
Dysfunction of Glutamate Delta-1 Receptor-Cerebellin 1 Trans-Synaptic Signaling in the Central Amygdala in Chronic Pain

 , , , ,
, , , ,
Abstract
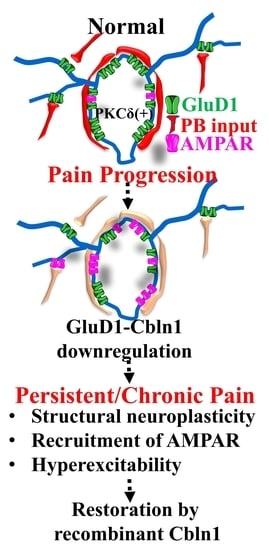
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Animals
2.2. Immunohistochemistry
2.3. Whole-Cell Electrophysiology
2.4. Synaptoneurosome Preparation and Western Blot Analysis
2.5. Stereotaxic Surgery
2.5.1. Stereotaxic AAV Injection
2.5.2. Cannulation
2.6. Pain Induction Models
2.6.1. CFA Inflammatory Pain Model
2.6.2. Spinal Nerve Ligation Pain Model
2.7. Drug Administration
2.7.1. Recombinant Cerebellin 1 (Cbln1) Administration
2.7.2. Chemogenetics
2.8. Behavioral Assessment Tests in Mouse
2.8.1. Von Frey Filament Test
2.8.2. Tail Flick Test
2.8.3. Hotplate Test
2.8.4. Gabapentin Induced Conditioned Place Preference
2.8.5. Formalin Test
2.8.6. Fear Conditioning
2.9. Behavioral Assessment Tests in Rats
2.9.1. Vocalization
2.9.2. Von Frey Test
2.9.3. Elevated plus Maze
2.10. Statistics
3. Results
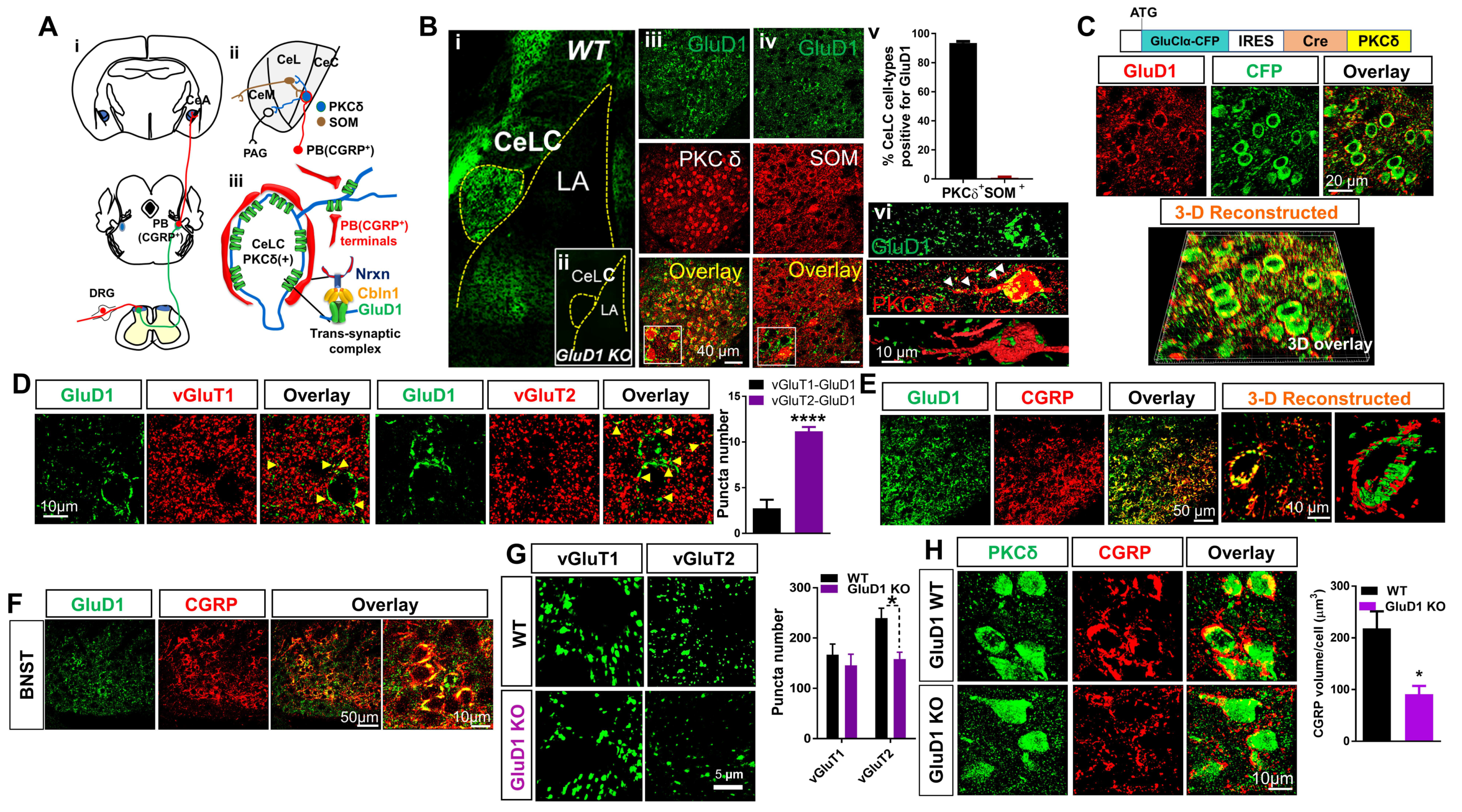
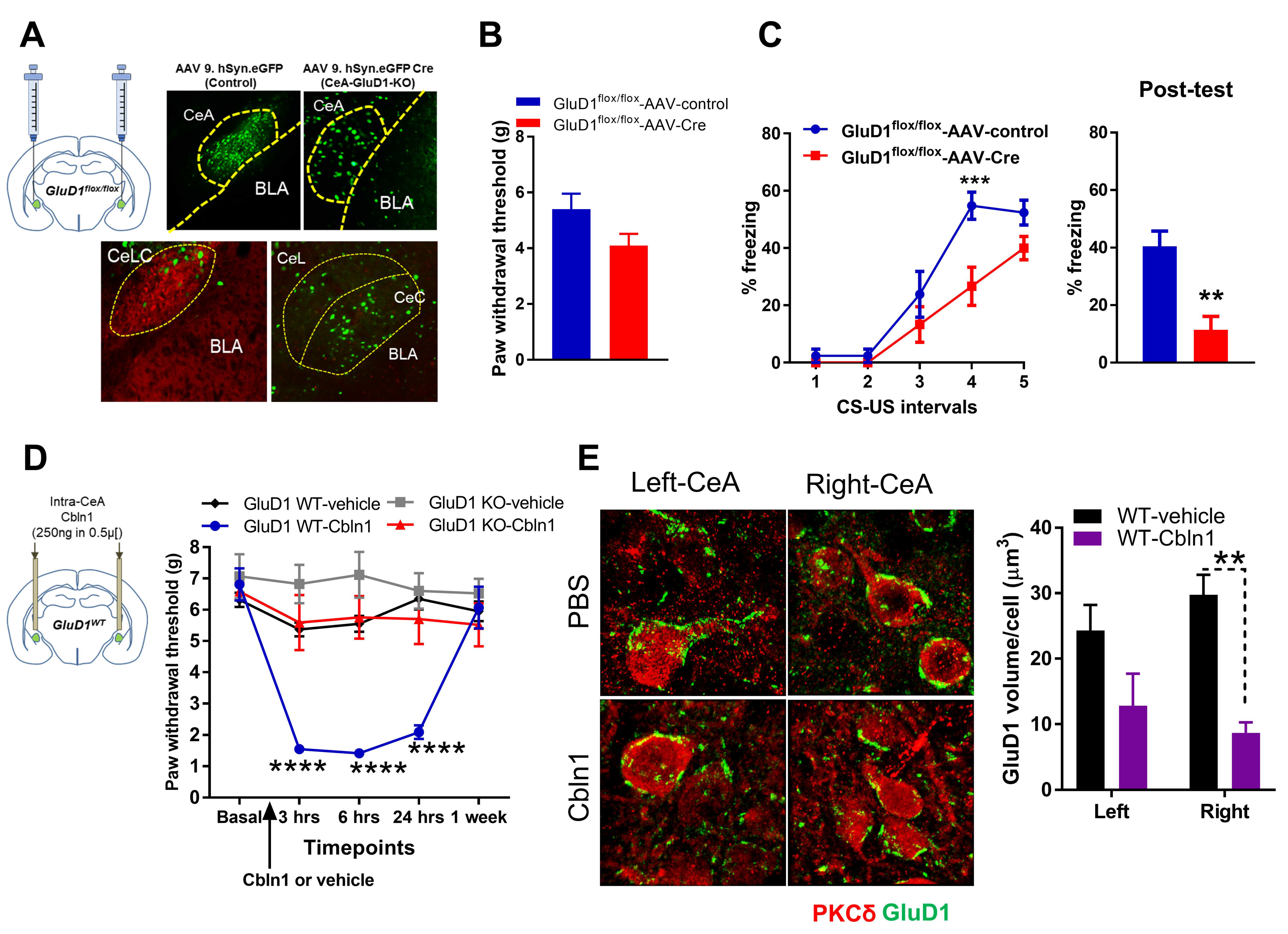
3.1. Unique Cell- and Input-Specific Expression of GluD1 in the Central Amygdala
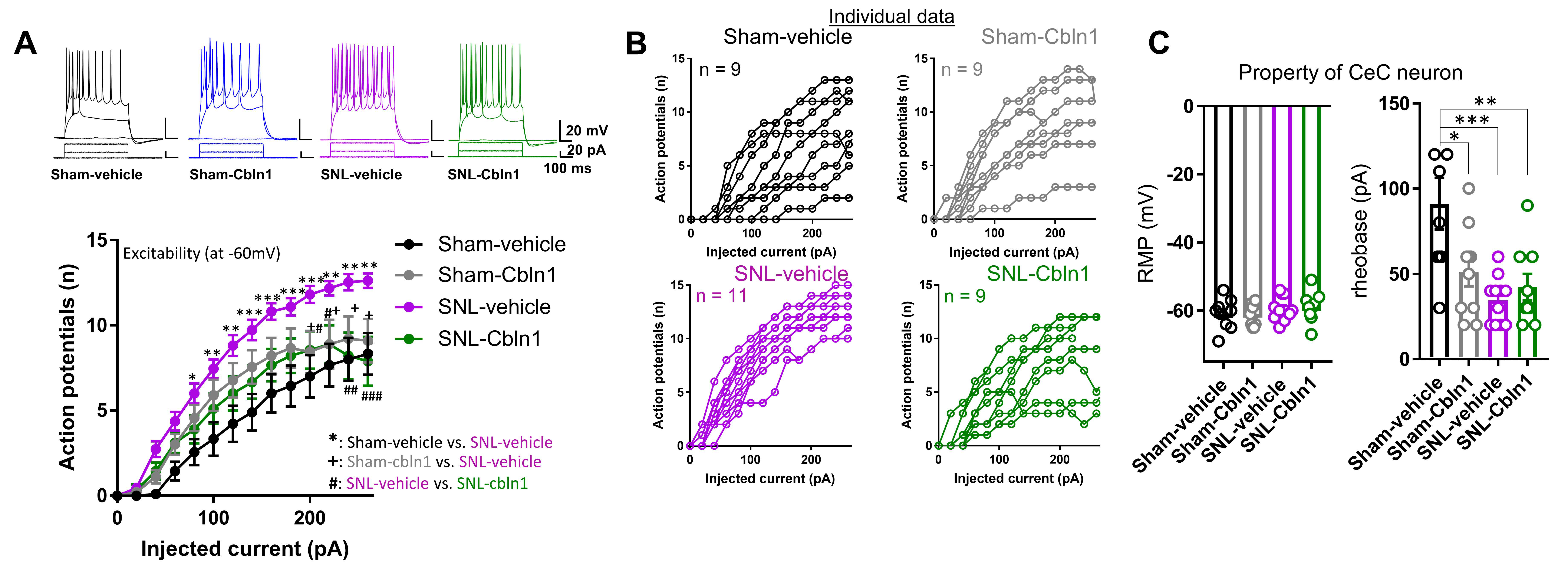
3.2. GluD1 Is Necessary for Normal Excitatory Neurotransmission at the Parabrachio-Amygdala Synapses
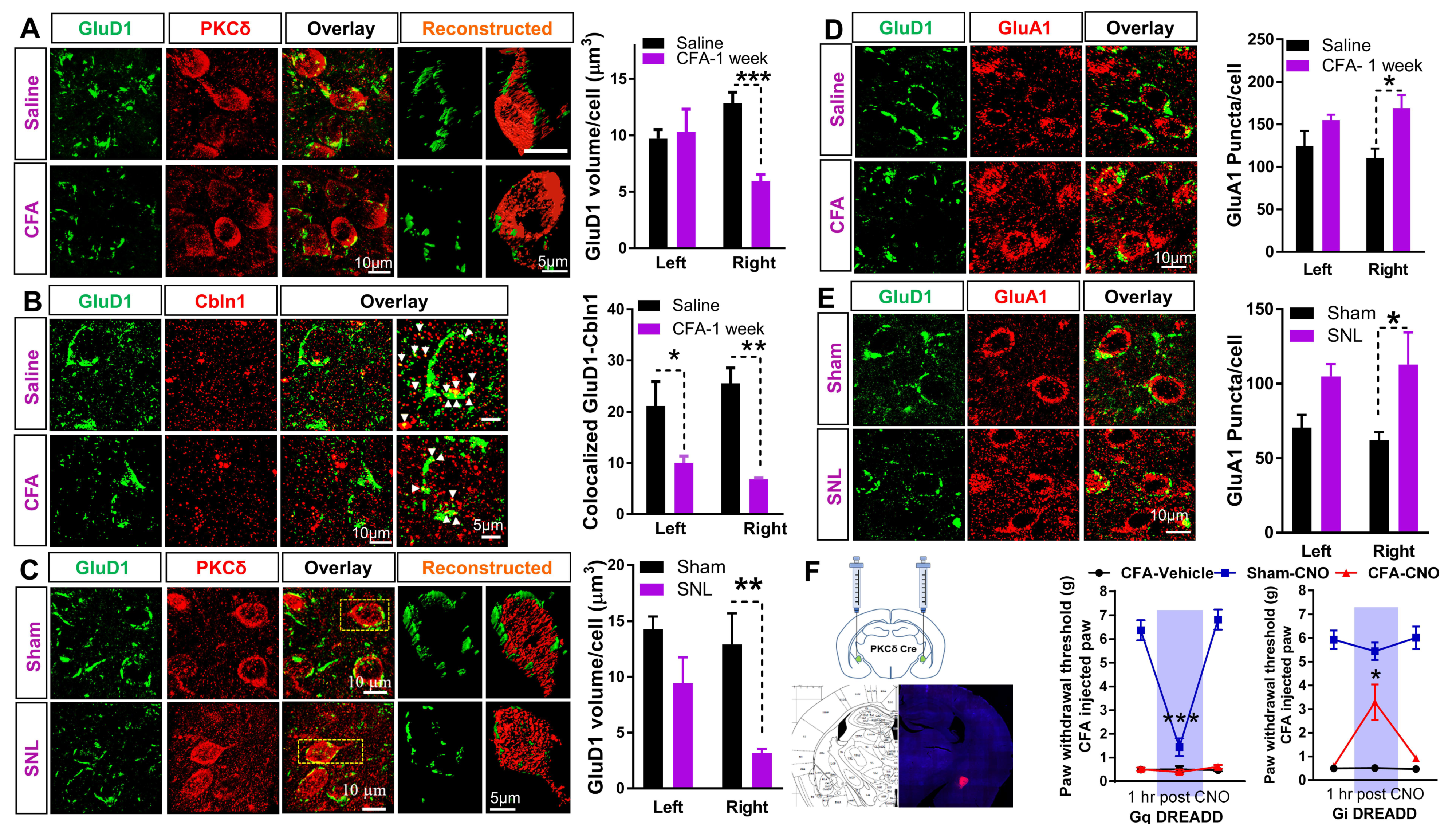
3.3. Downregulation of GluD1-Cbln1 in the CeLC in Inflammatory and Neuropathic Pain Models
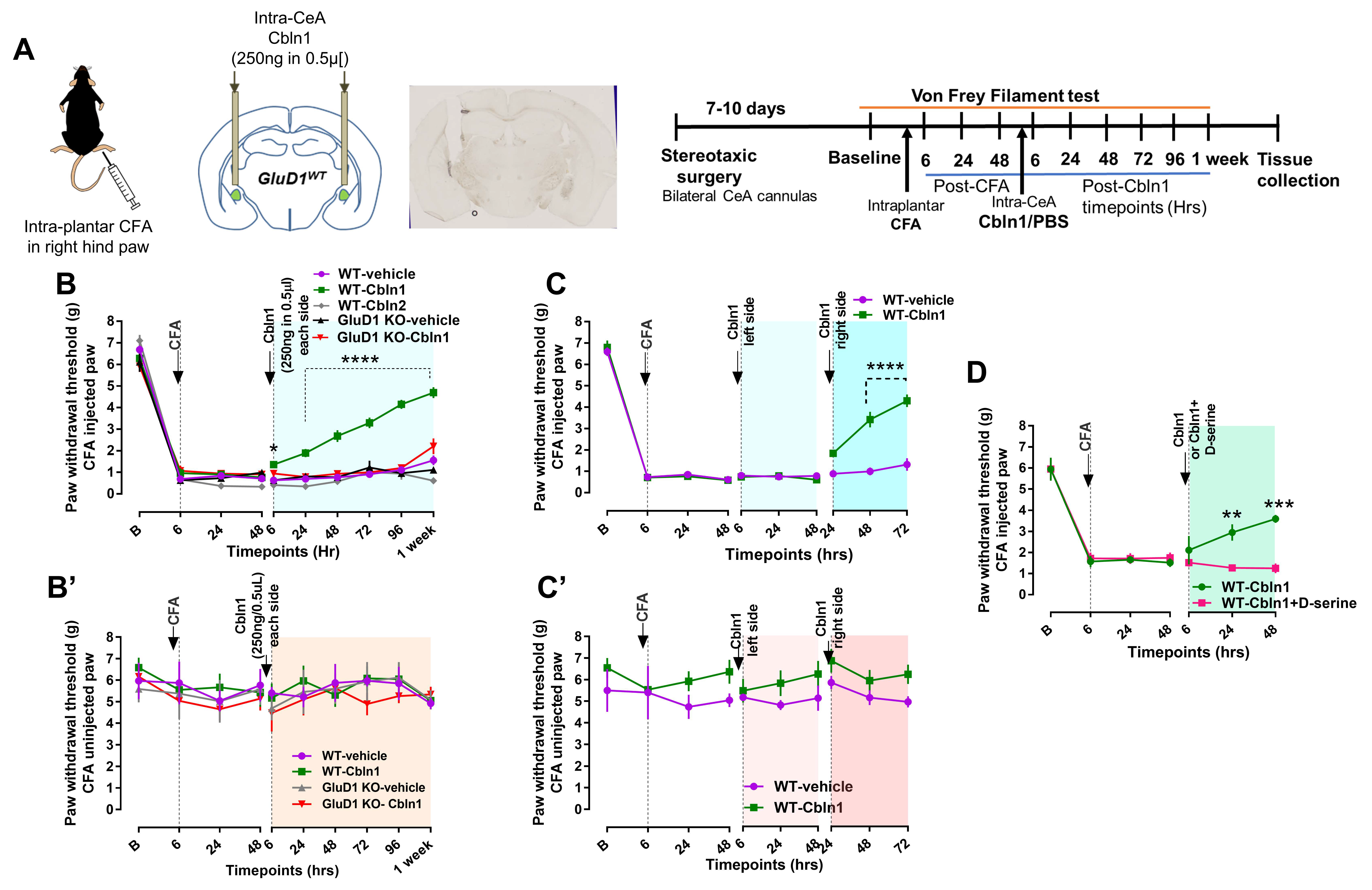
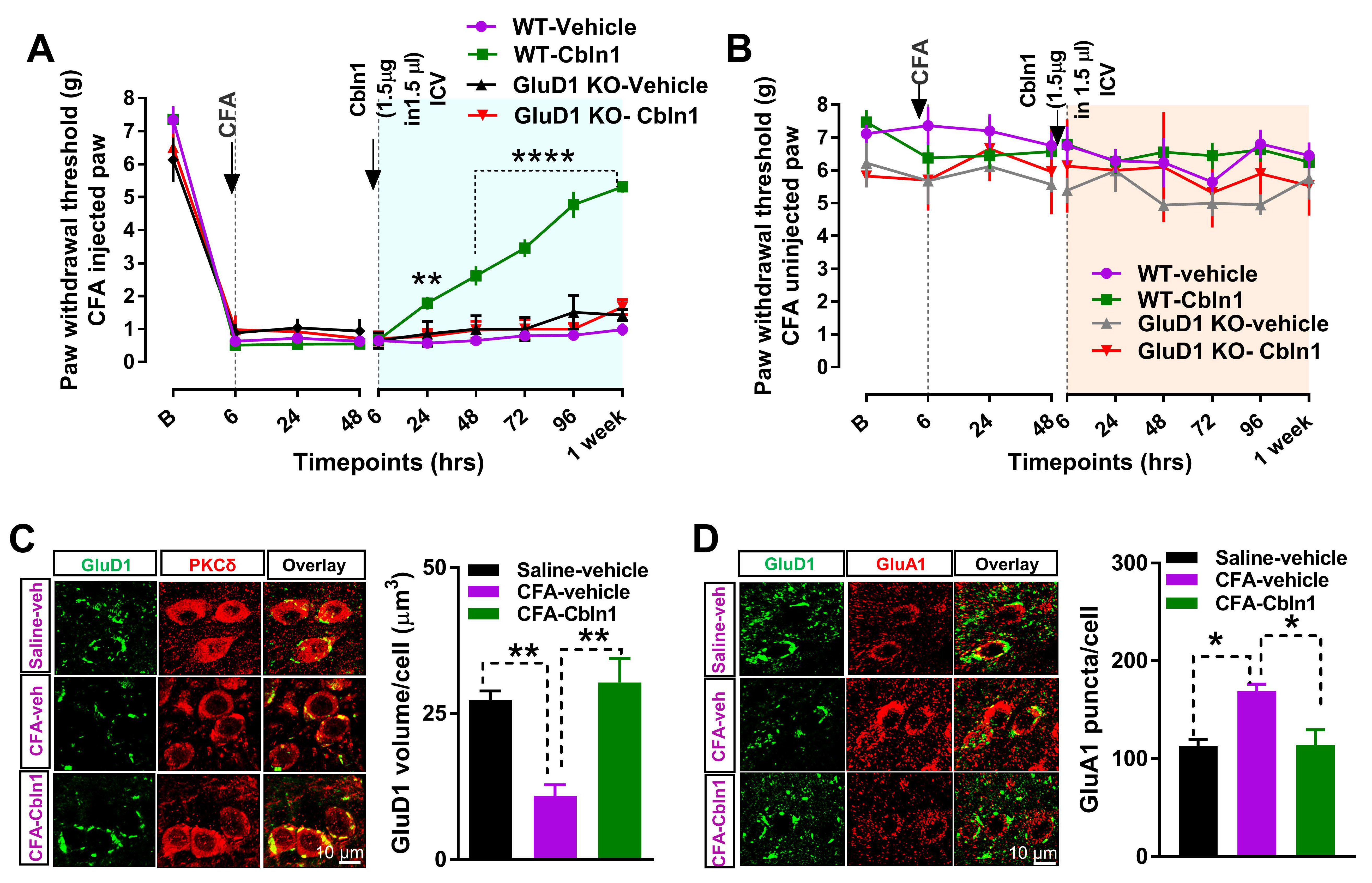
3.4. Recombinant Cbln1 Rescues Mechanical Hypersensitivity in an Inflammatory Pain Model with a Unique Time Course
3.5. Recombinant Cbln1 Normalizes Synapse Structure in the CeLC in an Inflammatory Pain Model
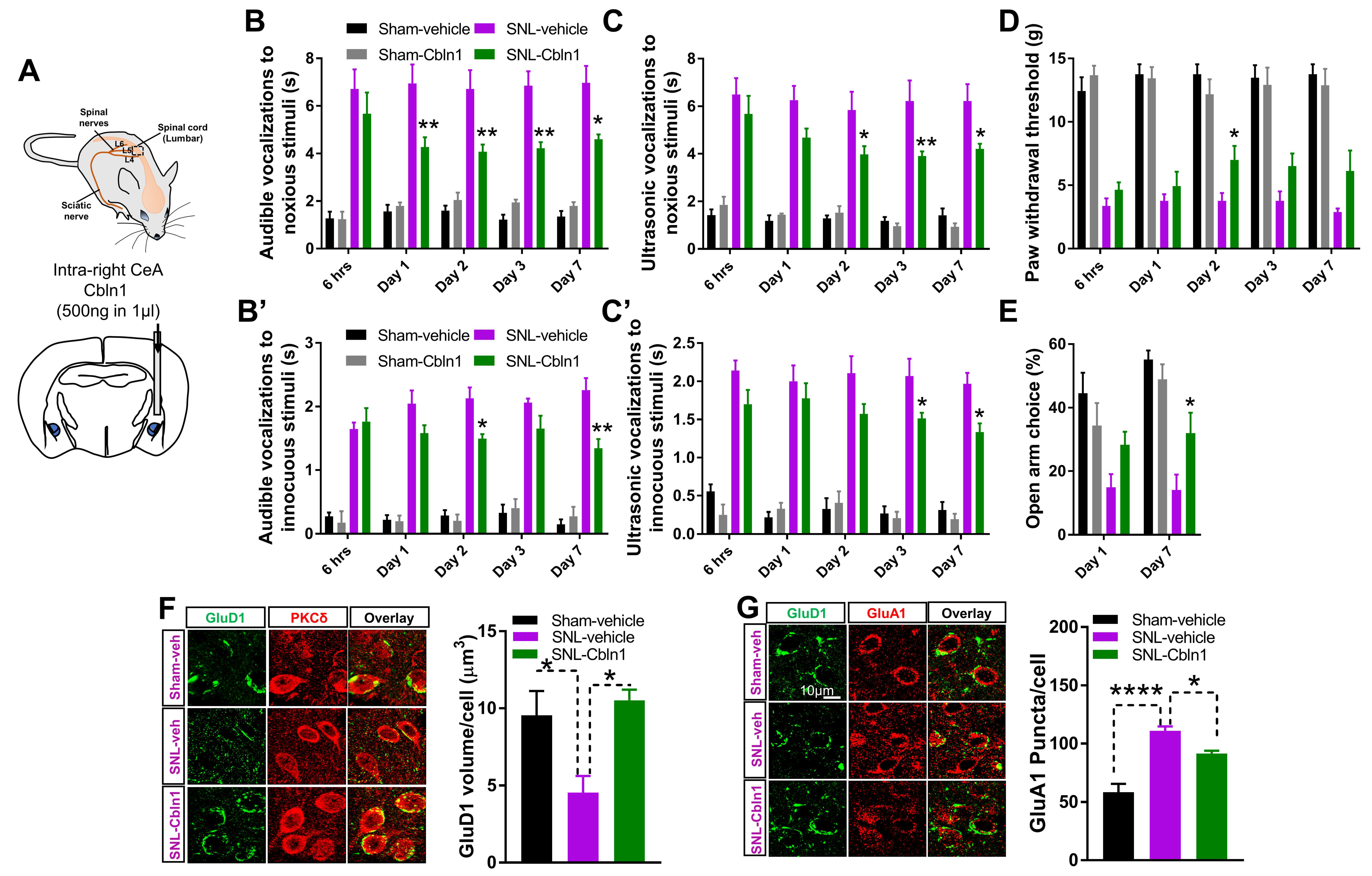
3.6. Recombinant Cbln1 Mitigates Averse and Affective Behaviors in a Neuropathic Pain Model
3.7. Cbln1 Release in the Central Amygdala in Normal Animals may Serve as a Nociceptive Signal
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability statement
Acknowledgments
Conflicts of Interest
References
- Gauriau, C.; Bernard, J.F. Pain pathways and parabrachial circuits in the rat. Exp. Physiol. 2002, 87, 251–258. [Google Scholar] [CrossRef]
- Braz, J.M.; Nassar, M.A.; Wood, J.N.; Basbaum, A.I. Parallel “pain” pathways arise from subpopulations of primary afferent noci-ceptor. Neuron 2005, 47, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, V. Amygdala pain mechanisms. Handb. Exp. Pharmacol. 2015, 227, 261–284. [Google Scholar]
- Neugebauer, V.; Li, W.; Bird, G.C.; Han, J.S. The Amygdala and Persistent Pain. Neuroscientist 2004, 10, 221–234. [Google Scholar] [CrossRef]
- Spike, R.C.; Puskar, Z.; Andrew, D.; Todd, A.J. A quantitative and morphological study of projection neurons in lamina I of the rat lumbar spinal cord. Eur. J. Neurosci. 2003, 18, 2433–2448. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Neugebauer, V. Amygdala Plasticity and Pain. Pain Res. Manag. 2017, 2017, 8296501. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, Y.; Gereau, R.W. Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J. Neurosci. 2007, 27, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Crock, L.W.; Kolber, B.J.; Morgan, C.D.; Sadler, K.E.; Vogt, S.K.; Bruchas, M.R.; Gereau, R.W. Central amygdala metabotropic gluta-mate receptor 5 in the modulation of visceral pain. J. Neurosci. 2012, 32, 14217–14226. [Google Scholar] [CrossRef] [PubMed]
- Han, J.S.; Li, W.; Neugebauer, V. Critical Role of Calcitonin Gene-Related Peptide 1 Receptors in the Amygdala in Synaptic Plasticity and Pain Behavior. J. Neurosci. 2005, 25, 10717–10728. [Google Scholar] [CrossRef]
- Han, J.S.; Neugebauer, V. Synaptic plasticity in the amygdala in a visceral pain model in rats. Neurosci. Lett. 2004, 361, 254–257. [Google Scholar] [CrossRef]
- Ikeda, R.; Takahashi, Y.; Inoue, K.; Kato, F. NMDA receptor-independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain 2007, 127, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Kato, F.; Sugimura, Y.K.; Takahashi, Y. Pain-Associated Neural Plasticity in the Parabrachial to Central Amygdala Circuit: Pain Changes the Brain, and the Brain Changes the Pain. Adv. Exp. Med. Biol. 2018, 1099, 157–166. [Google Scholar] [PubMed]
- Kolber, B.J.; Montana, M.C.; Carrasquillo, Y.; Xu, J.; Heinemann, S.F.; Muglia, L.J.; Gereau, R.W. Activation of metabotropic glutamate receptor 5 in the amygdala modulates pain-like behavior. J. Neurosci. 2010, 30, 8203–8213. [Google Scholar] [CrossRef] [PubMed]
- Neugebauer, V.; Li, W.; Bird, G.C.; Bhave, G.; Gereau, R.W. Synaptic plasticity in the amygdala in a model of arthritic pain: Differential roles of metabotropic glutamate receptors 1 and 5. J. Neurosci. 2003, 23, 52–63. [Google Scholar] [CrossRef]
- Veinante, P.; Yalcin, I.; Barrot, M. The amygdala between sensation and affect: A role in pain. J. Mol. Psychiatry 2013, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.C.; Lash, L.L.; Han, J.S.; Zou, X.; Willis, W.D.; Neugebauer, V. Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J. Physiol. 2005, 564, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-J.; Chen, C.-C.; Yang, H.-W.; Chang, Y.-T.; Bai, S.-W.; Chen, C.-C.; Yen, C.-T.; Min, M.-Y. Role of Extracellular Signal-Regulated Kinase in Synaptic Transmission and Plasticity of a Nociceptive Input on Capsular Central Amygdaloid Neurons in Normal and Acid-Induced Muscle Pain Mice. J. Neurosci. 2011, 31, 2258–2270. [Google Scholar] [CrossRef]
- Sato, M.; Ito, M.; Nagase, M.; Sugimura, Y.K.; Takahashi, Y.; Watabe, A.M.; Kato, F. The lateral parabrachial nucleus is actively in-volved in the acquisition of fear memory in mice. Mol. Brain 2015, 8, 1–15. [Google Scholar] [CrossRef]
- Sugimura, Y.K.; Takahashi, Y.; Watabe, A.M.; Kato, F. Synaptic and network consequences of monosynaptic nociceptive inputs of parabrachial nucleus origin in the central amygdala. J. Neurophysiol. 2016, 115, 2721–2739. [Google Scholar] [CrossRef]
- Watabe, A.M.; Ochiai, T.; Nagase, M.; Takahashi, Y.; Sato, M.; Kato, F. Synaptic potentiation in the nociceptive amygdala following fear learning in mice. Mol. Brain 2013, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Zhang, W.; Mahimainathan, L.; Narasimhan, M.; Kiritoshi, T.; Fan, X.; Wang, J.; Green, T.A.; Neugebauer, V. 5-HT2C Receptor Knockdown in the Amygdala Inhibits Neuropathic-Pain-Related Plasticity and Behaviors. J. Neurosci. 2017, 37, 1378–1393. [Google Scholar] [CrossRef]
- Nakao, A.; Takahashi, Y.; Nagase, M.; Ikeda, R.; Kato, F. Role of Capsaicin-Sensitive C-Fiber Afferents in Neuropathic Pain-Induced Synaptic Potentiation in the Nociceptive Amygdala. Mol. Pain 2012, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- De Lacalle, S.; Saper, C.B. Calcitonin gene-related peptide-like immunoreactivity marks putative visceral sensory pathways in human brain. Neuroscience 2000, 100, 115–130. [Google Scholar] [CrossRef]
- Dobolyi, A.; Irwin, S.; Makara, G.; Usdin, T.B.; Palkovits, M. Calcitonin gene-related peptide-containing pathways in the rat forebrain. J. Comp. Neurol. 2005, 489, 92–119. [Google Scholar] [CrossRef]
- Han, S.; Soleiman, M.T.; Soden, M.; Zweifel, L.; Palmiter, R.D. Elucidating an Affective Pain Circuit that Creates a Threat Memory. Cell 2015, 162, 363–374. [Google Scholar] [CrossRef]
- Kruger, L.; Mantyh, P.W.; Sternini, C.; Brecha, N.C.; Mantyh, C.R. Calcitonin gene-related peptide (CGRP) in the rat central nervous system: Patterns of immunoreactivity and receptor binding sites. Brain Res. 1988, 463, 223–244. [Google Scholar] [CrossRef]
- Schwaber, J.S.; Sternini, C.; Brecha, N.C.; Rogers, W.T.; Card, J.P. Neurons containing calcitonin gene-related peptide in the para-brachial nucleus project to the central nucleus of the amygdala. J. Comp. Neurol. 1988, 270, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-N.; Sheets, P.L. Spared nerve injury differentially alters parabrachial monosynaptic excitatory inputs to molecularly specific neurons in distinct subregions of the central amygdala. Pain 2019, 161, 166–176. [Google Scholar] [CrossRef]
- Wilson, T.D.; Valdivia, S.; Khan, A.; Ahn, H.-S.; Adke, A.; Gonzalez, S.M.; Sugimura, Y.K.; Carrasquillo, Y. Dual and Opposing Functions of the Central Amygdala in the Modulation of Pain. Cell Rep. 2019, 29, 332–346.e5. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Veinante, P. Cell-type specific parallel circuits in the bed nucleus of the stria terminalis and the central nucleus of the amygdala of the mouse. Brain Struct. Funct. 2019, 224, 1067–1095. [Google Scholar] [CrossRef] [PubMed]
- Adke, A.P.; Khan, A.; Ahn, H.-S.; Becker, J.J.; Wilson, T.D.; Valdivia, S.; Sugimura, Y.K.; Gonzalez, S.M.; Carrasquillo, Y. Cell-Type Specificity of Neuronal Excitability and Morphology in the Central Amygdala. Eneuro 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Naur, P.; Hansen, K.B.; Kristensen, A.S.; Dravid, S.M.; Pickering, D.S.; Olsen, L.; Vestergaard, B.; Egebjerg, J.; Gajhede, M.; Traynelis, S.F.; et al. Ionotropic glutamate-like receptor delta2 binds D-serine and glycine. Proc. Natl. Acad. Sci. USA 2007, 104, 14116–14121. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Rimerman, R.; Scofield, M.A.; Dravid, S.M. Mutations in the transmembrane domain M3 generate spontaneously open orphan glutamate delta 1 receptor. Brain Res. 2011, 1382, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Eapen, V. Balance within the Neurexin Trans-Synaptic Connexus Stabilizes Behavioral Control. Front. Hum. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kuroyanagi, T.; Yokoyama, M.; Hirano, T. Postsynaptic glutamate receptor delta family contributes to presynaptic terminal differentiation and establishment of synaptic transmission. Proc. Natl. Acad. Sci. USA 2009, 106, 4912–4916. [Google Scholar] [CrossRef]
- Matsuda, K.; Miura, E.; Miyazaki, T.; Kakegawa, W.; Emi, K.; Narumi, S.; Fukazawa, Y.; Ito-Ishida, A.; Kondo, T.; Shigemoto, R.; et al. Cbln1 is a ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science 2010, 328, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.; Yokoyama, M.; Yamashita, M.; Hirano, T. Induction of excitatory and inhibitory presynaptic differentiation by GluD1. Biochem. Biophys. Res. Commun. 2012, 417, 157–161. [Google Scholar] [CrossRef]
- Uemura, T.; Lee, S.J.; Yasumura, M.; Takeuchi, T.; Yoshida, T.; Ra, M.; Taguchi, R.; Sakimura, K.; Mishina, M. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 2010, 141, 1068–1079. [Google Scholar] [CrossRef]
- Yasumura, M.; Yoshida, T.; Lee, S.J.; Uemura, T.; Joo, J.Y.; Mishina, M. Glutamate receptor delta1 induces preferentially inhibitory presynaptic differentiation of cortical neurons by interacting with neurexins through cerebellin precursor protein subtypes. J. Neurochem. 2012, 121, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shelkar, G.; Gandhi, P.J.; Gawande, D.Y.; Hoover, A.; Villalba, R.M.; Pavuluri, R.; Smith, Y.; Dravid, S.M. Striatal glutamate delta-1 receptor regulates behavioral flexibility and thalamostriatal connectivity. Neurobiol. Dis. 2020, 137, 104746. [Google Scholar] [CrossRef]
- Tao, W.; Diaz-Alonso, J.; Sheng, N.; Nicoll, R.A. Postsynaptic delta1 glutamate receptor assembles and maintains hippocampal synapses via Cbln2 and neurexin. Proc. Natl. Acad. Sci. USA 2018, 115, E5373–E5381. [Google Scholar] [CrossRef]
- Hepp, R.; Hay, A.; Aguado, C.; Lujan, R.; Dauphinot, L.; Potier, M.-C.; Nomura, S.; Poirel, O.; El Mestikawy, S.; Lambolez, B.; et al. Glutamate receptors of the delta family are widely expressed in the adult brain. Brain Struct. Funct. 2014, 220, 2797–2815. [Google Scholar] [CrossRef] [PubMed]
- Konno, K.; Matsuda, K.; Nakamoto, C.; Uchigashima, M.; Miyazaki, T.; Yamasaki, M.; Sakimura, K.; Yuzaki, M.; Watanabe, M. Enriched expression of GluD1 in higher brain regions and its involvement in parallel fiber-interneuron synapse formation in the cere-bellum. J. Neurosci. 2014, 34, 7412–7424. [Google Scholar] [CrossRef] [PubMed]
- Miura, E.; Iijima, T.; Yuzaki, M.; Watanabe, M. Distinct expression of Cbln family mRNAs in developing and adult mouse brains. Eur. J. Neurosci. 2006, 24, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, S.; Konno, K.; Abe, M.; Motohashi, J.; Kohda, K.; Sakimura, K.; Watanabe, M.; Yuzaki, M. Roles of Cbln1 in Non-Motor Functions of Mice. J. Neurosci. 2016, 36, 11801–11816. [Google Scholar] [CrossRef]
- Yadav, R.; Hillman, B.G.; Gupta, S.C.; Suryavanshi, P.; Bhatt, J.M.; Pavuluri, R.; Stairs, D.J.; Dravid, S.M. Deletion of Glutamate Delta-1 Receptor in Mouse Leads to Enhanced Working Memory and Deficit in Fear Conditioning. PLoS ONE 2013, 8, e60785. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Maison, S.F.; Wu, X.; Hirose, K.; Jones, S.M.; Bayazitov, I.; Tian, Y.; Mittleman, G.; Matthews, D.B.; Zakharenko, S.S.; et al. Orphan glutamate receptor delta1 subunit required for high-frequency hearing. Mol. Cell Biol. 2007, 27, 4500–4512. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shelkar, G.; Zhao, F.; Clausen, R.P.; Dravid, S.M. Modulation of Burst Firing of Neurons in Nucleus Reticularis of the Thalamus by GluN2C-Containing NMDA Receptors. Mol. Pharmacol. 2019, 96, 193–203. [Google Scholar] [CrossRef]
- Navratilova, E.; Ji, G.; Phelps, C.; Qu, C.; Hein, M.; Yakhnitsa, V.; Neugebauer, V.; Porreca, F. Kappa opioid signaling in the central nucleus of the amygdala promotes disinhibition and aversiveness of chronic neuropathic pain. Pain 2019, 160, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Yakhnitsa, V.; Ji, G.; Neugebauer, V. Small conductance calcium activated potassium (SK) channel dependent and independent effects of riluzole on neuropathic pain-related amygdala activity and behaviors in rats. Neuropharmacology 2018, 138, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.N.; Bobnar, H.J.; Kolber, B.J. Left and right hemispheric lateralization of the amygdala in pain. Prog. Neurobiol. 2020, 196, 101891. [Google Scholar] [CrossRef]
- Carrasquillo, Y.; Gereau, R.W. Hemispheric lateralization of a molecular signal for pain modulation in the amygdala. Mol. Pain 2008, 4, 1744–8069. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Neugebauer, V. Hemispheric Lateralization of Pain Processing by Amygdala Neurons. J. Neurophysiol. 2009, 102, 2253–2264. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Chung, J.M. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992, 50, 355–363. [Google Scholar] [CrossRef]
- Ji, G.; Yakhnitsa, V.; Kiritoshi, T.; Presto, P.; Neugebauer, V. Fear extinction learning ability predicts neuropathic pain behaviors and amygdala activity in male rats. Mol. Pain 2018, 14. [Google Scholar] [CrossRef]
- Dixon, W. Staircase bioassay: The up-and-down method. Neurosci. Biobehav. Rev. 1991, 15, 47–50. [Google Scholar] [CrossRef]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- D’Hanis, W.; Linke, R.; Yilmazer-Hanke, D. Topography of thalamic and parabrachial calcitonin gene-related peptide (CGRP) immunoreactive neurons projecting to subnuclei of the amygdala and extended amygdala. J. Comp. Neurol. 2007, 505, 268–291. [Google Scholar] [CrossRef] [PubMed]
- Sadler, K.E.; McQuaid, N.A.; Cox, A.C.; Behun, M.N.; Trouten, A.M.; Kolber, B.J. Divergent functions of the left and right central amygdala in visceral nociception. Pain 2017, 158, 747–759. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, L.; Zhu, T.; Deng, S.; Ma, B.; Lv, H.; Shan, X.; Cheng, H.; Jiang, K.; Zhang, T.; et al. Reconsolidation of a post-ingestive nutrient memory requires mTOR in the central amygdala. Mol. Psychiatry 2020, 1–17. [Google Scholar] [CrossRef]
- Yuzaki, M. The C1q complement family of synaptic organizers: Not just complementary. Curr. Opin. Neurobiol. 2017, 45, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Yuzaki, M. Two Classes of Secreted Synaptic Organizers in the Central Nervous System. Annu. Rev. Physiol. 2018, 80, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Yuzaki, M.; Aricescu, A.R. A GluD Coming-Of-Age Story. Trends Neurosci. 2017, 40, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Li, H.; Li, X.; Xue, Z.; Sun, Y.; Ma, D.; Guan, Y.; Li, J.; Tian, M.; Wang, Y. Downregulation of neuroligin1 ameliorates postoperative pain through inhibiting neuroligin1/postsynaptic density 95-mediated synaptic targeting of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor GluA1 subunits in rat dorsal horns. Mol. Pain 2018, 14, 1744806918766745. [Google Scholar] [CrossRef]
- Lin, T.B.; Lai, C.Y.; Hsieh, M.C.; Jiang, J.L.; Cheng, J.K.; Chau, Y.P.; Ruan, T.; Chen, G.D.; Peng, H.Y. Neuropathic Allodynia Involves Spinal Neurexin-1beta-dependent Neuroligin-1/Postsynaptic Density-95/NR2B Cascade in Rats. Anesthesiology 2015, 123, 909–926. [Google Scholar] [CrossRef]
- Zhao, J.-Y.; Duan, X.-L.; Yang, L.; Liu, J.-P.; He, Y.-T.; Guo, Z.; Hu, X.-D.; Suo, Z.-W. Activity-dependent Synaptic Recruitment of Neuroligin 1 in Spinal Dorsal Horn Contributed to Inflammatory Pain. Neuroscience 2018, 388, 1–10. [Google Scholar] [CrossRef]
- Iijima, T.; Emi, K.; Yuzaki, M. Activity-dependent repression of Cbln1 expression: Mechanism for developmental and homeo-static regulation of synapses in the cerebellum. J. Neurosci. 2009, 29, 5425–5434. [Google Scholar] [CrossRef] [PubMed]
- Iijima, T.; Wu, K.; Witte, H.; Hanno-Iijima, Y.; Glatter, T.; Richard, S.; Scheiffele, P. SAM68 Regulates Neuronal Activity-Dependent Alternative Splicing of Neurexin-1. Cell 2011, 147, 1601–1614. [Google Scholar] [CrossRef]
- Wei, P.; Pattarini, R.; Rong, Y.; Guo, H.; Bansal, P.K.; Kusnoor, S.; Deutch, A.Y.; Parris, J.; Morgan, I.J. The Cbln family of proteins interact with multiple signaling pathways. J. Neurochem. 2012, 121, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Seigneur, E.; Sudhof, T.C. Cerebellins are differentially expressed in selective subsets of neurons throughout the brain. J. Comp. Neurol. 2017, 525, 3286–3311. [Google Scholar] [CrossRef]
- Miyazawa, Y.; Takahashi, Y.; Watabe, A.M.; Kato, F. Predominant synaptic potentiation and activation in the right central amygdala are independent of bilateral parabrachial activation in the hemilateral trigeminal inflammatory pain model of rats. Mol. Pain 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Naur, P.; Kurtkaya, N.L.; Kristensen, A.S.; Gajhede, M.; Kastrup, J.S.; Traynelis, S.F. Modulation of the dimer interface at ionotropic glutamate-like receptor delta2 by D-serine and extracellular calcium. J. Neurosci. 2009, 29, 907–917. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandhi, P.J.; Gawande, D.Y.; Shelkar, G.P.; Gakare, S.G.; Kiritoshi, T.; Ji, G.; Misra, B.; Pavuluri, R.; Liu, J.; Neugebauer, V.; et al. Dysfunction of Glutamate Delta-1 Receptor-Cerebellin 1 Trans-Synaptic Signaling in the Central Amygdala in Chronic Pain. Cells 2021, 10, 2644. https://doi.org/10.3390/cells10102644
Gandhi PJ, Gawande DY, Shelkar GP, Gakare SG, Kiritoshi T, Ji G, Misra B, Pavuluri R, Liu J, Neugebauer V, et al. Dysfunction of Glutamate Delta-1 Receptor-Cerebellin 1 Trans-Synaptic Signaling in the Central Amygdala in Chronic Pain. Cells. 2021; 10(10):2644. https://doi.org/10.3390/cells10102644
Chicago/Turabian StyleGandhi, Pauravi J., Dinesh Y. Gawande, Gajanan P. Shelkar, Sukanya G. Gakare, Takaki Kiritoshi, Guangchen Ji, Bishal Misra, Ratnamala Pavuluri, Jinxu Liu, Volker Neugebauer, and et al. 2021. "Dysfunction of Glutamate Delta-1 Receptor-Cerebellin 1 Trans-Synaptic Signaling in the Central Amygdala in Chronic Pain" Cells 10, no. 10: 2644. https://doi.org/10.3390/cells10102644
APA StyleGandhi, P. J., Gawande, D. Y., Shelkar, G. P., Gakare, S. G., Kiritoshi, T., Ji, G., Misra, B., Pavuluri, R., Liu, J., Neugebauer, V., & Dravid, S. M. (2021). Dysfunction of Glutamate Delta-1 Receptor-Cerebellin 1 Trans-Synaptic Signaling in the Central Amygdala in Chronic Pain. Cells, 10(10), 2644. https://doi.org/10.3390/cells10102644