
Roles of Crosstalk between Astrocytes and Microglia in Triggering Neuroinflammation and Brain Edema Formation in 1,2-Dichloroethane-Intoxicated Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents
2.3. Experimental Procedures and Treatment
2.4. Analysis
2.4.1. Brain Water Content
2.4.2. Histological Observation
2.4.3. Western Blot
2.4.4. ELISA
2.5. Statistical Analysis
3. Results
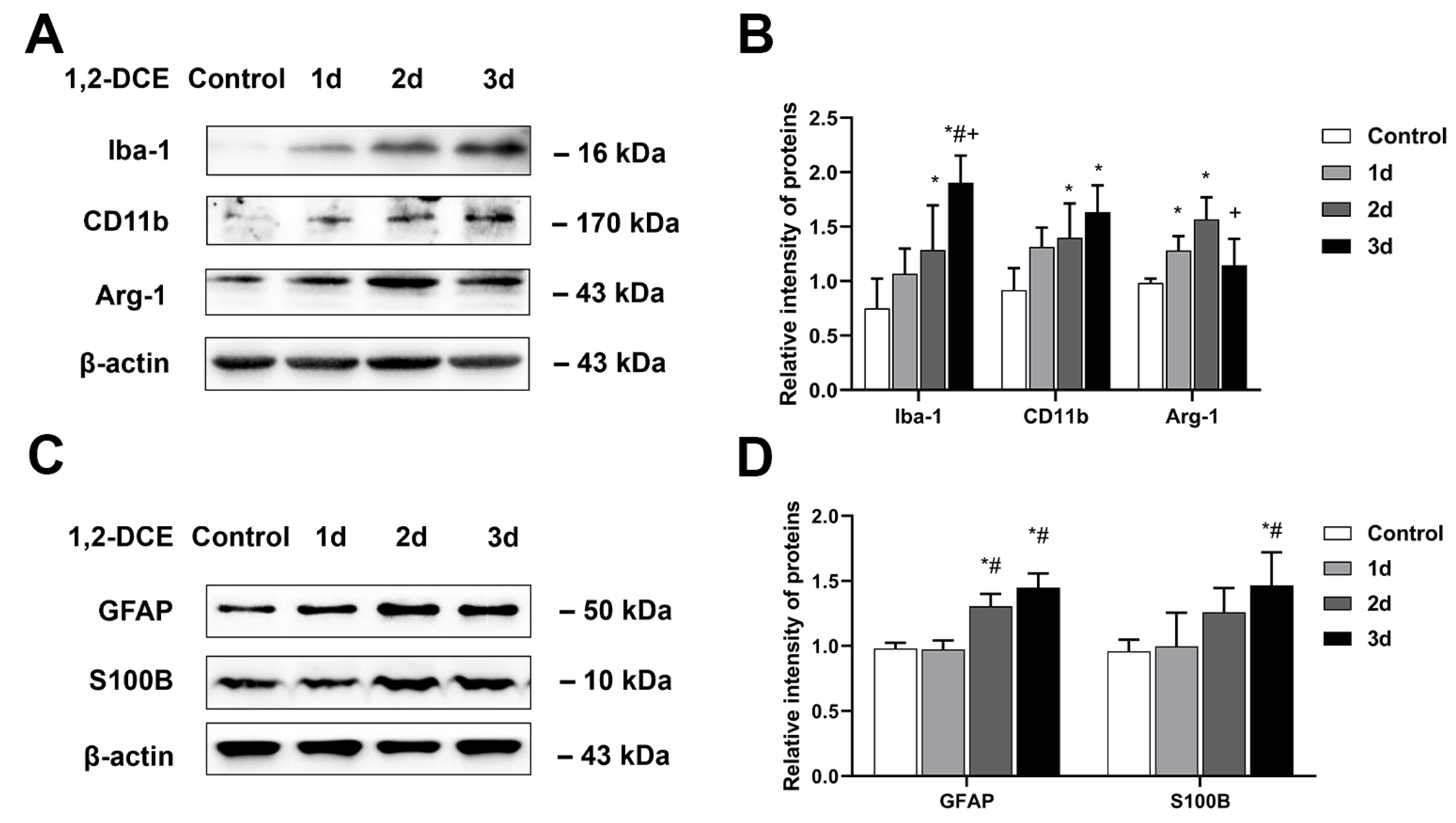
3.1. Effects of 1,2-DCE on Microglial Polarization during the Process of Brain Edema Formation in Mice
3.2. Roles of Activated Microglia in 1,2-DCE-Induced Neuroinflammation and Brain Edema in Mice
3.3. Role of TLR4/MyD88/NF-κB Signaling Pathway in Microglial Activation in 1,2-DCE-Intoxicated Mice
3.4. Roles of Reactive Astrocytes in Microglial Activation and Neuroinflammation in 1,2-DCE-Intoxicated Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, G.; Yuan, Y.; Zhang, J.; Gao, L.; Tan, X.; Yang, G.; Lv, X.; Jin, Y. Roles of aquaporins and matrix metalloproteinases in mouse brain edema formation induced by subacute exposure to 1,2-dichloroethane. Neurotoxicol. Teratol. 2014, 44, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Janssen, D.; van der Ploeg, J.; Pries, F. Genetics and biochemistry of 1,2-dichloroethane degradation. Biodegradation 1994, 5, 249–257. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jin, X.; Wang, T.; Liao, Y.; Guo, J.; Wang, G.; Zhao, F.; Jin, Y. Neuroinflammatory Reactions in the Brain of 1,2-DCE-Intoxicated Mice during Brain Edema. Cells 2019, 8, 987. [Google Scholar] [CrossRef]
- Zhao, Z.; Bao, X.Q.; Zhang, Z.; Liu, H.; Zhang, D. Phloroglucinol derivative compound 21 attenuates cuprizone-induced multiple sclerosis mice through promoting remyelination and inhibiting neuroinflammation. Sci. China Life Sci. 2020, 63, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Chugh, H.; Sakharkar, M.K.; Dhawan, U.; Chidambaram, S.B.; Chandra, R. Neuroinflammation Mechanisms and Phytotherapeutic Intervention: A Systematic Review. ACS Chem. Neurosci. 2020, 11, 3707–3731. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef]
- Meng, X.; Baine, J.M.; Yan, T.; Wang, S. Comprehensive Analysis of Lysine Lactylation in Rice (Oryza sativa) Grains. J. Agric. Food Chem. 2021, 69, 8287–8297. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Luo, X.G.; Chen, S.D. The changing phenotype of microglia from homeostasis to disease. Transl. Neurodegener. 2012, 1, 9. [Google Scholar] [CrossRef]
- Loane, D.J.; Byrnes, K.R. Role of microglia in neurotrauma. Neurotherapeutics 2010, 7, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Kirkley, K.S.; Popichak, K.A.; Afzali, M.F.; Legare, M.E.; Tjalkens, R.B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. NeuroInflamm. 2017, 14, 99. [Google Scholar] [CrossRef]
- Schirmer, L.; Schafer, D.P.; Bartels, T.; Rowitch, D.H.; Calabresi, P.A. Diversity and Function of Glial Cell Types in Multiple Sclerosis. Trends Immunol. 2021, 42, 228–247. [Google Scholar] [CrossRef]
- Wang, T.; Liao, Y.; Sun, Q.; Tang, H.; Wang, G.; Zhao, F.; Jin, Y. Upregulation of Matrix Metalloproteinase-9 in Primary Cultured Rat Astrocytes Induced by 2-Chloroethanol Via MAPK Signal Pathways. Front. Cell Neurosci. 2017, 11, 218. [Google Scholar] [CrossRef]
- Campisi, M.; Shin, Y.; Osaki, T.; Hajal, C.; Chiono, V.; Kamm, R.D. 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials 2018, 180, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, D.; Fan, Q.; Rong, W.; Zheng, J.; Gao, C.; Li, G.; Zeng, N.; Guo, T.; Zeng, L.; et al. 1,2-Dichloroethane impairs glucose and lipid homeostasis in the livers of NIH Swiss mice. Toxicology 2017, 380, 38–49. [Google Scholar] [CrossRef]
- Sanz, P.; Garcia-Gimeno, M.A. Reactive Glia Inflammatory Signaling Pathways and Epilepsy. Int. J. Mol. Sci. 2020, 21, 4096. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Qi, Y.; Gao, L.; Li, G.; Lv, X.; Jin, Y.P. Effects of subacute exposure to 1,2-dichloroethane on mouse behavior and the related mechanisms. Hum. Exp. Toxicol. 2013, 32, 983–991. [Google Scholar] [CrossRef]
- Jin, X.; Liao, Y.; Tan, X.; Guo, J.; Wang, G.; Zhao, F.; Jin, Y. Involvement of the p38 MAPK signaling pathway in overexpression of matrix metalloproteinase-9 during the course of brain edema in 1,2-dichloroethane-intoxicated mice. Neurotoxicology 2018, 69, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yuan, Y.; Gao, L.; Tan, X.; Yang, G.; Zhao, F.; Jin, Y. Disruption of Intracellular ATP Generation and Tight Junction Protein Expression during the Course of Brain Edema Induced by Subacute Poisoning of 1,2-Dichloroethane. Front. Neurosci. 2018, 12, 12. [Google Scholar] [CrossRef]
- Rodgers, J. Trypanosomiasis and the brain. Parasitology 2010, 137, 1995–2006. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Ma, E.L.; Smith, A.D.; Desai, N.; Cheung, L.; Hanscom, M.; Stoica, B.A.; Loane, D.J.; Shea-Donohue, T.; Faden, A.I. Bidirectional brain-gut interactions and chronic pathological changes after traumatic brain injury in mice. Brain Behav. Immun. 2017, 66, 56–69. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 2018, 21, 1380–1391. [Google Scholar] [CrossRef]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol. 2016, 173, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Cunningham, C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019, 40, 358–374. [Google Scholar] [CrossRef] [PubMed]
- Trias, E.; Ibarburu, S.; Barreto-Nunez, R.; Barbeito, L. Significance of aberrant glial cell phenotypes in pathophysiology of amyotrophic lateral sclerosis. Neurosci. Lett. 2017, 636, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Bardi, G.T.; Smith, M.A.; Hood, J.L. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine 2018, 105, 63–72. [Google Scholar] [CrossRef]
- Lisi, L.; Ciotti, G.M.; Braun, D.; Kalinin, S.; Curro, D.; Dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Han, X.; Li, Q.; Yang, Q.W.; Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 2017, 13, 420–433. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Tse, J.K.Y. Gut Microbiota, Nitric Oxide, and Microglia as Prerequisites for Neurodegenerative Disorders. ACS Chem. Neurosci. 2017, 8, 1438–1447. [Google Scholar] [CrossRef]
- Scholz, R.; Sobotka, M.; Caramoy, A.; Stempfl, T.; Moehle, C.; Langmann, T. Minocycline counter-regulates pro-inflammatory microglia responses in the retina and protects from degeneration. J. Neuroinflamm. 2015, 12, 209. [Google Scholar] [CrossRef]
- Du, X.; Xu, Y.; Chen, S.; Fang, M. Inhibited CSF1R Alleviates Ischemia Injury via Inhibition of Microglia M1 Polarization and NLRP3 Pathway. Neural Plast. 2020, 2020, 8825954. [Google Scholar] [CrossRef]
- Wang, X.; Li, S.; Ma, J.; Wang, C.; Chen, A.; Xin, Z.; Zhang, J. Effect of Gastrodin on Early Brain Injury and Neurological Outcome After Subarachnoid Hemorrhage in Rats. Neurosci. Bull. 2019, 35, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Zusso, M.; Lunardi, V.; Franceschini, D.; Pagetta, A.; Lo, R.; Stifani, S.; Frigo, A.C.; Giusti, P.; Moro, S. Ciprofloxacin and levofloxacin attenuate microglia inflammatory response via TLR4/NF-kB pathway. J. Neuroinflamm. 2019, 16, 148. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Deng, Y.; Li, F.; Yin, C.; Shi, J.; Gong, Q. Icariside II attenuates lipopolysaccharide-induced neuroinflammation through inhibiting TLR4/MyD88/NF-kappaB pathway in rats. Biomed. Pharmacother. 2019, 111, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.H.; Kim, Y.K.; Kim, M.R.; Jang, J.H.; Lee, S. Emerging Roles of Vascular Cell Adhesion Molecule-1 (VCAM-1) in Immunological Disorders and Cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Sharp, F.R. Implications of MMP-9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front. Cell Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef]
- Gawdi, R.; Emmady, P.D. Physiology, Blood Brain Barrier; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013, 4, e525. [Google Scholar] [CrossRef]
- Wang, G.; Li, Z.; Li, S.; Ren, J.; Suresh, V.; Xu, D.; Zang, W.; Liu, X.; Li, W.; Wang, H.; et al. Minocycline Preserves the Integrity and Permeability of BBB by Altering the Activity of DKK1-Wnt Signaling in ICH Model. Neuroscience 2019, 415, 135–146. [Google Scholar] [CrossRef]
- Ling, J.; Kumar, R. Crosstalk between NFkB and glucocorticoid signaling: A potential target of breast cancer therapy. Cancer Lett. 2012, 322, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jin, X.; Liao, Y.; Sun, Q.; Luo, C.; Wang, G.; Zhao, F.; Jin, Y. Association of NF-kappaB and AP-1 with MMP-9 Overexpression in 2-Chloroethanol Exposed Rat Astrocytes. Cells 2018, 7, 96. [Google Scholar] [CrossRef]
- Macaisa, C.M.; Watabe, T.; Liu, Y.; Romanov, V.; Kanai, Y.; Horitsugi, G.; Kato, H.; Shimosegawa, E.; Hatazawa, J. Preserved Cerebral Oxygen Metabolism in Astrocytic Dysfunction: A Combination Study of (15)O-Gas PET with (14)C-Acetate Autoradiography. Brain Sci. 2019, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Ikram, M.; Ullah, N.; Alam, S.I.; Park, H.Y.; Badshah, H.; Choe, K.; Kim, M.O. Neurological Enhancement Effects of Melatonin against Brain Injury-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration via AMPK/CREB Signaling. Cells 2019, 8, 760. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Liu, M.; Xu, S.; Hong, H.; Chen, M.; Tian, L.; Xie, J.; Deng, P.; Zhou, C.; Zhang, L.; et al. Inhibition of SERPINA3N-dependent neuroinflammation is essential for melatonin to ameliorate trimethyltin chloride-induced neurotoxicity. J. Pineal Res. 2019, 67, e12596. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Wang, T.; Jin, X.; Wang, G.; Zhao, F.; Jin, Y. Roles of Crosstalk between Astrocytes and Microglia in Triggering Neuroinflammation and Brain Edema Formation in 1,2-Dichloroethane-Intoxicated Mice. Cells 2021, 10, 2647. https://doi.org/10.3390/cells10102647
Yang J, Wang T, Jin X, Wang G, Zhao F, Jin Y. Roles of Crosstalk between Astrocytes and Microglia in Triggering Neuroinflammation and Brain Edema Formation in 1,2-Dichloroethane-Intoxicated Mice. Cells. 2021; 10(10):2647. https://doi.org/10.3390/cells10102647
Chicago/Turabian StyleYang, Jinhan, Tong Wang, Xiaoxia Jin, Gaoyang Wang, Fenghong Zhao, and Yaping Jin. 2021. "Roles of Crosstalk between Astrocytes and Microglia in Triggering Neuroinflammation and Brain Edema Formation in 1,2-Dichloroethane-Intoxicated Mice" Cells 10, no. 10: 2647. https://doi.org/10.3390/cells10102647
APA StyleYang, J., Wang, T., Jin, X., Wang, G., Zhao, F., & Jin, Y. (2021). Roles of Crosstalk between Astrocytes and Microglia in Triggering Neuroinflammation and Brain Edema Formation in 1,2-Dichloroethane-Intoxicated Mice. Cells, 10(10), 2647. https://doi.org/10.3390/cells10102647