
Molecular Determinants for RNA Release into Extracellular Vesicles


Abstract
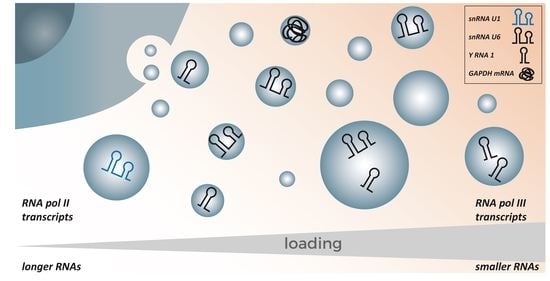
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Isolation of Extracellular Vesicles
2.3. SDS-PAGE and Western Blot Analysis
2.4. Electron Microscopy
2.5. Nano-Flow Cytometry (NanoFCM)
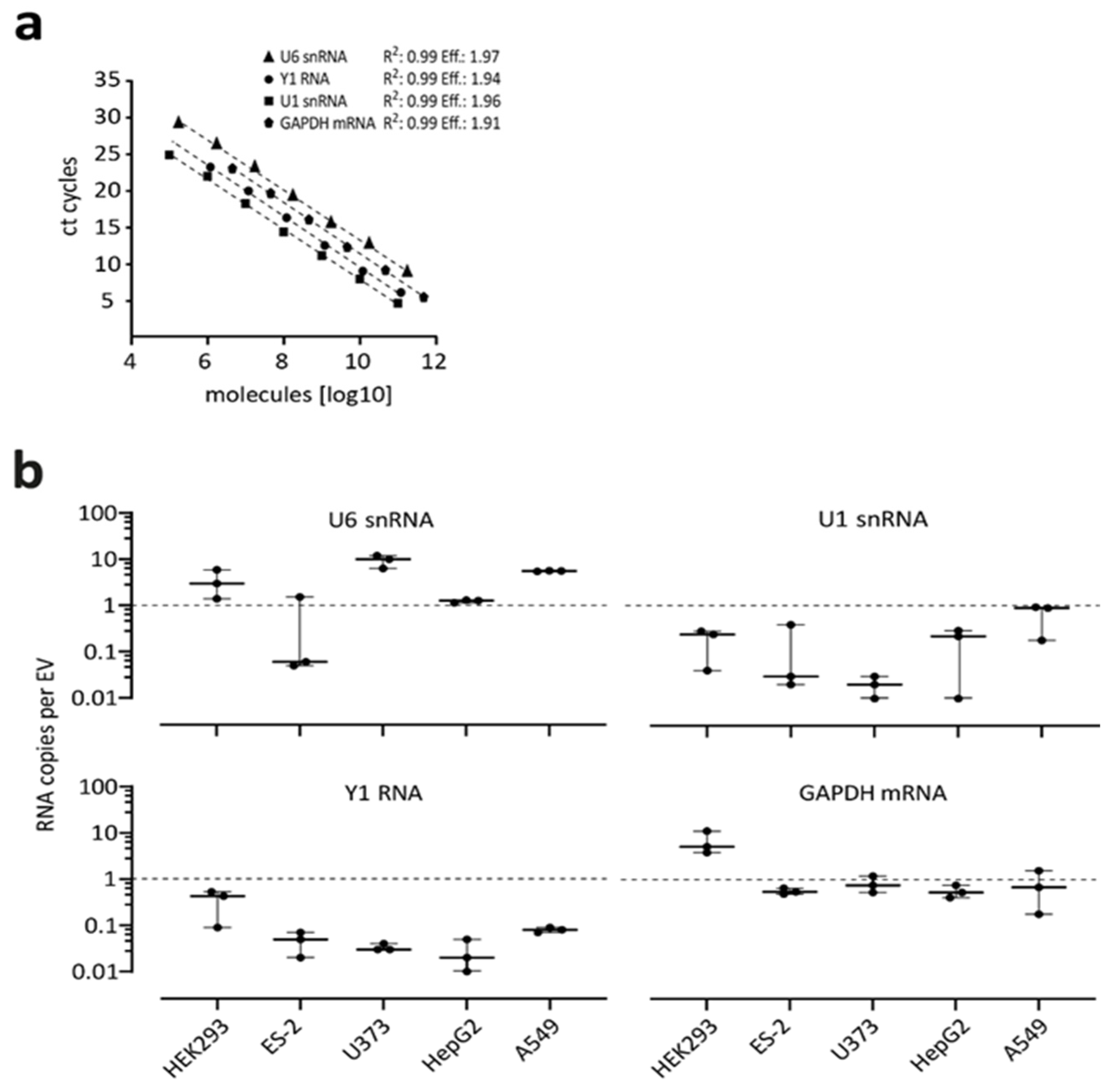
2.6. RNA Isolation, Relative and Absolute qPCR
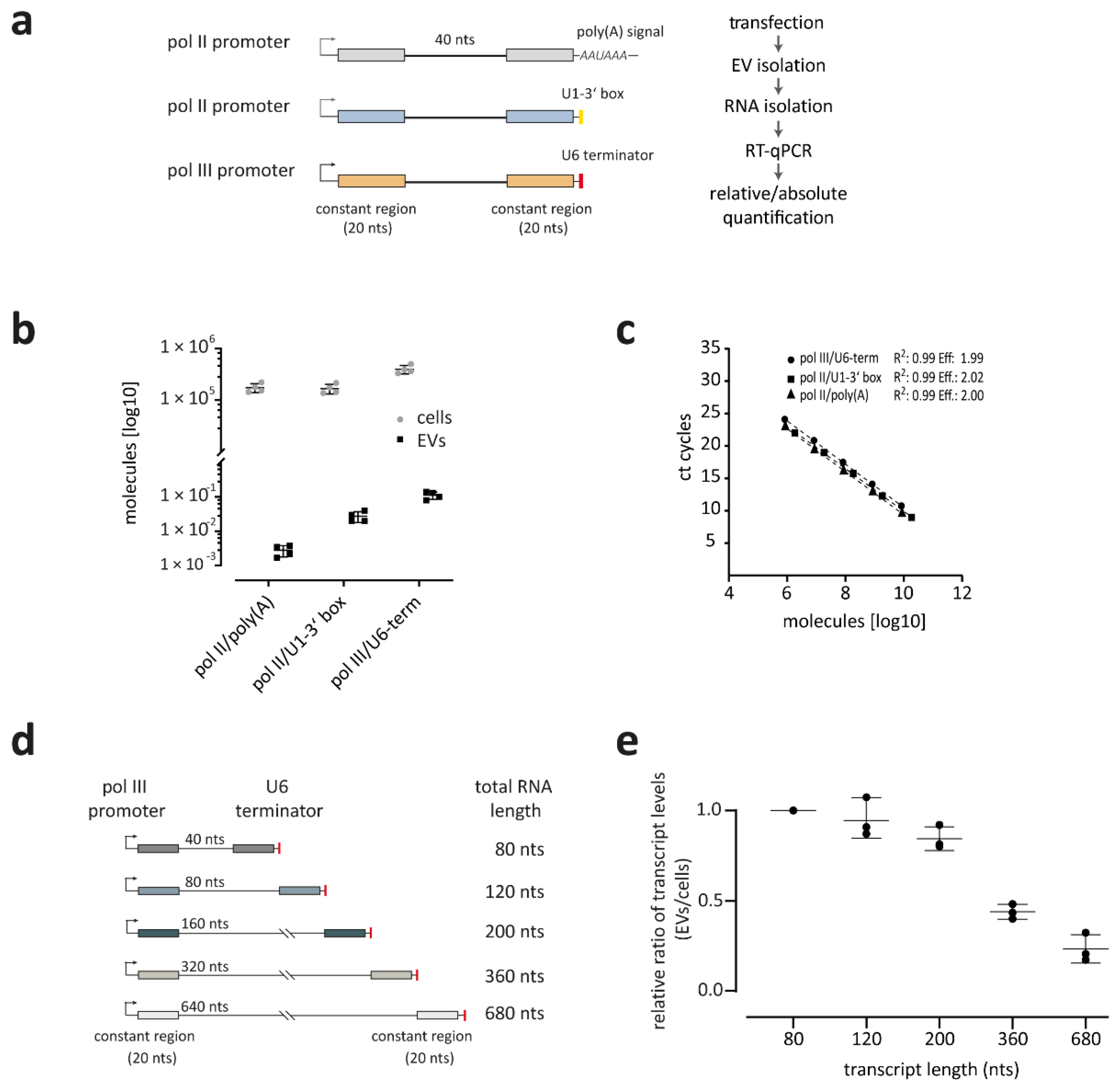
2.7. Plasmid Constructs
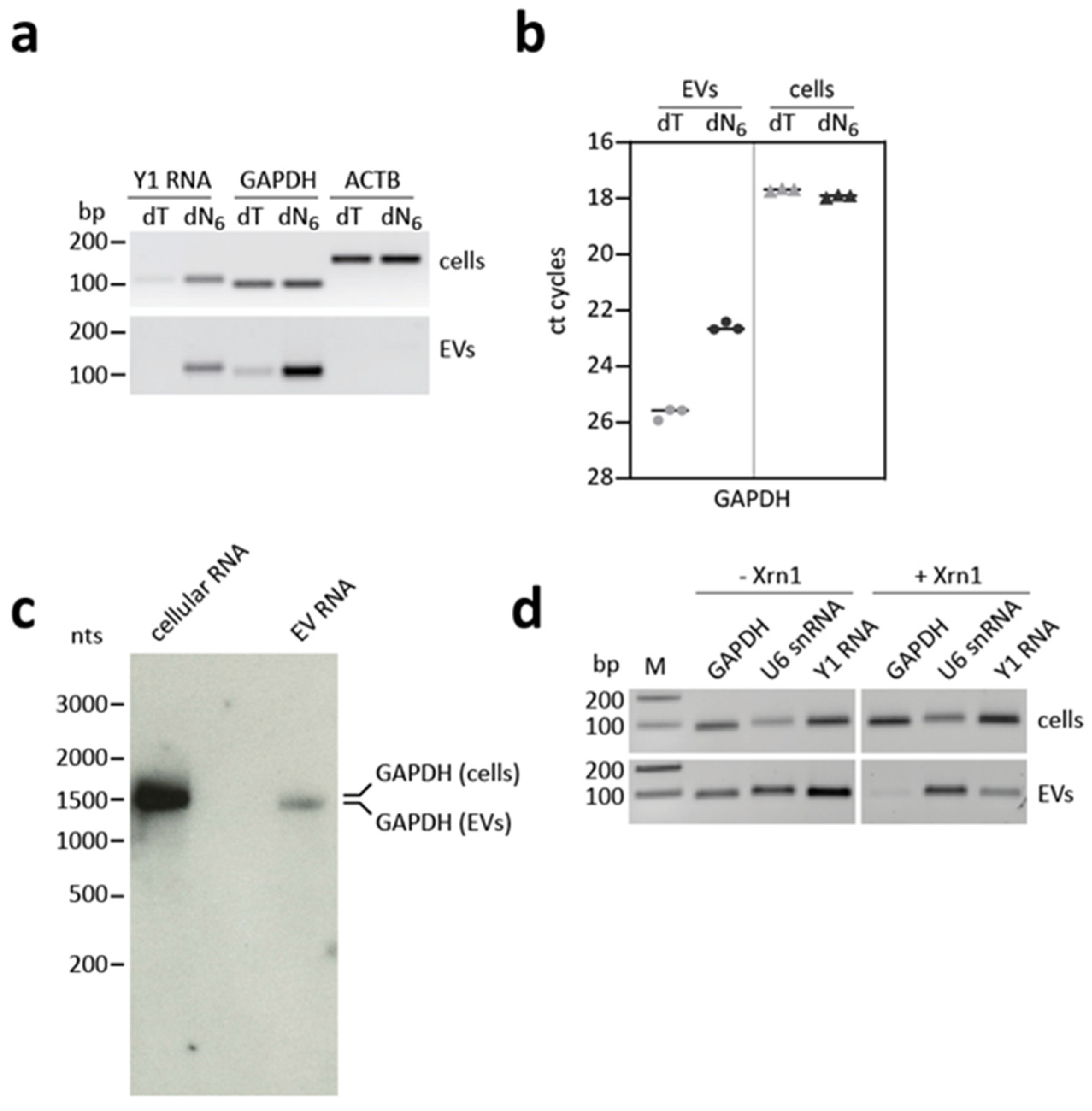
2.8. Northern Blot
3. Results
3.1. Isolation and Characterization of Extracellular Vesicles
3.2. RNA Polymerase III Transcripts Are Preferentially Loaded into EVs
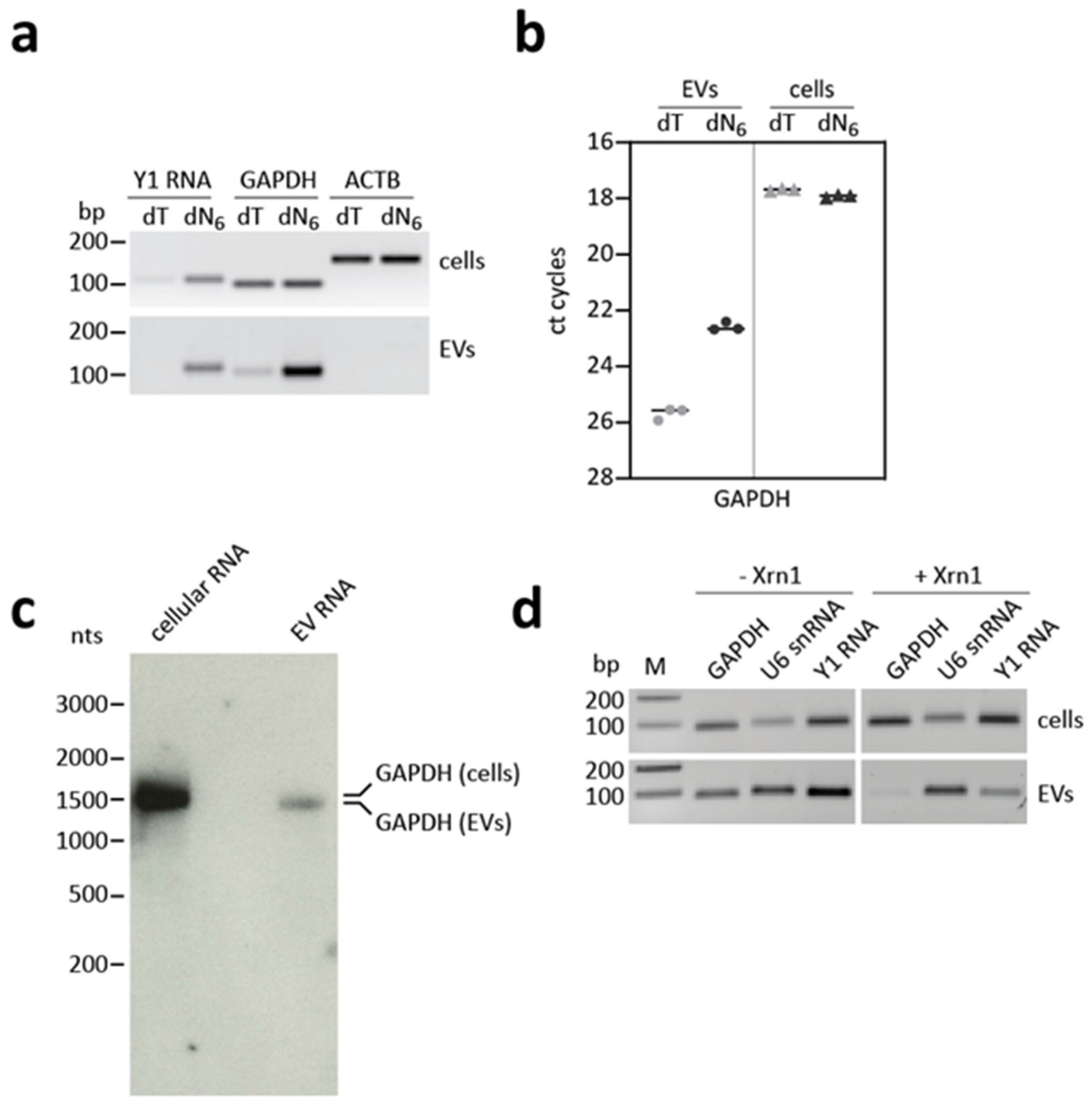
3.3. Smaller Transcripts Are Preferentially Loaded into EVs
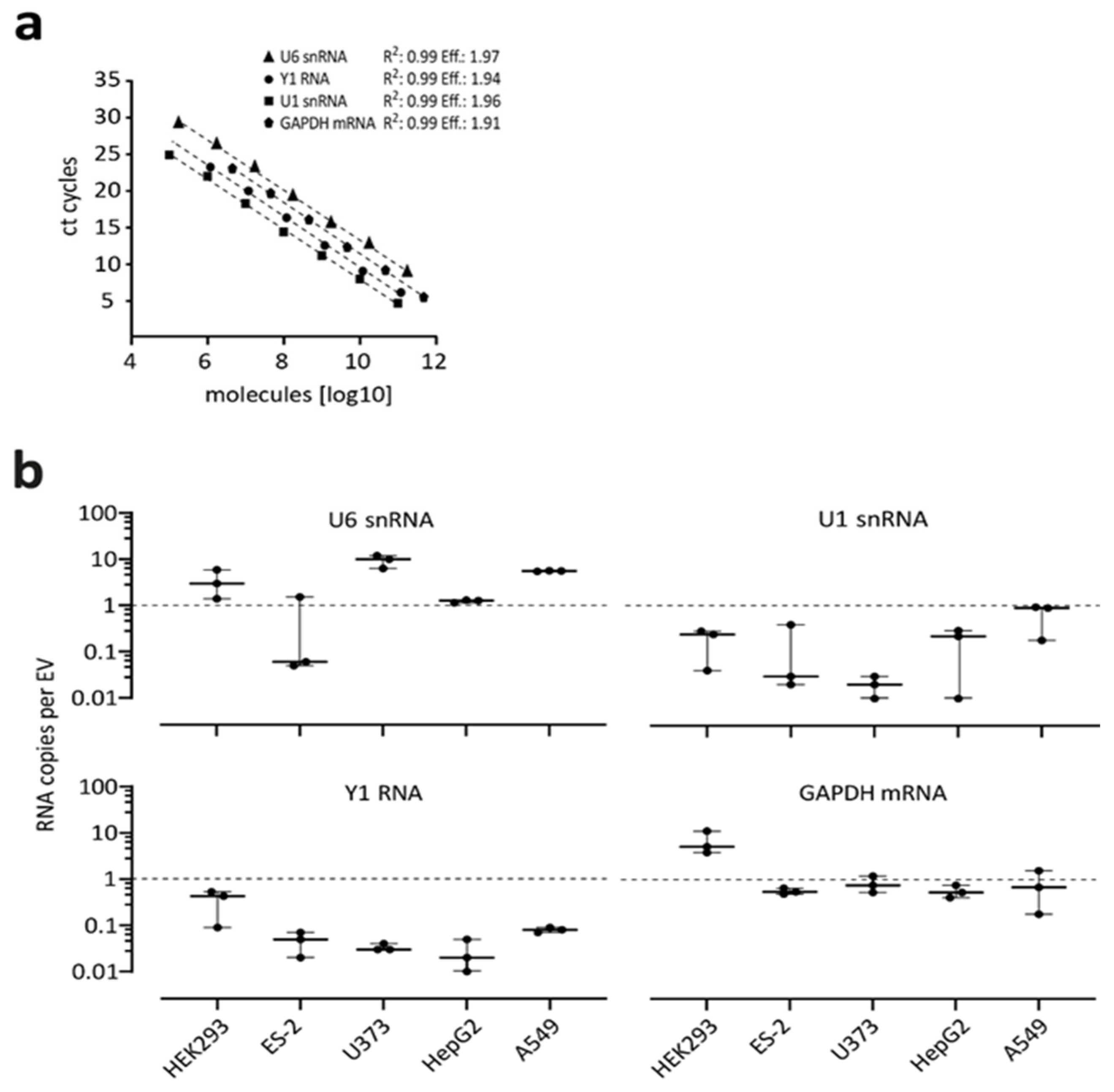
3.4. Absolute Quantification of Endogenous RNAs in EVs
3.5. GAPDH mRNA Shows an Aberrant Cap and Poly(A) Status
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simons, M.; Raposo, G. Exosomes-vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanada, M.; Bachmann, M.H.; Hardy, J.W.; Frimannson, D.O.; Bronsart, L.; Wang, A.; Sylvester, M.D.; Schmidt, T.L.; Kaspar, R.L.; Butte, M.J.; et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015, 112, E1433-42. [Google Scholar] [CrossRef] [Green Version]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular vesicles: Composition, biological relevance, and methods of study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Witwer, K.W.; Soekmadji, C.; Hill, A.F.; Wauben, M.H.; Buzás, E.I.; Di Vizio, D.; Falcon-Perez, J.M.; Gardiner, C.; Hochberg, F.; Kurochkin, I.V.; et al. Updating the MISEV minimal requirements for extracellular vesicle studies: Building bridges to reproducibility. J. Extracell. Vesicles 2017, 6, 1396823. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteomics 2010, 73, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [Green Version]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of exosome composition. Cell 2019, 177, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Kesidou, D.; da Costa, M.; Paula, A.; de Windt, L.J.; Brittan, M.; Beqqali, A.; Baker, A.H. Extracellular vesicle miRNAs in the promotion of cardiac neovascularisation. Front. Physiol. 2020, 11, 579892. [Google Scholar] [CrossRef]
- Park, S.; Lee, K.; Park, I.B.; Kim, N.H.; Cho, S.; Rhee, W.J.; Oh, Y.; Choi, J.; Nam, S.; Lee, D.H. The profiles of microRNAs from urinary extracellular vesicles (EVs) prepared by various isolation methods and their correlation with serum EV microRNAs. Diabetes Res. Clin. Pract. 2020, 160, 108010. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; Monique, A.J.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Würdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolte-’t Hoen, E.N.M.; Buermans, H.P.J.; Waasdorp, M.; Stoorvogel, W.; Wauben, M.H.M.; Peter, A.C. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res. 2012, 40, 9272–9285. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zheng, Q.; Bao, C.; Li, S.; Guo, W.; Zhao, J.; Chen, D.; Gu, J.; He, X.; Huang, S. Circular RNA is enriched and stable in exosomes: A promising biomarker for cancer diagnosis. Cell Res. 2015, 25, 981–984. [Google Scholar] [CrossRef] [Green Version]
- van Balkom, B.W.M.; Eisele, A.S.; Pegtel, D.M.; Bervoets, S.; Verhaar, M.C. Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting. J. Extracell. Vesicles 2015, 4, 26760. [Google Scholar] [CrossRef] [PubMed]
- Preußer, C.; Hung, L.-H.; Schneider, T.; Schreiner, S.; Hardt, M.; Moebus, A.; Santoso, S.; Bindereif, A. Selective release of circRNAs in platelet-derived extracellular vesicles. J. Extracell. Vesicles 2018, 7, 1424473. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M.Z. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batagov, A.O.; Kurochkin, I.V. Exosomes secreted by human cells transport largely mRNA fragments that are enriched in the 3′-untranslated regions. Biol. Direct 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarroya-Beltri, C.; Baixauli, F.; Gutiérrez-Vázquez, C.; Sánchez-Madrid, F.; Mittelbrunn, M. Sorting it out: Regulation of exosome loading. Semin. Cancer Biol. 2014, 28, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, L.; Giurato, G.; Cicchini, C.; Montaldo, C.; Mancone, C.; Tarallo, R.; Battistelli, C.; Alonzi, T.; Weisz, A.; Tripodi, M. The RNA-binding protein SYNCRIP is a component of the hepatocyte exosomal machinery controlling microRNA sorting. Cell Rep. 2016, 17, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Shurtleff, M.J.; Temoche-Diaz, M.M.; Karfilis, K.V.; Ri, S.; Schekman, R. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife 2016, 5. [Google Scholar] [CrossRef]
- Groot, M.; Lee, H. Sorting mechanisms for microRNAs into extracellular vesicles and their associated diseases. Cells 2020, 9, 1044. [Google Scholar] [CrossRef] [PubMed]
- Zietzer, A.; Hosen, M.R.; Wang, H.; Goody, P.R.; Sylvester, M.; Latz, E.; Nickenig, G.; Werner, N.; Jansen, F. The RNA-binding protein hnRNPU regulates the sorting of microRNA-30c-5p into large extracellular vesicles. J. Extracell. Vesicles 2020, 9, 1786967. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [Green Version]
- Gámbaro, F.; Li Calzi, M.; Fagúndez, P.; Costa, B.; Greif, G.; Mallick, E.; Lyons, S.; Ivanov, P.; Witwer, K.; Cayota, A.; et al. Stable tRNA halves can be sorted into extracellular vesicles and delivered to recipient cells in a concentration-dependent manner. RNA Biol. 2020, 17, 1168–1182. [Google Scholar] [CrossRef] [PubMed]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Batagov, A.O.; Schinelli, S.; Wang, J.; Wang, Y.; El Fatimy, R.; Rabinovsky, R.; Balaj, L.; Chen, C.C.; Hochberg, F.; et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 2017, 8, 1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albanese, M.; Chen, Y.-F.A.; Hüls, C.; Gärtner, K.; Tagawa, T.; Mejias-Perez, E.; Keppler, O.T.; Göbel, C.; Zeidler, R.; Shein, M.; et al. Micro RNAs are minor constituents of extracellular vesicles and are hardly delivered to target cells. bioRvix 2020. [Google Scholar] [CrossRef]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef]
- Olejniczak, S.H.; La Rocca, G.; Gruber, J.J.; Thompson, C.B. Long-lived microRNA-Argonaute complexes in quiescent cells can be activated to regulate mitogenic responses. Proc. Natl. Acad. Sci. USA 2013, 110, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Mateescu, B.; Kowal, E.J.K.; van Balkom, B.W.M.; Bartel, S.; Bhattacharyya, S.N.; Buzás, E.I.; Buck, A.H.; de Candia, P.; Chow, F.W.N.; Das, S.; et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA - an ISEV position paper. J. Extracell. Vesicles 2017, 6, 1286095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- de Miguel Pérez, D.; Rodriguez Martínez, A.; Ortigosa Palomo, A.; Delgado Ureña, M.; Garcia Puche, J.L.; Robles Remacho, A.; Exposito Hernandez, J.; Lorente Acosta, J.A.; Ortega Sánchez, F.G.; Serrano, M.J. Extracellular vesicle-miRNAs as liquid biopsy biomarkers for disease identification and prognosis in metastatic colorectal cancer patients. Sci. Rep. 2020, 10, 3974. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.; Busatto, S.; Pham, A.; Tian, M.; Suh, A.; Carson, K.; Quintero, A.; Lafrence, M.; Malik, H.; Santana, M.X.; et al. Extracellular vesicle-based drug delivery systems for cancer treatment. Theranostics 2019, 9, 8001–8017. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; He, C.; Hao, Y.; Wang, L.; Li, L.; Zhu, G. Prospects and challenges of extracellular vesicle-based drug delivery system: Considering cell source. Drug Deliv. 2020, 27, 585–598. [Google Scholar] [CrossRef] [Green Version]
- Gopal, A.; Zhou, Z.H.; Knobler, C.M.; Gelbart, W.M. Visualizing large RNA molecules in solution. RNA 2012, 18, 284–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Reddy, R. Gamma-monomethyl phosphate: A cap structure in spliceosomal U6 small nuclear RNA. Proc. Natl. Acad. Sci. USA. 1989, 86, 8280–8283. [Google Scholar] [CrossRef] [Green Version]
- Köhn, M.; Pazaitis, N.; Hüttelmaier, S. Why YRNAs? About versatile RNAs and their functions. Biomolecules 2013, 3, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schäfer, R.; Beerling, E.; Schiffelers, R.M.; de Wit, E.; Berenguer, J.; Ellenbroek, S.I.J.; et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosbach, M.-L.; Pfafenrot, C.; von Strandmann, E.P.; Bindereif, A.; Preußer, C. Molecular Determinants for RNA Release into Extracellular Vesicles. Cells 2021, 10, 2674. https://doi.org/10.3390/cells10102674
Mosbach M-L, Pfafenrot C, von Strandmann EP, Bindereif A, Preußer C. Molecular Determinants for RNA Release into Extracellular Vesicles. Cells. 2021; 10(10):2674. https://doi.org/10.3390/cells10102674
Chicago/Turabian StyleMosbach, Marie-Luise, Christina Pfafenrot, Elke Pogge von Strandmann, Albrecht Bindereif, and Christian Preußer. 2021. "Molecular Determinants for RNA Release into Extracellular Vesicles" Cells 10, no. 10: 2674. https://doi.org/10.3390/cells10102674
APA StyleMosbach, M.-L., Pfafenrot, C., von Strandmann, E. P., Bindereif, A., & Preußer, C. (2021). Molecular Determinants for RNA Release into Extracellular Vesicles. Cells, 10(10), 2674. https://doi.org/10.3390/cells10102674