNA pol II, RNA polymerase II.
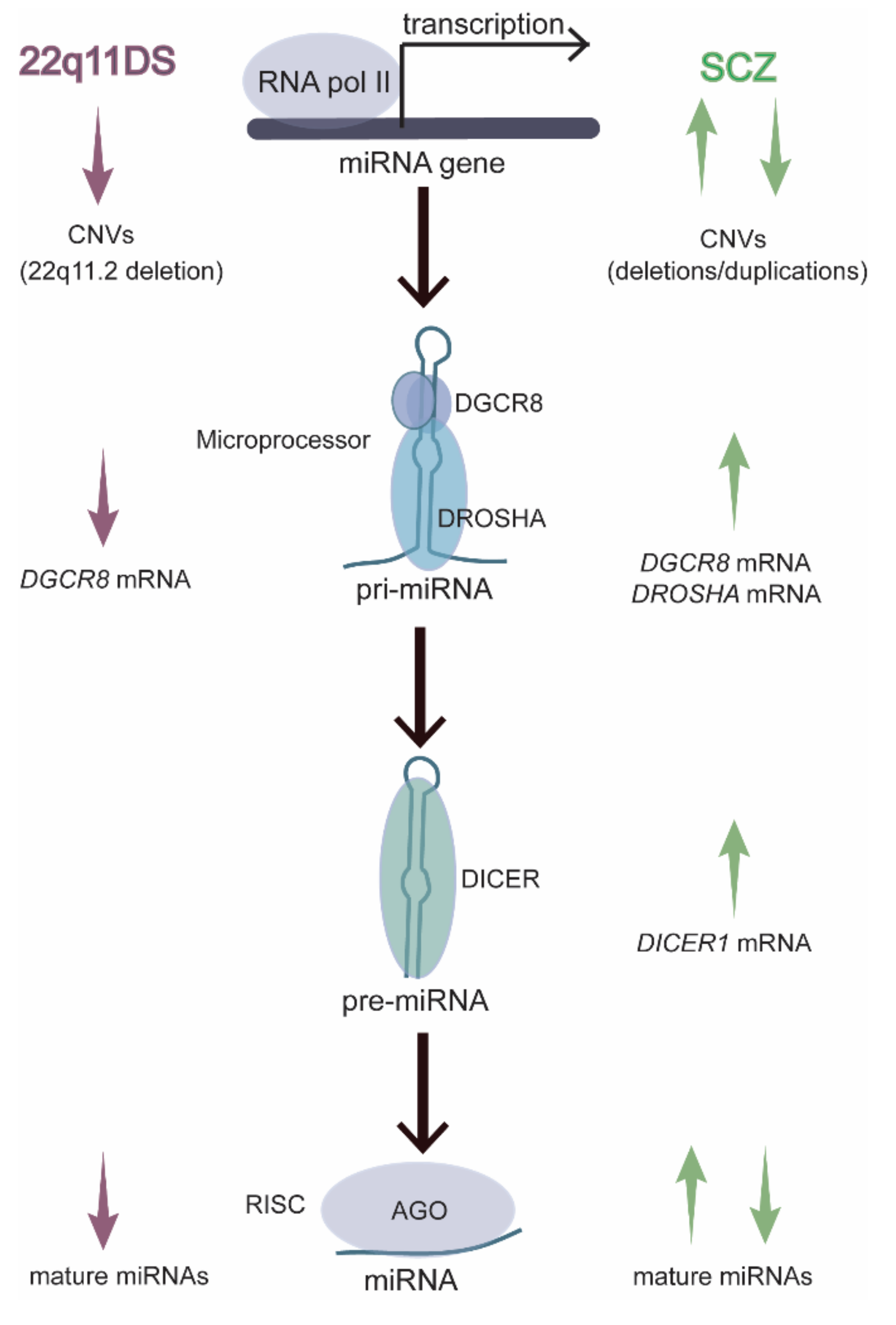
Figure 2.
Anatomy of a miRNA. The miR-132-3p canonical sequence targets several sites within the DNMT3A-3′ UTR, including the one shown (interaction predicted by miRbase). Targeting is mostly dictated by the seed sequence (shaded in blue) within the miRNA: nucleotides 2-7 or 2-8 (5′ to 3′). Additional targeting can occur within the 3′ supplementary region (shaded in pink), which is separated from the seed sequence by a noncomplementary linker sequence. The linker sequence varies in length, depending on the target and the position of the 3′ supplementary region. The final nucleotides near the 3′ end of the miRNA form the miRNA’s 3′ tail. The 3′ tail rarely contributes to mRNA targeting but undergoes frequent editing by exonucleases, which trim the miRNA, and nucleotidyl transferases, which add nucleotides to the tail of the miRNA.
Figure 2.
Anatomy of a miRNA. The miR-132-3p canonical sequence targets several sites within the DNMT3A-3′ UTR, including the one shown (interaction predicted by miRbase). Targeting is mostly dictated by the seed sequence (shaded in blue) within the miRNA: nucleotides 2-7 or 2-8 (5′ to 3′). Additional targeting can occur within the 3′ supplementary region (shaded in pink), which is separated from the seed sequence by a noncomplementary linker sequence. The linker sequence varies in length, depending on the target and the position of the 3′ supplementary region. The final nucleotides near the 3′ end of the miRNA form the miRNA’s 3′ tail. The 3′ tail rarely contributes to mRNA targeting but undergoes frequent editing by exonucleases, which trim the miRNA, and nucleotidyl transferases, which add nucleotides to the tail of the miRNA.
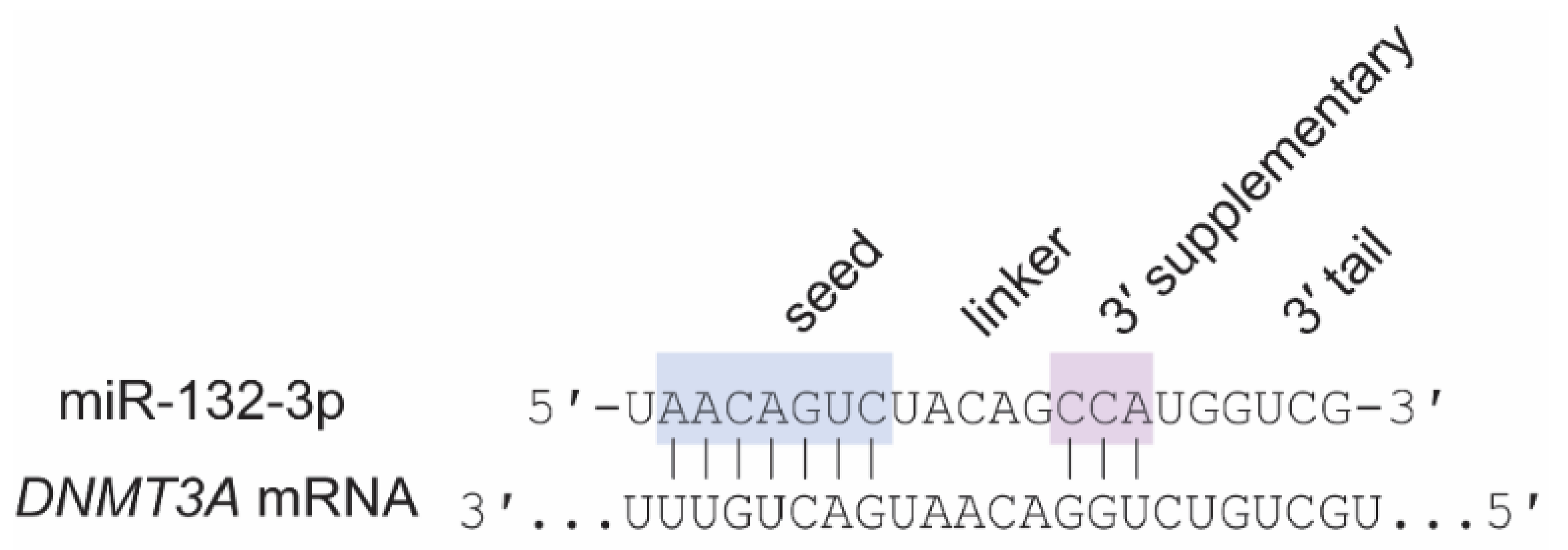
Figure 3.
Overview of human postnatal neurodevelopment and the stages of SCZ. The prodromal phase of SCZ typically overlaps with adolescent development, and first-episode psychosis (and diagnosis) typically occurs in late adolescence or early adulthood. The chronic disease phase persists throughout adulthood. The onset of early SCZ symptoms during adolescence overlaps with many facets of brain maturation, including intracortical myelination, synaptic pruning, maturation of GABAergic and dopaminergic signaling in the PFC, increases in oxidative stress markers, and maturation of both the innate and adaptive immune systems. Dysregulation of any (or all) of these processes might contribute to symptom onset.
Figure 3.
Overview of human postnatal neurodevelopment and the stages of SCZ. The prodromal phase of SCZ typically overlaps with adolescent development, and first-episode psychosis (and diagnosis) typically occurs in late adolescence or early adulthood. The chronic disease phase persists throughout adulthood. The onset of early SCZ symptoms during adolescence overlaps with many facets of brain maturation, including intracortical myelination, synaptic pruning, maturation of GABAergic and dopaminergic signaling in the PFC, increases in oxidative stress markers, and maturation of both the innate and adaptive immune systems. Dysregulation of any (or all) of these processes might contribute to symptom onset.
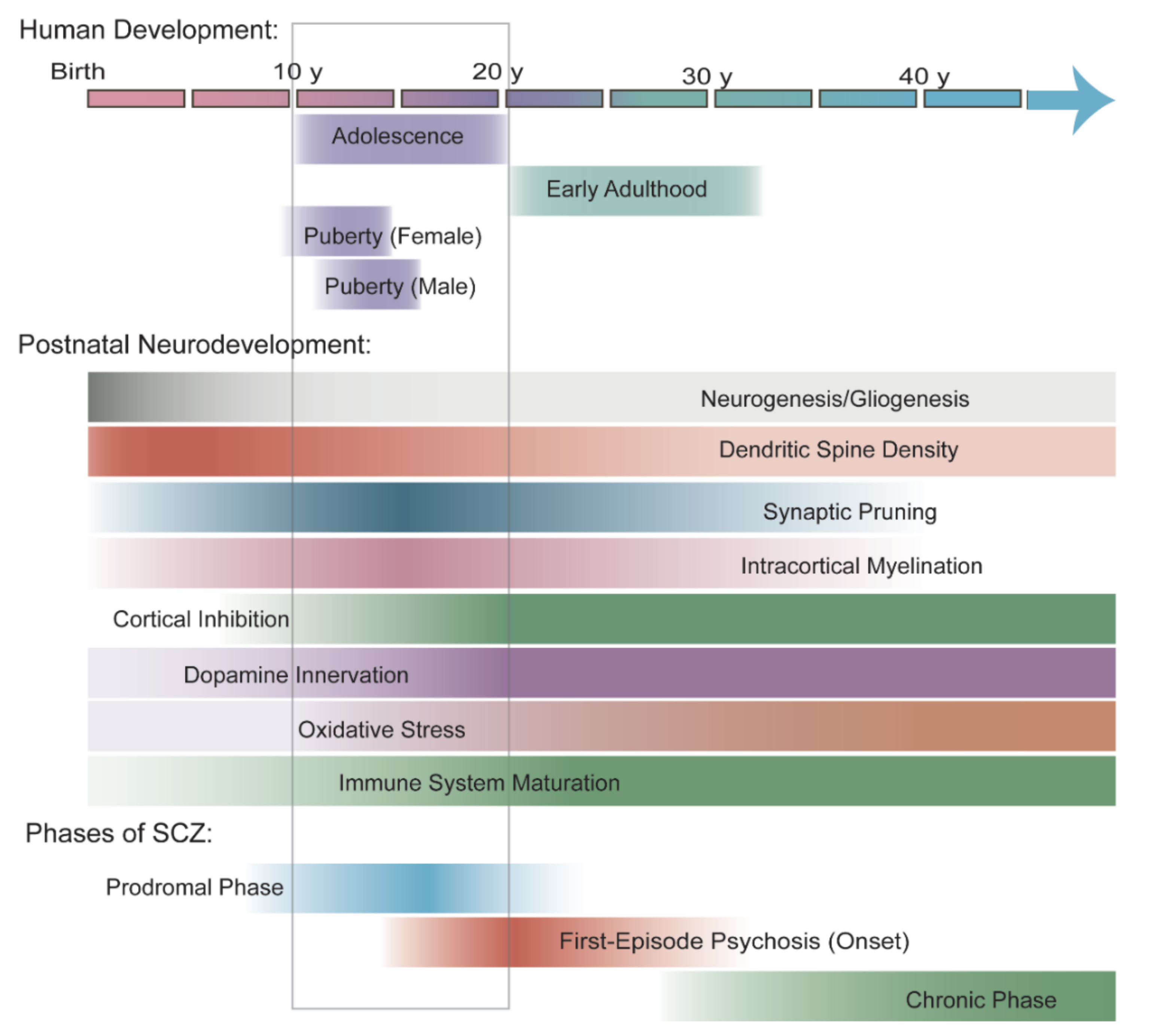
Figure 4.
Overview of mouse postnatal neurodevelopment and the timing of SCZ-associated phenotypes in 22q11DS mouse models. Mouse adolescence begins with weaning at ~P22 and ends with the onset of full sexual maturity at ~P60. Increases in miR-29a-3p and miR-132-3p before and during adolescence inhibit ocular dominance plasticity and limit DNMT3A-driven CH-methylation in the visual cortex, providing an example of how temporal regulation of miRNA levels drives late brain maturation in mouse models. Several miRNA-dependent phenotypes also display an age-dependent onset in mouse models of 22q11DS that mimics the developmentally delayed onset of SCZ in patients. In the hippocampus, elevated LTP at Schaffer collateral (CA3–CA1) synapses is absent at P60 but apparent by P120. The exact timing of this phenotype’s onset is unclear, however. Deficits in auditory thalamocortical synapses gradually appear between P60 and P120. Ventricular enlargement is developmentally delayed; however, the ciliary motility deficit in ependymal cells that underlies this phenotype is observed by P120.
Figure 4.
Overview of mouse postnatal neurodevelopment and the timing of SCZ-associated phenotypes in 22q11DS mouse models. Mouse adolescence begins with weaning at ~P22 and ends with the onset of full sexual maturity at ~P60. Increases in miR-29a-3p and miR-132-3p before and during adolescence inhibit ocular dominance plasticity and limit DNMT3A-driven CH-methylation in the visual cortex, providing an example of how temporal regulation of miRNA levels drives late brain maturation in mouse models. Several miRNA-dependent phenotypes also display an age-dependent onset in mouse models of 22q11DS that mimics the developmentally delayed onset of SCZ in patients. In the hippocampus, elevated LTP at Schaffer collateral (CA3–CA1) synapses is absent at P60 but apparent by P120. The exact timing of this phenotype’s onset is unclear, however. Deficits in auditory thalamocortical synapses gradually appear between P60 and P120. Ventricular enlargement is developmentally delayed; however, the ciliary motility deficit in ependymal cells that underlies this phenotype is observed by P120.
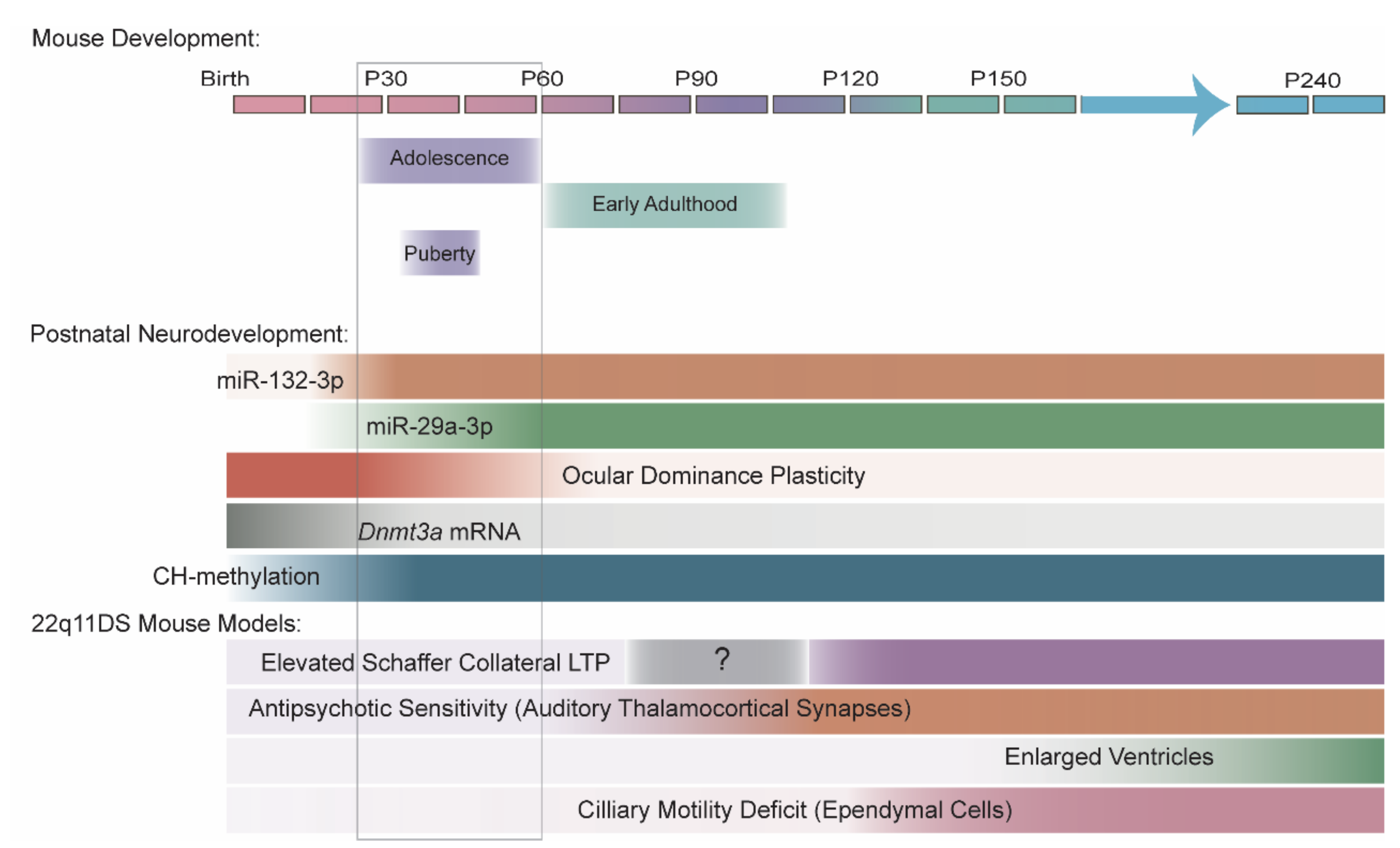

{kind=link}
{kind=link}
{kind=link}
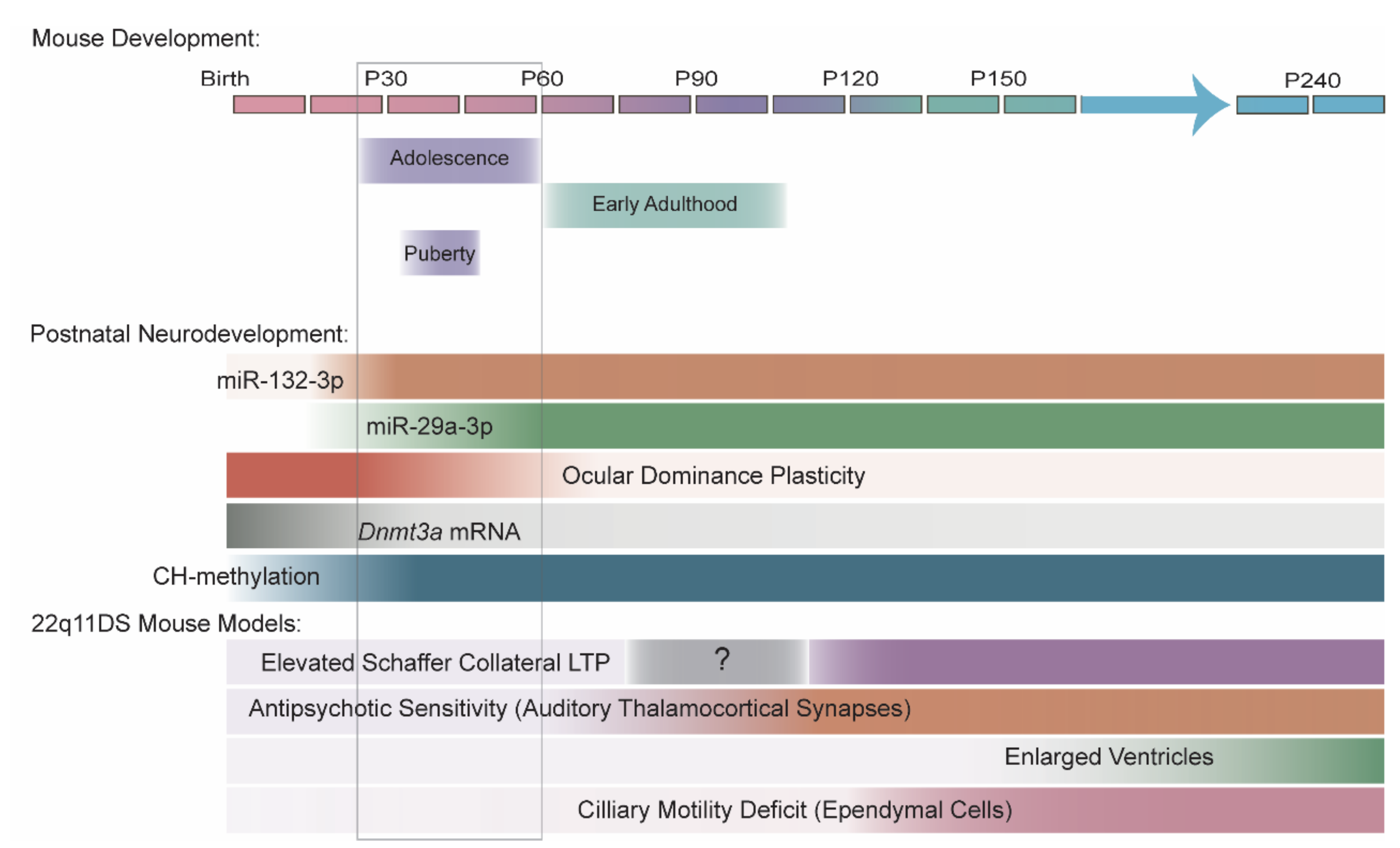{kind=link}
Table 1.
Global patterns in miRNA levels with age.
| Species | Reference | Region | Method | Age Range | Adolescent Time Points? | Global Patterns in miRNA Levels |
|---|---|---|---|---|---|---|
| Human | Somel et al., 2010 [45] | Superior frontal gyrus of PFC | Small RNA-seq | 0–98 y | Yes (one 13 y) | Highly dynamic with age, with inflection points at ~4 y and ~20 y |
| Somel et al., 2011 [46] | PFC and cerebellum | Microarray | 0–98 y | No | None noted | |
| Moreau et al., 2013 [47] | Cerebrum | Microarray | GW14–adult a | No | Increase with age | |
| Beveridge et al., 2014 [48] | DLPFC (BA 46) | Microarray | 2 mo–78 y | Yes | Decrease with age, with inflection point at late adolescence | |
| Ziats and Rennert 2014 [44] | OPFC, DLPFC, MPFC, VLPFC, hippocampus and cerebellum | Small RNA-seq | 4 mo–19 y | Yes | Most dynamic during transition from infancy to early childhood | |
| Hu et al., 2019 [11] | DLPFC (BA 46) | Small RNA-seq | Second trimester–74 y | Yes | Most dynamic/peak expression before puberty | |
| Pig | Podolska et al., 2011 [49] | Cortex and cerebellum | Microarray | F50–3 mo | Yes (3 mo) | None noted |
| Rat | Krichevsky et al., 2003 [50] | Forebrain | Array b | E12–adult | No | Highly dynamic with age |
| Yao et al., 2012 [51] | Dorsolateral cortex | Small RNA-seq | E17–P28 | Yes (P28) | Highly dynamic across most time points | |
| Mouse | Miska et al., 2004 [52] | Whole brain | Microarray c | E12.5–4 mo | No | Increase with age |
| Eda et al., 2011 [53] | Cerebrum, cerebellum, hippocampus | Microarray | E16.5–19 mo | Yes (1 mo) | Most dynamic in early postnatal brain (P6–1 mo) | |
| Fertuzinhos et al., 2014 [54] | Primary somatosensory cortex | Small RNA-seq | P4–P180 | No | None noted | |
| Swahari et al., 2021 [55] | Cerebellum | Small RNA-seq | P18 and P250 | No | None noted |
a Adult samples were commercially derived and elderly. b First microRNA array consisted of a nylon membrane spotted with oligos. c First miRNA microarray. Abbreviations: BA, Brodmann’s area; DLPFC, dorsolateral prefrontal cortex; F, fetal day; GW, gestational week; MPFC, medial prefrontal cortex; OFPC, orbitofrontal prefrontal cortex; PFC, prefrontal cortex; VLPFC, ventrolateral prefrontal cortex.
Table 2.
Changes in specific miRNAs with age in primate and pig brains.
| Study Information | miR-29a/b/c-3p | miR-132-3p | miR-137-3p | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | Reference | Method | p-Value | Age Range | Region | Direction | Differential Age Range | Direction | Differential Age Range | Direction | Differential Age Range |
| Human | Somel et al., 2010 [45] | Small RNA-seq | Yes | 2 d–98 y | Superior frontal gyrus (BA9) | Increased (a) NR (b/c) | 2 d–98 y (a) | NR | -- | NR | -- |
| Moreau et al., 2013 [47] | Microarray | No | fetal (GW14–GW24), early postnatal (5 d–4 y), adult * | Cerebrum | Increased (a) Increased (b) NR (c) | Postnatal-adult (a) postnatal (5 d–4 y) (b) | Increased | Fetal-postnatal | Increased | Fetal (GW14–GW20) | |
| Beveridge et al., 2014 [48] | Microarray | Yes | 2 mo–78 y | DLPFC (BA46) | Decreased (a/c) NR (b) | 2 mo–78 y | Decreased | 2 mo–78 y | Decreased | 2 mo–78 y | |
| Ziats and Rennert 2014 [44] | Small RNA-seq | Yes | 4 mo–19 y | DLPFC (BA9, 46) | Increased (b) NS (a/c) | Infancy-early childhood (b) | NS | -- | NS | -- | |
| cerebellum (CBC) | NS | -- | NS | -- | Decreased | Infancy-early childhood | |||||
| OPFC (BA11), MPFC (BA32-34), VLPFC (BA44-45), hippocampus | NS | -- | NS | -- | NS | -- | |||||
| Macaque | Somel et al., 2010 [45] | Small RNA-seq | Yes | 16 d–28 y | Superior frontal gyrus (cortex) | Increased (a) NR (b/c) | 16 d–28 y (a) | NR | -- | NR | -- |
| Pig | Podolska et al., 2011 [49] | Microarray | No | F50, F100, 3 mo | Cortex | Increased (a/b/c) | F100–3 mo | NR | -- | NR | -- |
| Cerebellum | Increased (a/b/c) | F100–3 mo | NR | -- | NR | -- | |||||
* Adult samples were commercially derived and elderly. (a/b/c) indicates specific miR-29 variant reported in each study. Abbreviations: BA, Brodmann’s area; DLPFC, dorsolateral prefrontal cortex; GW, gestational weeks; MPFC, medial prefrontal cortex; NE, not examined; NR, not reported (unclear if NE or NS); NS, not significant; OFPC, orbitofrontal prefrontal cortex; PFC, prefrontal cortex; VLPFC, ventrolateral prefrontal cortex.
Table 3.
Changes in specific miRNAs with age in rodent brain.
| Study Information | miR-29a/b/c-3p | miR-132-3p | miR-137-3p | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | Reference | Method | p-Value | Age Range | Region | Direction | Differential Age Range | Direction | Differential Age Range | Direction | Differential Age Range |
| Rat | Yao et al., 2012 [51] | Small RNA-seq | No | E17–P28 | Whole cortex (E10 and E13) dorsolateral cortex (E17–P28) | Increased (a/b/c) | P3–P28 | Increased | P3–P28 | Peak at P0 | Increased (E10–P0) Decreased (P0–P28) |
| Sangiao-Alvarellos et al., 2013 [78] | RT-qPCR | Yes | P1–adult (>P75) | Hypothalamus | NE | — | Increased | Neonatal-juvenile | NE | — | |
| Mouse | Eda et al., 2011 [53] | Microarray | No | E16.5–19 mo | Cerebrum | Increased (a/b/c) | E16.5–3 mo | Increased | E16.5–1 mo | Increased | E16.5–P6 |
| P2–19 mo | Hippocampus | Increased (a/b/c) | P2–6 mo | Increased | P2–6 mo | ND | — | ||||
| E16.5–19 mo | Cerebellum | ND | — | Increased | E16.5–1 mo | Two peaks | Increased (E16.5–P2) Decreased (P2–6 mo), Increased (6 mo–19 mo) | ||||
| Tognini et al., 2011 [79] | RT-qPCR | Yes | P7–P35 | Visual cortex | NE | — | Increased | P7–P30 | NE | — | |
| Miller et al., 2012 [10] | RT-qPCR | Yes | E15–P60 | PFC | NE | — | Increased | P7–P28 | NE | — | |
| Fertuzinhos et al., 2014 [54] | Small RNA-seq | Yes | P4–P180 | S1 cortex | Increased (a) ND (b/c) | P4–P180 | Increased | P4–P180 | ND | — | |
| Li et al., 2014 [80] | Microarray | No | E12.5–E18.5, P60 | Cortex | Increased (a) ND (b/c) | E18.5–P60 | Increased | E12.5–P60 | NR | — | |
| RT-qPCR | Yes | E12.5–P60 | Cortex | Increased (a) NE (b/c) | P1–P60 (a) | NE | — | NE | — | ||
| E18.5–P60 | Hippocampus | Increased (a) NE (b/c) | P1–P60 (a) | NE | — | NE | — | ||||
| Mazziotti et al., 2017 [81] | Small RNA-seq | Yes | P10, P28 | V1 cortex | Increased (a/c), NS (b) | P10–P28 | Increased | P10–P28 | NS | — | |
| Li et al., 2019 [82] | RT-qPCR | Yes | P10–P120 | Hypothalamus | Increased (a/b/c) | P10–P120 | NE | — | NE | — | |
| Swahari et al., 2021 [55] | RT-qPCR | No | P0–P60 | Cortex | Increased (a/b/c) | P0–P40 (a/c), P0–P60 (b) | NE | — | NE | — | |
| Cerebellum | Increased (b) NE (a/c) | P0–P60 (b) | NE | — | NE | — | |||||
| Small RNA-seq | No | P18, P250 | Cerebellum | Increased (a/b/c) | P18–P250 | NR | — | Increased | P18–P250 | ||
| Napoli et al., 2020 [83] | RT-qPCR | Yes | P10–P200 | Visual cortex | Increased (a) NE (b/c) | P10–P60 | NE | — | NE | — | |
(a/b/c) indicates specific miR-29 variant reported in each study. Abbreviations: F, fetal day; NE, not examined; NR, not reported (unclear if NE or NS); NS, not significant; S1, primary somatosensory; V1, primary visual.
Table 4.
MiRNA sequences of interest.
| MiRNA | Sequence (Human/hsa-) | Sequence (Mouse/mmu-) | Match a | Targets of Interest |
|---|---|---|---|---|
| miR-25-3p | CAUUGCACUUGUCUCGGUCUGA | CAUUGCACUUGUCUCGGUCUGA | Yes | ATP2A2 (Serca2)b |
| miR-29a-3p | UAGCACCAUCUGAAAUCGGUUA | UAGCACCAUCUGAAAUCGGUUA | Yes | DNMT3A |
| miR-29b-3p | UAGCACCAUUUGAAAUCAGUGUU | UAGCACCAUUUGAAAUCAGUGUU | Yes | DNMT3A |
| miR-29c-3p | UAGCACCAUUUGAAAUCGGUUA | UAGCACCAUUUGAAAUCGGUUA | Yes | DNMT3A |
| miR-132-3p | UAACAGUCUACAGCCAUGGUCG | UAACAGUCUACAGCCAUGGUCG | Yes | DNMT3A |
| miR-137-3pb | UUAUUGCUUAAGAAUACGCGUAG | UUAUUGCUUAAGAAUACGCGUAG | Yes | GRIA1b, GRIN2Ab |
| miR-185-5p | UGGAGAGAAAGGCAGUUCCUGA | UGGAGAGAAAGGCAGUUCCUGA | Yes | ATP2A2 (Serca2)b |
| miR-338-3p | UCCAGCAUCAGUGAUUUUGUUG | UCCAGCAUCAGUGAUUUUGUUG | Yes | DRD2b |
| miR-382-3p | AAUCAUUCACGGACAACACUU | UCAUUCACGGACAACACUUUUU | No | DRD1 |
| miR-674-3p | no human ortholog | CACAGCUCCCAUCUCAGAACAA | No | DRD1 |
The seed sequence is underlined in each miRNA sequence. a Yes: indicates miRNA sequence is identical in human and mouse; No: indicates species difference. b Ripke et al., 2014: 108 loci associated with SCZ.
Table 5.
MiRNA levels in postmortem brains from patients with SCZ.
| Brain Region | Reference | Method | miR-29a/b/c-3p | miR-132-3p | miR-137-3p |
|---|---|---|---|---|---|
| DLPFC (BA9) | Perkins et al., 2007 [6] | Microarray, RT-qPCR (miR-29b) | Decreased (a/b/c) | NS | NS |
| Beveridge et al., 2010 [7] | Microarray | Increased (c) NS (a/b) | NS | NR | |
| Moreau et al., 2011 [8] | RT-qPCR | NS | NS | NE | |
| DLPFC (BA46) | Kim et al., 2010 [9] | RT-qPCR | NS | Increased | NS |
| Miller et al., 2012 [10] | Microarray RT-qPCR (miR-132-3p) | NS | Decreased | NS | |
| Hu et al., 2019 [11] | Small RNA-seq | NS | NS | NS | |
| STG (BA22) | Beveridge et al., 2008 [12] | Microarray | NS | NS | NE |
| Beveridge et al., 2010 [7] | Microarray | NS | NS | NR | |
| Amygdala | Liu et al., 2018 [13] | Small RNA-seq | NS | Decreased | NS |
(a/b/c) indicates specific miR-29 variant reported in each study. Abbreviations: BA, Brodmann’s area; DLPFC, dorsolateral prefrontal cortex; NE, not examined; NR, not reported (unclear if NE or NS); NS, not significant; STG, superior temporal gyrus.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Thomas, K.T.; Zakharenko, S.S. MicroRNAs in the Onset of Schizophrenia. Cells 2021, 10, 2679. https://doi.org/10.3390/cells10102679
AMA Style
Thomas KT, Zakharenko SS. MicroRNAs in the Onset of Schizophrenia. Cells. 2021; 10(10):2679. https://doi.org/10.3390/cells10102679
Chicago/Turabian StyleThomas, Kristen T., and Stanislav S. Zakharenko. 2021. "MicroRNAs in the Onset of Schizophrenia" Cells 10, no. 10: 2679. https://doi.org/10.3390/cells10102679
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.