
Evaluation of an RNAseq-Based Immunogenomic Liquid Biopsy Approach in Early-Stage Prostate Cancer

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
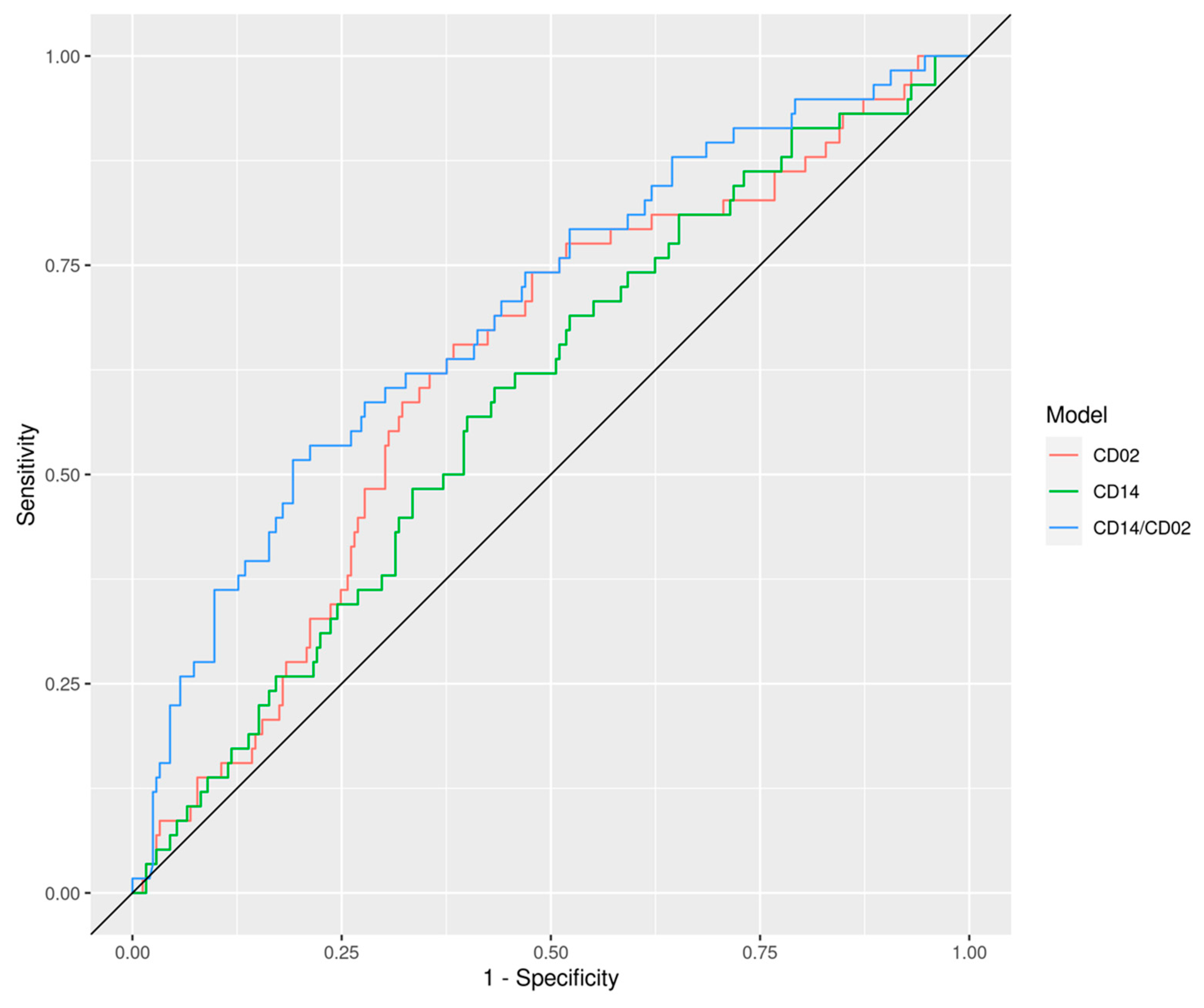
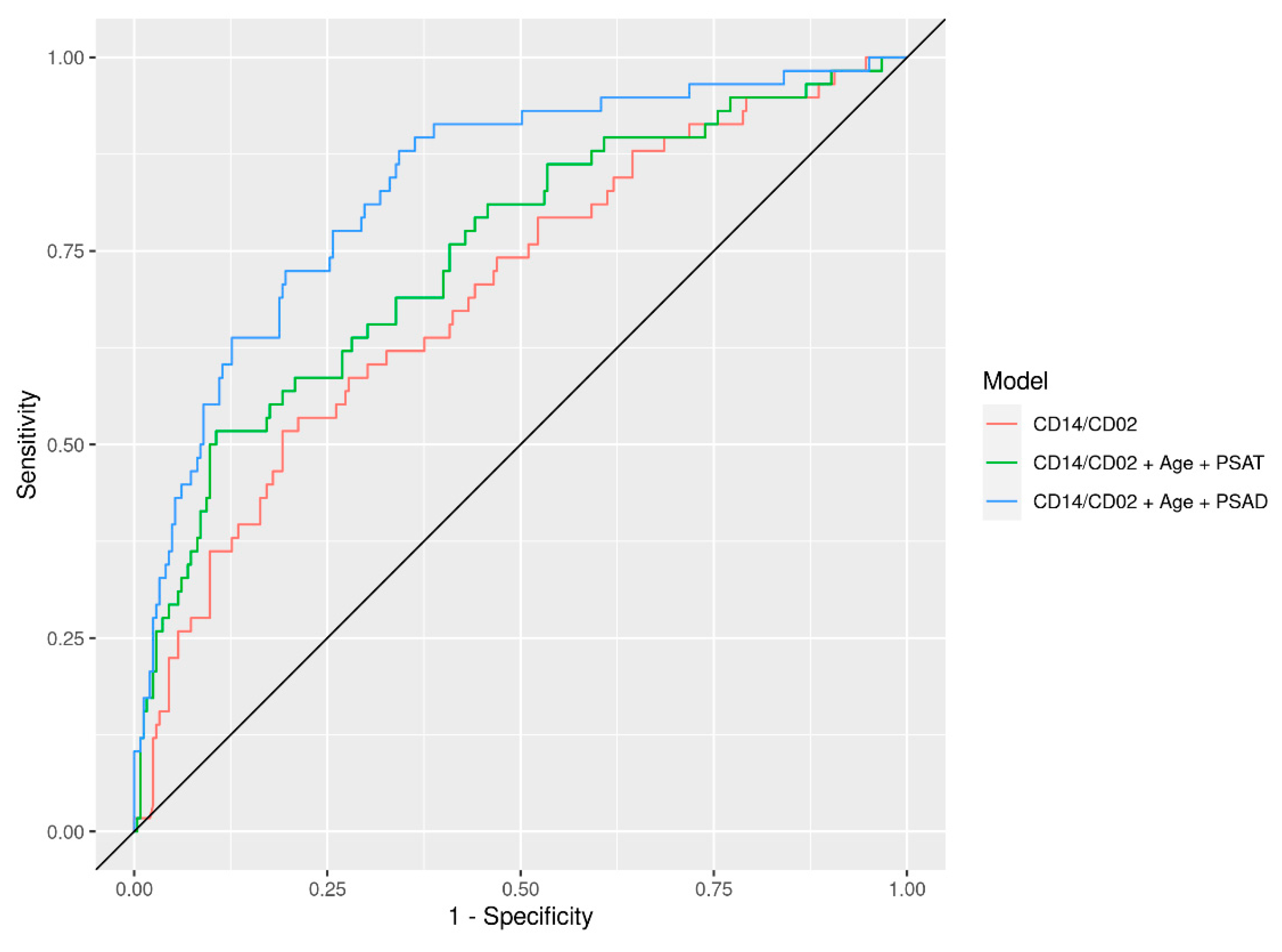
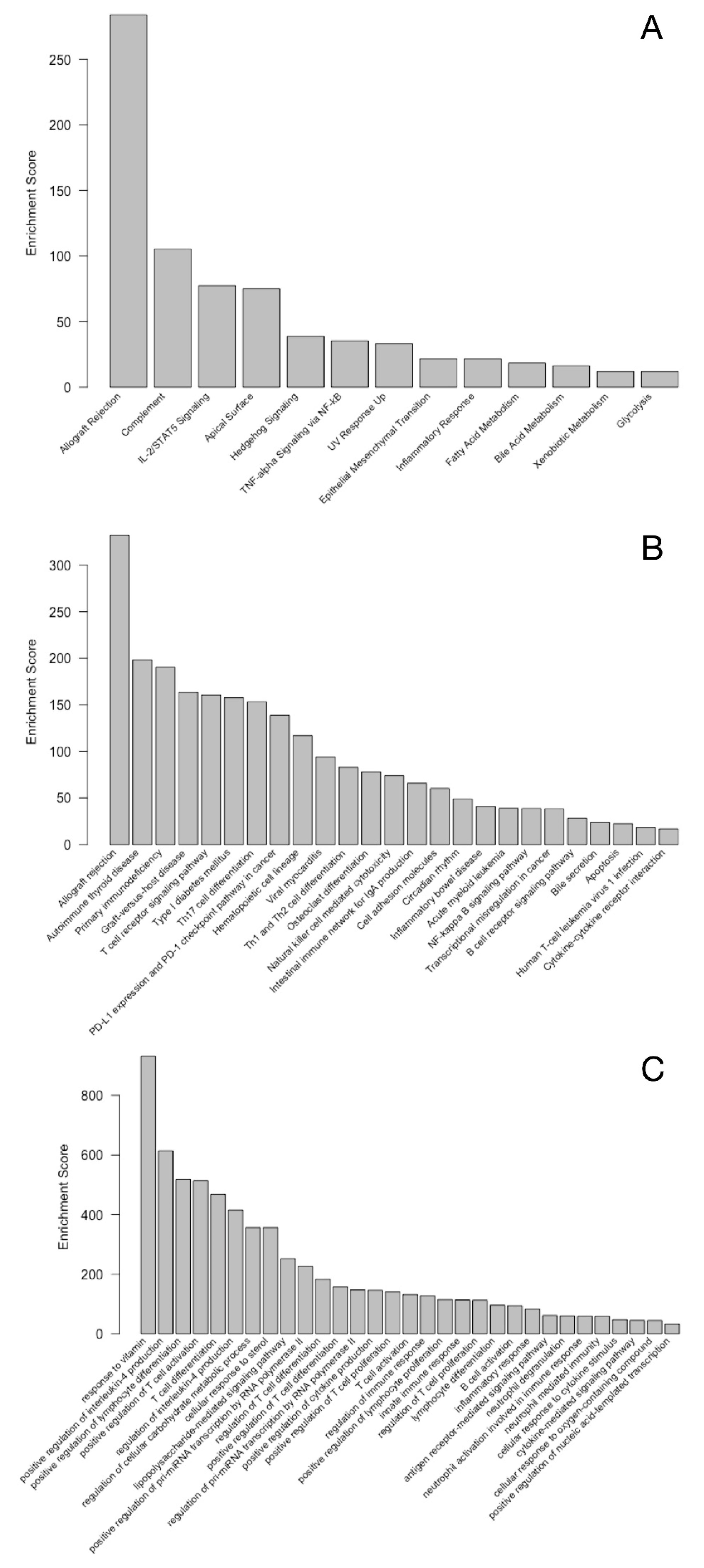
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hou, J.-S.; He, Y.-Y.; Du, L.-L.; Duan, Z.-M.; Chen, X.; Li, M. Gene expression profile and long noncoding RNA analysis in Candida albicans insoluble β-glucan-stimulated CD14+ monocytes and THP-1 cells. Microb. Pathog. 2021, 157, 104963. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.L.; Ding, J.; Taylor, J.R.; Lohman, K.; Soranzo, N.; De La Fuente, A.; Liu, T.F.; Johnson, C.; Barr, R.G.; Register, T.C.; et al. Transcriptomic profiles of aging in purified human immune cells. BMC Genom. 2015, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Limon, P.; Ortega-Castro, R.; Barbarroja, N.; Perez-Sanchez, C.; Jamin, C.; Patiño-Trives, A.M.; Luque-Tevar, M.; Ibáñez-Costa, A.; Perez-Sanchez, L.; de la Rosa, I.A.; et al. Molecular Characterization of Monocyte Subsets Reveals Specific and Distinctive Molecular Signatures Associated With Cardiovascular Disease in Rheumatoid Arthritis. Front. Immunol. 2019, 10, 1111. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Felts, S.J.; Van Keulen, V.P.; Scheid, A.D.; Block, M.S.; Markovic, S.N.; Pease, L.R.; Zhang, Y. Integrative Genome-Wide Analysis of Long Noncoding RNAs in Diverse Immune Cell Types of Melanoma Patients. Cancer Res. 2018, 78, 4411–4423. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.; Diehn, M.; Alizadeh, A.A.; Brown, P.O. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genom. 2006, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Okubo, K.; Utiyama, H.; Hirano, T.; Yoshii, J.; Matsubara, K. Expression profile of active genes in granu-locytes. Blood 1998, 92, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.-I.; Nagai, S.; Sese, J.; Suzuki, T.; Obata, A.; Sato, T.; Toyoda, N.; Dong, H.-Y.; Kurachi, M.; Nagahata, T.; et al. Gene expression profile in human leukocytes. Blood 2003, 101, 3509–3513. [Google Scholar] [CrossRef]
- Allantaz, F.; Cheng, D.T.; Bergauer, T.; Ravindran, P.; Rossier, M.F.; Ebeling, M.; Badi, L.; Reis, B.; Bitter, H.; D’Asaro, M.; et al. Expression Profiling of Human Immune Cell Subsets Identifies miRNA-mRNA Regulatory Relationships Correlated with Cell Type Specific Expression. PLoS ONE 2012, 7, e29979. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.W.; Galsky, M.D.; Scher, H.I.; Magidson, J.; Wassmann, K.; Lee, G.-S.M.; Katz, L.; Subudhi, S.K.; Anand, A.; Fleisher, M.; et al. A whole-blood RNA transcript-based prognostic model in men with castration-resistant prostate cancer: A prospective study. Lancet Oncol. 2012, 13, 1105–1113. [Google Scholar] [CrossRef]
- Olmos, D.; Brewer, D.; Clark, J.; Danila, D.C.; Parker, C.; Attard, G.; Fleisher, M.; Reid, A.H.; Castro, E.; Sandhu, S.K.; et al. Prognostic value of blood mRNA expression signatures in castration-resistant prostate cancer: A prospective, two-stage study. Lancet Oncol. 2012, 13, 1114–1124. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Gong, Y.; Chippada-Venkata, U.; Heck, M.M.; Retz, M.; Nawroth, R.; Galsky, M.; Tsao, C.-K.; Schadt, E.; De Bono, J.; et al. A robust blood gene expression-based prognostic model for castration-resistant prostate cancer. BMC Med. 2015, 13, 201. [Google Scholar] [CrossRef] [Green Version]
- Huen, N.-Y.; Pang, A.L.-Y.; Tucker, J.A.; Lee, T.-L.; Vergati, M.; Jochems, C.; Intrivici, C.; Cereda, V.; Chan, W.-Y.; Rennert, O.M.; et al. Up-regulation of proliferative and migratory genes in regulatory T cells from patients with metastatic castration-resistant prostate cancer. Int. J. Cancer 2013, 133, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Wallace, T.A.; Downey, R.F.; Seufert, C.J.; Schetter, A.; Dorsey, T.H.; Johnson, C.A.; Goldman, R.; Loffredo, C.A.; Yan, P.; Sullivan, F.J.; et al. Elevated HERV-K mRNA expression in PBMC is associated with a prostate cancer diagnosis particularly in older men and smokers. Carcinogenesis 2014, 35, 2074–2083. [Google Scholar] [CrossRef]
- Showe, M.K.; Vachani, A.; Kossenkov, A.V.; Yousef, M.; Nichols, C.; Nikonova, E.V.; Chang, C.; Kucharczuk, J.; Tran, B.; Wakeam, E.; et al. Gene Expression Profiles in Peripheral Blood Mononuclear Cells Can Distinguish Patients with Non–Small Cell Lung Cancer from Patients with Nonmalignant Lung Disease. Cancer Res. 2009, 69, 9202–9210. [Google Scholar] [CrossRef] [Green Version]
- Zander, T.; Hofmann, A.; Staratschek-Jox, A.; Classen, S.; Debey-Pascher, S.; Maisel, D.; Ansén, S.; Hahn, M.; Beyer, M.; Thomas, R.K.; et al. Blood-Based Gene Expression Signatures in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2011, 17, 3360–3367. [Google Scholar] [CrossRef] [Green Version]
- Kossenkov, A.V.; Dawany, N.; Evans, T.L.; Kucharczuk, J.C.; Albelda, S.M.; Showe, L.C.; Showe, M.K.; Vachani, A. Peripheral Immune Cell Gene Expression Predicts Survival of Patients with Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e34392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Lin, Y.; Zhan, M.; Mann, D.L.; A Stass, S.; Jiang, F. Differential miRNA expressions in peripheral blood mononuclear cells for diagnosis of lung cancer. Lab. Investig. 2015, 95, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Kossenkov, A.V.; Qureshi, R.; Dawany, N.B.; Wickramasinghe, J.; Liu, Q.; Majumdar, R.S.; Chang, C.; Widura, S.; Kumar, T.; Horng, W.-H.; et al. A Gene Expression Classifier from Whole Blood Distinguishes Benign from Malignant Lung Nodules Detected by Low-Dose CT. Cancer Res. 2018, 79, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baine, M.J.; Menning, M.; Smith, L.M.; Mallya, K.; Kaur, S.; Rachagani, S.; Chakraborty, S.; Sasson, A.R.; Brand, R.E.; Batra, S.K. Differential gene expression analysis of peripheral blood mononuclear cells reveals novel test for early detection of pancreatic cancer. Cancer Biomarkers 2012, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Batliwalla, F.M.; Baechler, E.C.; Xiao, X.; Li, W.; Balasubramanian, S.; Khalili, H.; Damle, A.; Ortmann, W.A.; Perrone, A.; Kantor, A.B.; et al. Peripheral blood gene expression profiling in rheumatoid arthritis. Genes Immun. 2005, 6, 388–397. [Google Scholar] [CrossRef]
- Ramilo, O.; Allman, W.; Chung, W.; Mejias, A.; Ardura, M.; Glaser, C.; Wittkowski, K.M.; Piqueras, B.; Banchereau, J.; Palucka, A.K.; et al. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood 2006, 109, 2066–2077. [Google Scholar] [CrossRef] [Green Version]
- Dale, D.C.; Boxer, L.; Liles, W.C. The phagocytes: Neutrophils and monocytes. Blood 2008, 112, 935–945. [Google Scholar] [CrossRef]
- Chow, A.; Brown, B.D.; Merad, M. Studying the mononuclear phagocyte system in the molecular age. Nat. Rev. Immunol. 2011, 11, 788–798. [Google Scholar] [CrossRef]
- Gutknecht, M.F.; Bouton, A.H. Functional significance of mononuclear phagocyte populations generated through adult hematopoiesis. J. Leukoc. Biol. 2014, 96, 969–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, D.; Spring, D.J.; Depinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef] [Green Version]
- Ross-Adams, H.; Lamb, A.; Dunning, M.; Halim, S.; Lindberg, J.; Massie, C.; Egevad, L.; Russell, R.; Ramos-Montoya, A.; Vowler, S.; et al. Integration of copy number and transcriptomics provides risk stratification in prostate cancer: A discovery and validation cohort study. EBioMedicine 2015, 2, 1133–1144. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Xu, D.; Zhang, Z.; Li, X.; Shi, J.; Sun, J.; Liu, H.-Z.; Li, X.; Zhou, M.; Zheng, T. The pan-cancer landscape of crosstalk between epithelial-mesenchymal transition and immune evasion relevant to prognosis and immunotherapy response. npj Precis. Oncol. 2021, 5, 1–10. [Google Scholar] [CrossRef]
- Efremova, M.; Rieder, D.; Klepsch, V.; Charoentong, P.; Finotello, F.; Hackl, H.; Kleiter, N.; Löwer, M.; Baier, G.; Krogsdam, A.; et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Raulet, D.H. The DNA Damage Response Arouses the Immune System. Cancer Res. 2006, 66, 3959–3962. [Google Scholar] [CrossRef] [Green Version]
- Fleuren, G.J.; Kuppen, P.J.K. Immune Surveillance. In Encyclopedia of Immunology, 2nd ed.; Delves, P., Roitt, I., Eds.; Academic Press: New York, NY, USA, 1998. [Google Scholar]
- Murphy, K.; Weaver, C. Janeway’s Immunobiology; Garland Science: New York, NY, USA, 2012. [Google Scholar]
- Aderem, A.; Underhill, D. Mechanisms of Phagocytosis in macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef]
- Foulds, G.A.; Vadakekolathu, J.; Abdel-Fatah, T.M.A.; Nagarajan, D.; Reeder, S.; Johnson, C.; Hood, S.; Moseley, P.; Chan, S.Y.T.; Pockley, A.; et al. Immune-Phenotyping and Transcriptomic Profiling of Peripheral Blood Mononuclear Cells From Patients With Breast Cancer: Identification of a 3 Gene Signature Which Predicts Relapse of Triple Negative Breast Cancer. Front. Immunol. 2018, 9, 2028. [Google Scholar] [CrossRef]
- Shill, D.K.; Roobol, M.J.; Ehdaie, B.; Vickers, A.J.; Carlsson, S.V. Active surveillance for prostate cancer. Transl. Androl. Urol. 2021, 10, 2809819. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.; Wojno, K.J.; Stone, S.; Evans, B.; Bernhisel, R.; Meek, S.; D’Anna, R.E.; Ferguson, J.; Glaser, J.; Morgan, T.M.; et al. Clinical outcomes in men with prostate cancer who selected active surveillance using a clinical cell cycle risk score. Pers. Med. 2019, 16, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egevad, L.; Delahunt, B.; Srigley, J.R.; Samaratunga, H. International Society of Urological Pathology (ISUP) grading of prostate cancer—An ISUP consensus on contemporary grading. APMIS 2016, 124, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.; Pachter, L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 2013, 10, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.Y. Lightgbm: A highly efficient gradient boost-ing decision tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3146–3154. [Google Scholar]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2005, 27, 861–874. [Google Scholar] [CrossRef]
- Delong, E.R.; Delong, D.M.; Clarke-Pearson, D.L. Comparing the Areas under Two or More Correlated Receiver Operating Characteristic Curves: A Nonparametric Approach. Biometrics 1988, 44, 837. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Level | Validation | Discovery | p | Missing | |
|---|---|---|---|---|---|
| n | 305 | 713 | |||
| age (mean (SD)) | 62.69 (7.60) | 64.09 (7.89) | 0.009 | 0 | |
| race (%) | AA | 38 (12.5) | 69 (9.7) | 0.006 | 0 |
| Caucasian | 227 (74.4) | 593 (83.2) | |||
| Other | 25 (8.2) | 29 (4.1) | |||
| Unknown | 15 (4.9) | 22 (3.1) | |||
| family history (%) | First | 59 (19.3) | 132 (18.5) | 0.764 | 0 |
| None | 204 (66.9) | 494 (69.3) | |||
| Positive | 7 (2.3) | 12 (1.7) | |||
| Second | 16 (5.2) | 27 (3.8) | |||
| Unknown | 19 (6.2) | 48 (6.7) | |||
| DRE (%) | Abnormal/Positive | 74 (24.3) | 114 (16.0) | 0.002 | 0 |
| Normal/Negative (T1c) | 203 (66.6) | 550 (77.1) | |||
| Unknown | 28 (9.2) | 49 (6.9) | |||
| volume (median (IQR)) | 40.20 (29.60, 54.08) | 40.85 (30.78, 57.85) | 0.264 | 9.6 | |
| psa_total (median (IQR)) | 4.97 (3.80, 7.10) | 5.12 (3.80, 7.30) | 0.3 | 1.2 | |
| psa_density (median (IQR)) | 0.12 (0.08, 0.19) | 0.12 (0.07, 0.19) | 0.929 | 10.8 | |
| cores_positive (median (IQR)) | 1.00 (0.00, 4.00) | 1.00 (0.00, 4.00) | 0.973 | 0 | |
| cores_percent (median (IQR)) | 4.00 (0.00, 30.00) | 5.00 (0.00, 35.00) | 0.862 | 0 | |
| site (%) | Unknown | 0 (0.0) | 55 (7.7) | <0.001 | 0 |
| CU | 147 (48.2) | 577 (80.9) | |||
| MIU | 62 (20.3) | 46 (6.5) | |||
| Urology Austin | 96 (31.5) | 35 (4.9) | |||
| Gleason Group (%) | 0 | 145 (47.5) | 337 (47.3) | 0.963 | 0 |
| 1 | 32 (10.5) | 78 (10.9) | |||
| 2 | 66 (21.6) | 166 (23.3) | |||
| 3 | 38 (12.5) | 77 (10.8) | |||
| 4 | 13 ( 4.3) | 27 ( 3.8) | |||
| 5 | 11 (3.6) | 28 (3.9) | |||
| Adverse Pathology (%) | 0 | 245 (80.9) | 577 (80.9) | 1 | 0.2 |
| 1 | 58 (19.1) | 136 (19.1) |
| Model | AUC (disc) | AUC (val) | AUC (val) | ||
|---|---|---|---|---|---|
| Age (42,60) | Age (60,66) | Age (66,87) | |||
| Age | 0.60 (0.54, 0.65) | 0.56 (0.47, 0.65) | |||
| PSAT | 0.72 (0.67, 0.77) | 0.67 (0.59, 0.75) | 0.63 (0.48, 0.77) | 0.74 (0.59, 0.89) | 0.66 (0.52, 0.82) |
| Vol | 0.60 (0.55, 0.66) | 0.72 (0.65, 0.80) | 0.75 (0.65, 0.85) | 0.82 (0.67, 0.96) | 0.62 (0.48, 0.76) |
| PSAD | 0.77 (0.72, 0.81) | 0.78 (0.71, 0.85) | 0.76 (0.64, 0.89) | 0.88 (0.80, 0.97) | 0.74 (0.61, 0.87) |
| Clinical (PSAT, Age) | 0.72 (0.67, 0.77) | 0.67 (0.59, 0.75) | 0.60 (0.46, 0.75) | 0.73 (0.58, 0.88) | 0.67 (0.54, 0.80) |
| Clinical (PSAT, Age, Race, DRE, FamH) | 0.73 (0.69, 0.78) | 0.73 (0.65, 0.80) | 0.70 (0.56, 0.83) | 0.72 (0.56, 0.88) | 0.72 (0.61, 0.83) |
| Clinical (PSAD, Age) | 0.78 (0.74, 0.82) | 0.78 (0.71, 0.85) | 0.73 (0.61, 0.86) | 0.89 (0.80, 0.97) | 0.76 (0.64, 0.88) |
| CD2 | 0.80 (0.76, 0.84) | 0.63 (0.55, 0.71) | 0.67 (0.53, 0.80) | 0.53 (0.35, 0.71) | 0.61 (0.49, 0.74) |
| CD14 | 0.82 (0.77, 0.86) | 0.59 (0.51, 0.67) | 0.60 (0.46, 0.74) | 0.54 (0.36, 0.73) | 0.63 (0.51, 0.75) |
| CD14/CD2 | 0.72 (0.67, 0.77) | 0.70 (0.62, 0.77) | 0.76 (0.65, 0.88) | 0.63 (0.47, 0.79) | 0.68 (0.55, 0.81) |
| CD14/CD2 + Clinical (PSAT, Age) | 0.99 (0.99, 1.00) | 0.75 (0.67, 0.82) | 0.73 (0.60, 0.86) | 0.72 (0.57, 0.88) | 0.76 (0.64, 0.88) |
| CD14/CD2 + Clinical (PSAT, Age, Race, DRE, FamH) | 0.97 (0.95, 0.99) | 0.76 (0.69, 0.83) | 0.78 (0.66, 0.90) | 0.72 (0.55, 0.89) | 0.75 (0.63, 0.86) |
| CD14/CD2 + Clinical (PSAD, Age) | 0.91 (0.88, 0.93) | 0.83 (0.77, 0.89) | 0.79 (0.67, 0.92) | 0.91 (0.84, 0.97) | 0.80 (0.70, 0.90) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Neste, L.; Wojno, K.J.; Henao, R.; Mane, S.; Korman, H.; Hafron, J.; Kernen, K.; Tinawi-Aljundi, R.; Putzi, M.; Kassis, A.I.; et al. Evaluation of an RNAseq-Based Immunogenomic Liquid Biopsy Approach in Early-Stage Prostate Cancer. Cells 2021, 10, 2567. https://doi.org/10.3390/cells10102567
Van Neste L, Wojno KJ, Henao R, Mane S, Korman H, Hafron J, Kernen K, Tinawi-Aljundi R, Putzi M, Kassis AI, et al. Evaluation of an RNAseq-Based Immunogenomic Liquid Biopsy Approach in Early-Stage Prostate Cancer. Cells. 2021; 10(10):2567. https://doi.org/10.3390/cells10102567
Chicago/Turabian StyleVan Neste, Leander, Kirk J. Wojno, Ricardo Henao, Shrikant Mane, Howard Korman, Jason Hafron, Kenneth Kernen, Rima Tinawi-Aljundi, Mathew Putzi, Amin I. Kassis, and et al. 2021. "Evaluation of an RNAseq-Based Immunogenomic Liquid Biopsy Approach in Early-Stage Prostate Cancer" Cells 10, no. 10: 2567. https://doi.org/10.3390/cells10102567
APA StyleVan Neste, L., Wojno, K. J., Henao, R., Mane, S., Korman, H., Hafron, J., Kernen, K., Tinawi-Aljundi, R., Putzi, M., Kassis, A. I., & Kantoff, P. W. (2021). Evaluation of an RNAseq-Based Immunogenomic Liquid Biopsy Approach in Early-Stage Prostate Cancer. Cells, 10(10), 2567. https://doi.org/10.3390/cells10102567