
Haskap Berry Phenolic Subclasses Differentially Impact Cellular Stress Sensing in Primary and Immortalized Dermal Fibroblasts

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Haskap Berry Phenolic Extracts and Fractions
2.2. Cell Culture
2.3. Cell Treatments
2.4. Cell Counts and Cell Viability
2.5. MitoTracker Orange Labeling of Intracellular Free Radicals
2.6. Protein Extraction and Quantification
2.7. Western Blotting
2.8. RNA Extraction
2.9. cDNA Synthesis and Reverse-Transcriptase-Quantitative PCR (RT-qPCR)
2.10. SIRT1 Activity Assay
2.11. siRNA Knockdown of SIRT1
2.12. Chromatin Immuno-Precipitation
3. Results
3.1. Haskap Berry Extracts and Fractions
3.2. Phenolic Extracts and Fractions Increased Population Doubling Times and Scavenged Intracellular Free Radicals
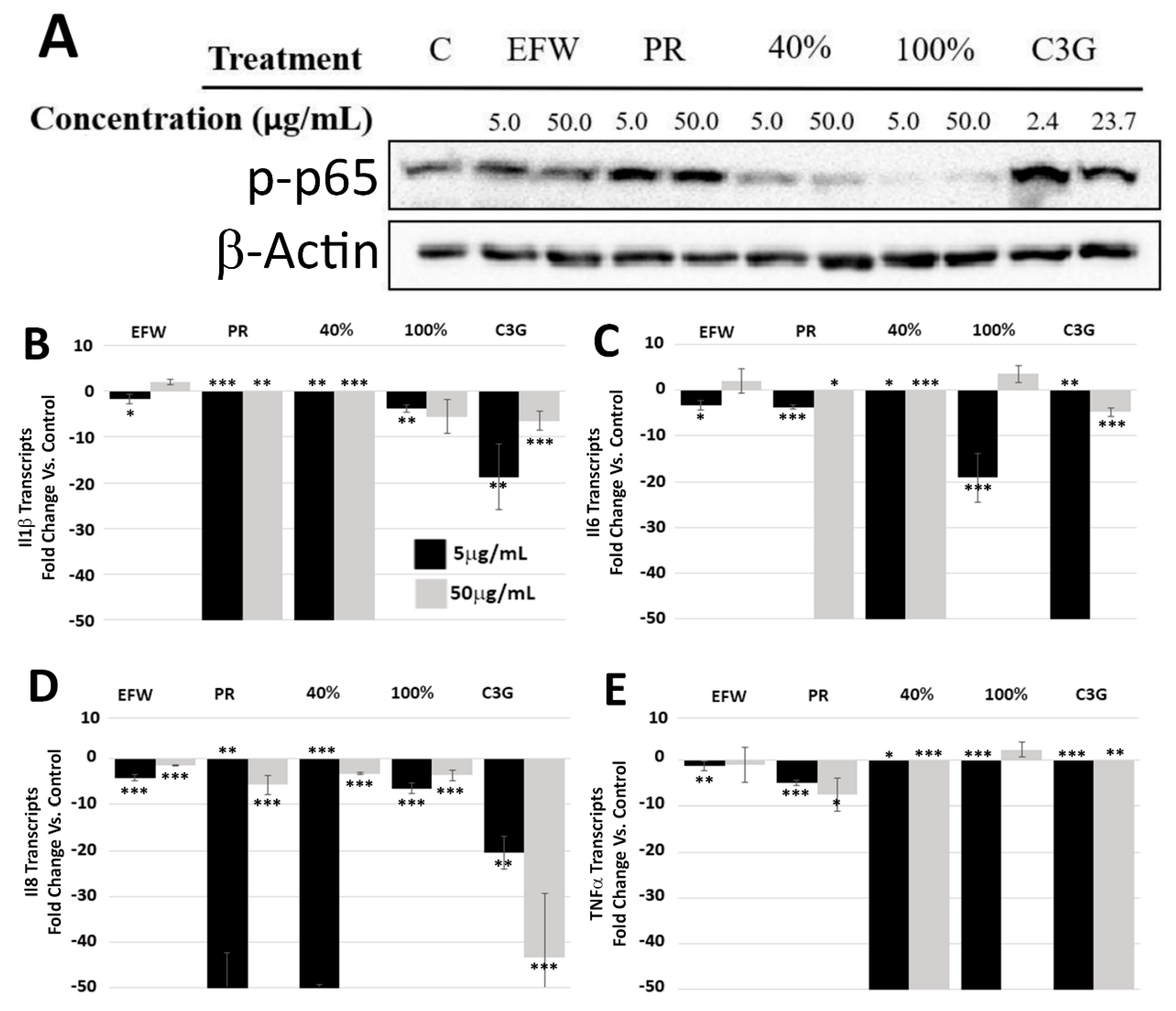
3.3. Phenolic Subclass Differentially Impacted NF-κB Activation
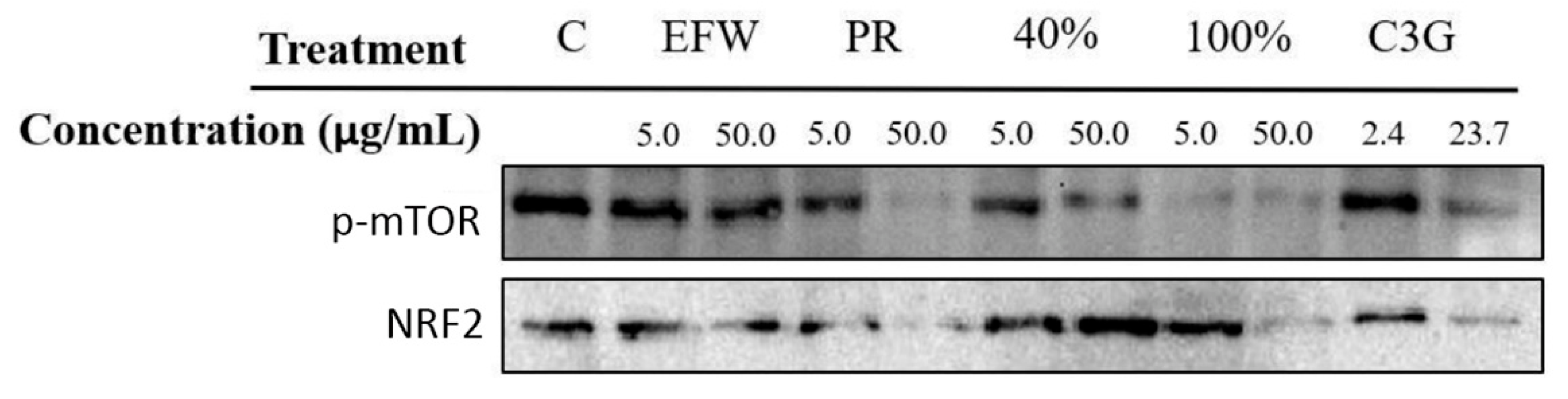
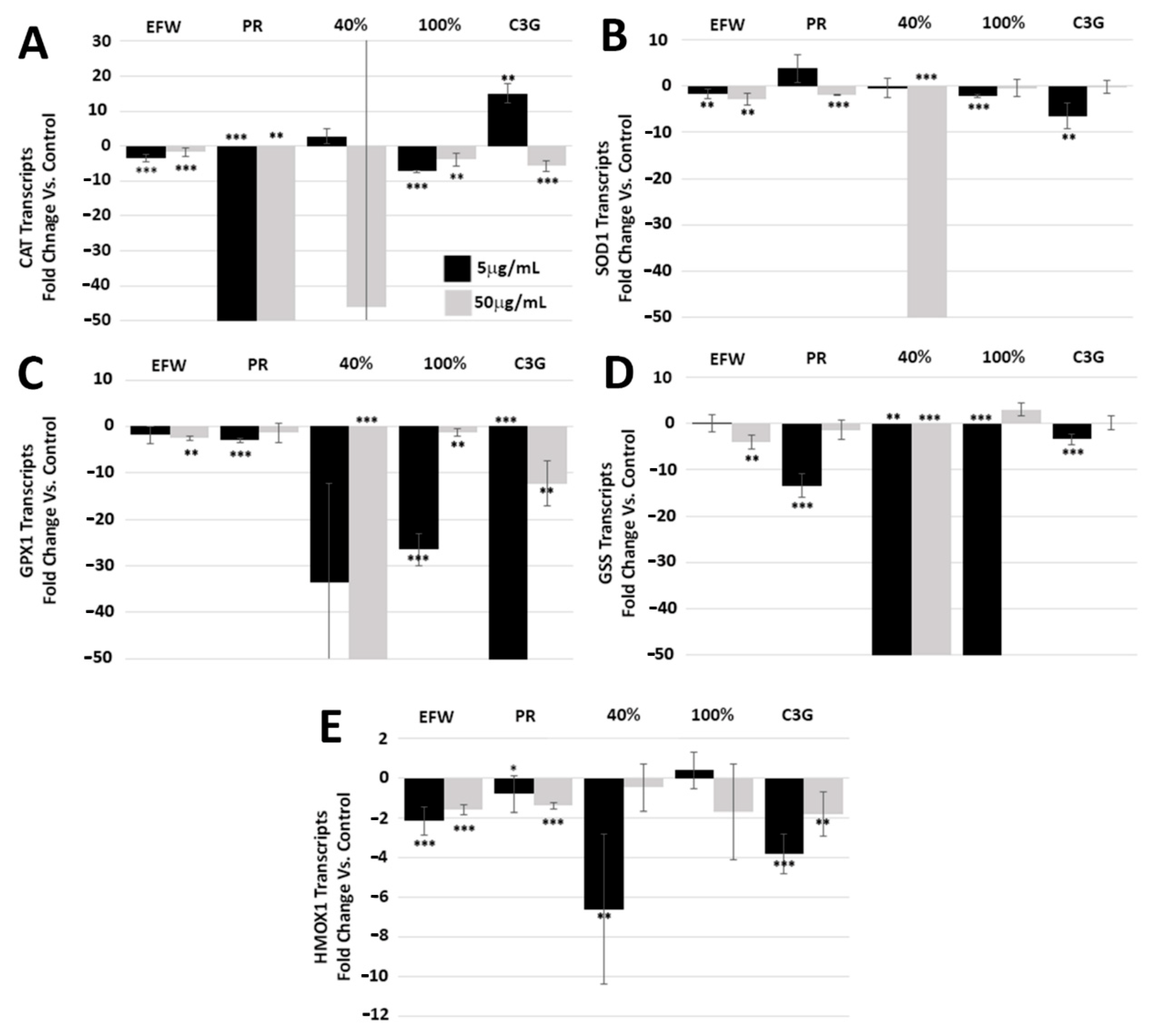
3.4. Phenolic Subclass Impacts Cellular Pathways That Promote Cellular Health
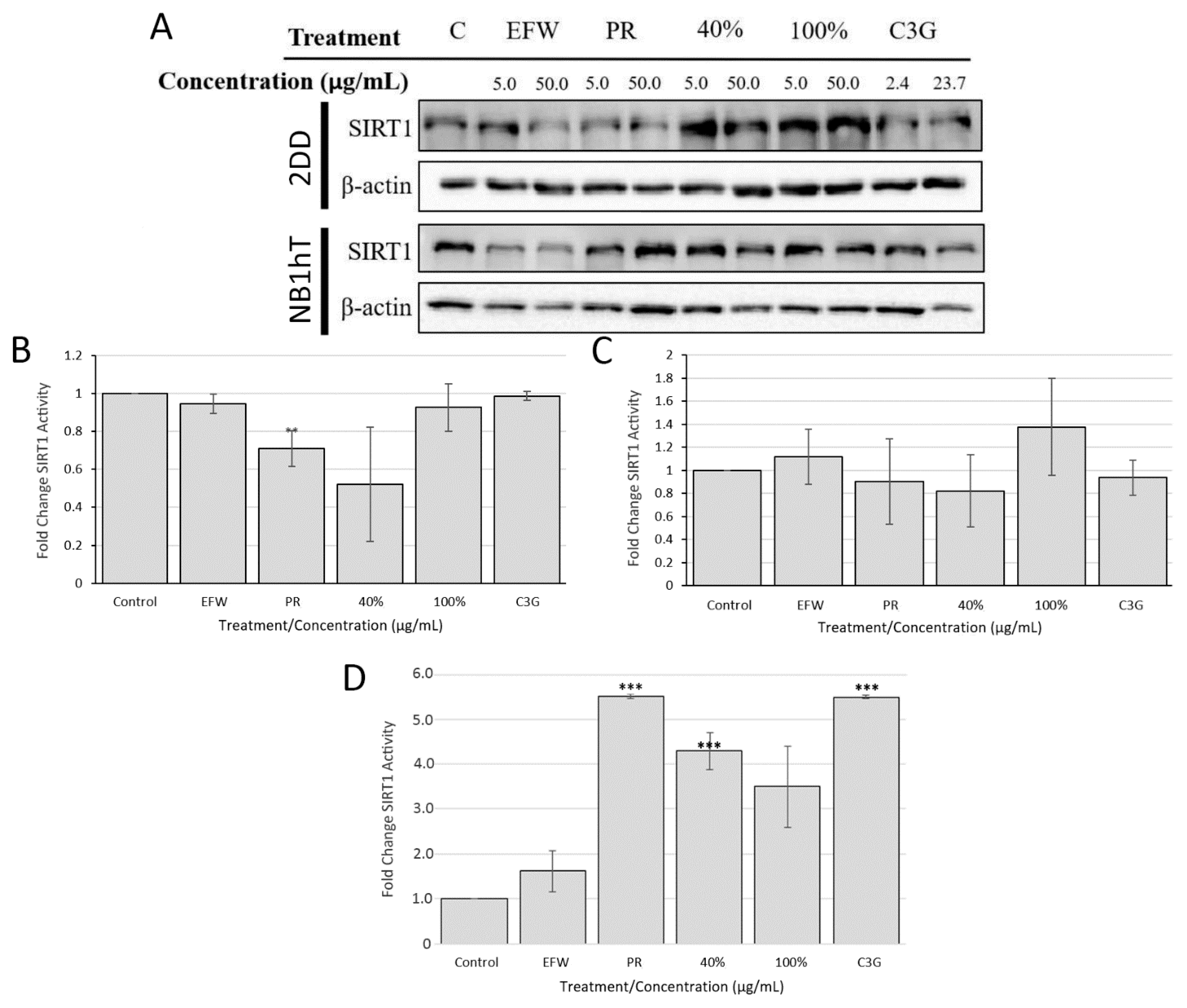
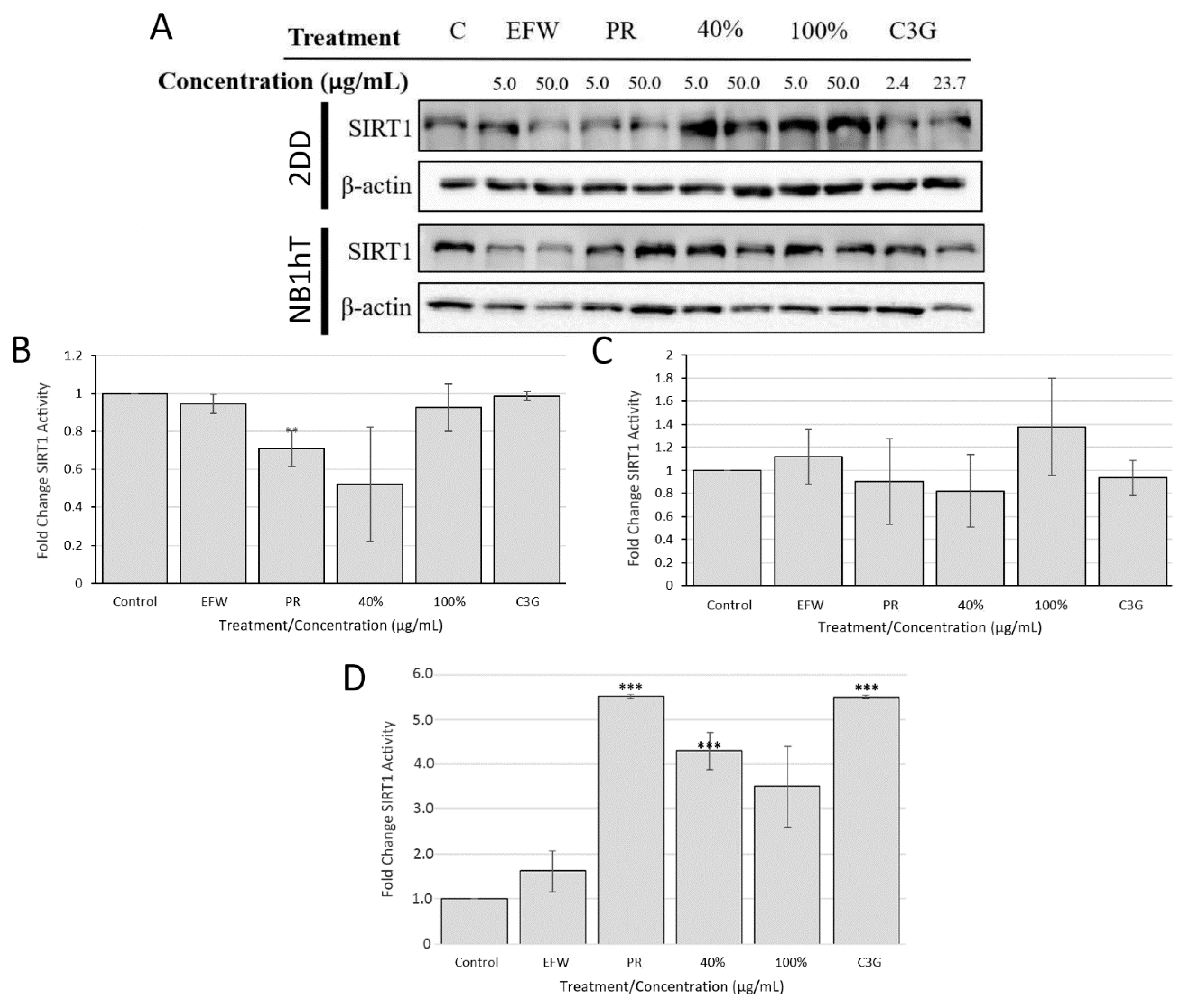
3.5. Sirtuin 1 Is Indirectly Impacted by Haskap Phenolics But Is Required for Downstream Effects
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gillespie, Z.E.; Pickering, J.; Eskiw, C.H. Better living through chemistry: Caloric restriction (CR) and CR mimetics alter genome function to promote increased health and lifespan. Front. Genet. 2016, 7, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandhorst, S.; Longo, V.D. Dietary restrictions and nutrition in the prevention and treatment of cardiovascular disease. Circ. Res. 2019, 124, 952–965. [Google Scholar] [CrossRef] [PubMed]
- de Magalhaes, J.P.; Wuttke, D.; Wood, S.H.; Plank, M.; Vora, C. Genome-environment interactions that modulate aging: Powerful targets for drug discovery. Pharmacol. Rev. 2012, 64, 88–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahfoufi, N.; Alsadi, N.; Jambi, M.; Matar, C. The immunomodulatory and anti-inflammatory role of polyphenols. Nutrients 2018, 10, 1618. [Google Scholar] [CrossRef] [Green Version]
- Serino, A.; Salazar, G. Protective role of polyphenols against vascular inflammation, aging and cardiovascular disease. Nutrients 2018, 11, 53. [Google Scholar] [CrossRef] [Green Version]
- Vina, J.; Borras, C.; Abdelaziz, K.M.; Garcia-Valles, R.; Gomez-Cabrera, M.C. The free radical theory of aging revisited: The cell signaling disruption theory of aging. Antioxid. Redox Signal. 2013, 19, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.L.; Sinha, S.; Lindner, A.B. The good, the bad, and the ugly of ROS: New insights on aging and aging-related diseases from eukaryotic and prokaryotic model organisms. Oxidative Med. Cell. Longev. 2018, 2018, 1941285. [Google Scholar] [CrossRef]
- Kubben, N.; Misteli, T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat. Rev. Mol. Cell Biol. 2017, 18, 559–560. [Google Scholar] [CrossRef]
- Ferguson, L.R.; Schlothauer, R.C. The potential role of nutritional genomics tools in validating high health foods for cancer control: Broccoli as example. Mol. Nutr. Food Res. 2012, 56, 126–146. [Google Scholar] [CrossRef] [PubMed]
- Caprioli, G.; Iannarelli, R.; Innocenti, M.; Bellumori, M.; Fiorini, D.; Sagratini, G.; Vittori, S.; Buccioni, M.; Santinelli, C.; Bramucci, M.; et al. Blue honeysuckle fruit (Lonicera caerulea L.) from eastern Russia: Phenolic composition, nutritional value and biological activities of its polar extracts. Food Funct. 2016, 7, 1892–1903. [Google Scholar] [CrossRef] [PubMed]
- Rupasinghe, H.P.; Boehm, M.M.; Sekhon-Loodu, S.; Parmar, I.; Bors, B.; Jamieson, A.R. Anti-inflammatory activity of haskap cultivars is polyphenols-dependent. Biomolecules 2015, 5, 1079–1098. [Google Scholar] [CrossRef] [PubMed]
- De Silva, A.B.K.H.; Rupasinghe, H.P.V. Polyphenols composition and anti-diabetic properties in vitro of haskap (Lonicera caerulea L.) berries in relation to cultivar and harvesting date. J. Food Compos. Anal. 2020, 88, 103402. [Google Scholar] [CrossRef]
- Pace, E.; Jiang, Y.; Clemens, A.; Crossman, T.; Rupasinghe, H.P.V. Impact of thermal degradation of cyanidin-3-O-glucoside of haskap berry on cytotoxicity of hepatocellular carcinoma HepG2 and breast cancer MDA-MB-231 cells. Antioxidants 2018, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- de Souza, D.R.; Willems, J.L.; Low, N.H. Phenolic composition and antioxidant activities of saskatoon berry fruit and pomace. Food Chem. 2019, 290, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Zehfus, L.R.; Eskiw, C.H.; Low, N.H. Phenolic profiles and antioxidant activities of Saskatchewan (Canada) bred haskap (Lonicera caerulea) berries. BioRxiv 2021. [Google Scholar] [CrossRef]
- Bridger, J.M.; Kill, I.R.; O’Fareell, M.; Hutchinson, C.J. Internal lamin structures within G1 nuclei of human dermal fibroblasts. J. Cell Sci. 1993, 104, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, Z.E.; MacKay, K.; Sander, M.; Trost, B.; Dawicki, W.; Wickramarathna, A.; Gordon, J.; Eramian, M.; Kill, I.R.; Bridger, J.M.; et al. Rapamycin reduces fibroblast proliferation without causing quiescence and induces STAT5A/B-mediated cytokine production. Nucleus 2015, 6, 490–506. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, Z.E.; Wang, C.; Vadan, F.; Yu, T.Y.; Ausio, J.; Kusalik, A.; Eskiw, C.H. Metformin induces the AP-1 transcription factor network in normal dermal fibroblasts. Sci. Rep. 2019, 9, 5369–5386. [Google Scholar] [CrossRef]
- Wang, C.; Eskiw, C.H. Cytoprotective effects of Avenathramide C against oxidative and inflammatory stress in normal human dermal fibroblasts. Sci. Rep. 2019, 9, 2932. [Google Scholar] [CrossRef]
- Kim, H.S.; Quon, M.J.; Kim, J.A. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2014, 2, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Perrelli, A.; Goitre, L.; Salzano, A.M.; Moglia, A.; Scaloni, A.; Retta, S.F. Biological activities, health benefits, and therapeutic properties of avenanthramides: From skin protection to prevention and treatment of cerebrovascular diseases. Oxidative Med. Cell. Longev. 2018, 2018, 6015351. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Stec, D.E.; Hinds, T.D., Jr. Natural product heme oxygenase inducers as treatment for nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2020, 21, 9493. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D.; Leeuwenburgh, C. Resveratrol and novel potent activators of SIRT1: Effects on aging and age-related diseases. Nutr. Rev. 2008, 66, 591–596. [Google Scholar] [CrossRef]
- Trost, B.; Moir, C.A.; Gillespie, Z.E.; Kusalik, A.; Mitchell, J.A.; Eskiw, C.H. Concordance between RNA-sequencing data and DNA microarray data in transcriptome analysis of proliferative and quiescent fibroblasts. R. Soc. Open Sci. 2015, 2, 150402. [Google Scholar] [CrossRef] [Green Version]
- Almendariz-Palacios, C.; Gillespie, Z.E.; Janzen, M.; Martinez, V.; Bridger, J.M.; Harkness, T.A.A.; Mousseau, D.D.; Eskiw, C.H. The nuclear lamina: Protein accumulation and disease. Biomedicines 2020, 8, 188. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, B.; Wang, X.; Wu, L.; Yang, Y.; Cheng, X.; Hu, Z.; Cai, X.; Yang, J.; Sun, X.; et al. Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2, and autophagy pathways. Sci. Rep. 2016, 6, 32206. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [Green Version]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.H.; Misteli, T. Repression of the antioxidant NRF2 pathway in premature aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef] [Green Version]
- Almendariz-Palacios, C.; Mousseau, D.D.; Eskiw, C.H.; Gillespie, Z.E. Still living better through chemistry: An update on caloric restriction and caloric restriction mimetics as tools to promote health and lifespan. Int. J. Mol. Sci. 2020, 21, 9220. [Google Scholar] [CrossRef] [PubMed]
- de Boer, V.C.; de Goffau, M.C.; Arts, I.C.; Hollman, P.C.; Keijer, J. SIRT1 stimulation by polyphenols is affected by their stability and metabolism. Mech. Ageing Dev. 2006, 127, 618–627. [Google Scholar] [CrossRef]
- Liu, S.; Sui, Q.; Zhao, Y.; Chang, X. lonicera caerulea berry polyphenols activate SIRT1, enhancing inhibition of RAW264.7 macrophage foam cell formation and promoting cholesterol efflux. J. Agric. Food Chem. 2019, 67, 7157–7166. [Google Scholar] [CrossRef] [PubMed]
- Palikova, I.; Heinrich, J.; Bednar, P.; Marhol, P.; Kren, V.; Cvak, L.; Valentova, K.; Ruzicka, F.; Hola, V.; Kolar, M.; et al. Constituents and antimicrobial properties of blue honeysuckle: A novel source for phenolic antioxidants. J. Agric. Food Chem. 2008, 56, 11883–11889. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, L.A.; Ions, L.J.; Ford, D. Could Sirt1-mediated epigenetic effects contribute to the longevity response to dietary restriction and be mimicked by other dietary interventions? Age 2009, 31, 327–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.H.; Manganiello, V.; Dyck, J.R. Resveratrol as a calorie restriction mimetic: Therapeutic implications. Trends Cell Biol. 2012, 22, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Ramis, M.R.; Esteban, S.; Miralles, A.; Tan, D.X.; Reiter, R.J. Caloric restriction, resveratrol and melatonin: Role of SIRT1 and implications for aging and related-diseases. Mech. Ageing Dev. 2015, 146–148, 28–41. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zehfus, L.R.; Gillespie, Z.E.; Almendáriz-Palacios, C.; Low, N.H.; Eskiw, C.H. Haskap Berry Phenolic Subclasses Differentially Impact Cellular Stress Sensing in Primary and Immortalized Dermal Fibroblasts. Cells 2021, 10, 2643. https://doi.org/10.3390/cells10102643
Zehfus LR, Gillespie ZE, Almendáriz-Palacios C, Low NH, Eskiw CH. Haskap Berry Phenolic Subclasses Differentially Impact Cellular Stress Sensing in Primary and Immortalized Dermal Fibroblasts. Cells. 2021; 10(10):2643. https://doi.org/10.3390/cells10102643
Chicago/Turabian StyleZehfus, Lily R., Zoe E. Gillespie, Carla Almendáriz-Palacios, Nicholas H. Low, and Christopher H. Eskiw. 2021. "Haskap Berry Phenolic Subclasses Differentially Impact Cellular Stress Sensing in Primary and Immortalized Dermal Fibroblasts" Cells 10, no. 10: 2643. https://doi.org/10.3390/cells10102643
APA StyleZehfus, L. R., Gillespie, Z. E., Almendáriz-Palacios, C., Low, N. H., & Eskiw, C. H. (2021). Haskap Berry Phenolic Subclasses Differentially Impact Cellular Stress Sensing in Primary and Immortalized Dermal Fibroblasts. Cells, 10(10), 2643. https://doi.org/10.3390/cells10102643