
New Insights into the Pivotal Role of Iron/Heme Metabolism in TLR4/NF-κB Signaling-Mediated Inflammatory Responses in Human Monocytes




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Cell Culture Reagents
2.2. Cell Culture and Treatment
2.3. Iron Assay Analysis
2.4. FACS Analysis for Ferrous Ion (Fe2+)
2.5. Western Blotting
2.6. Heme Quantification
2.7. Statistical Analyses
3. Results
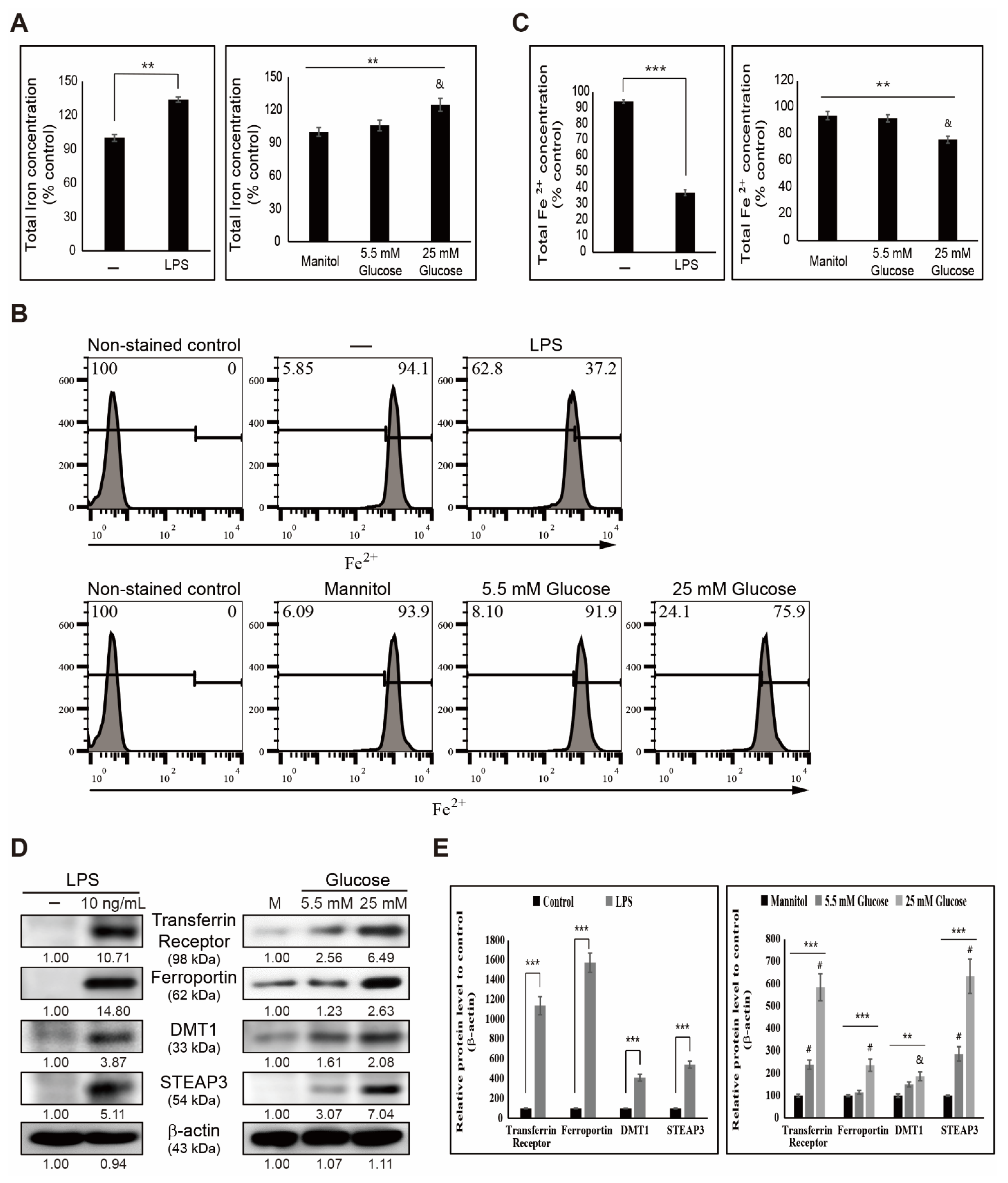
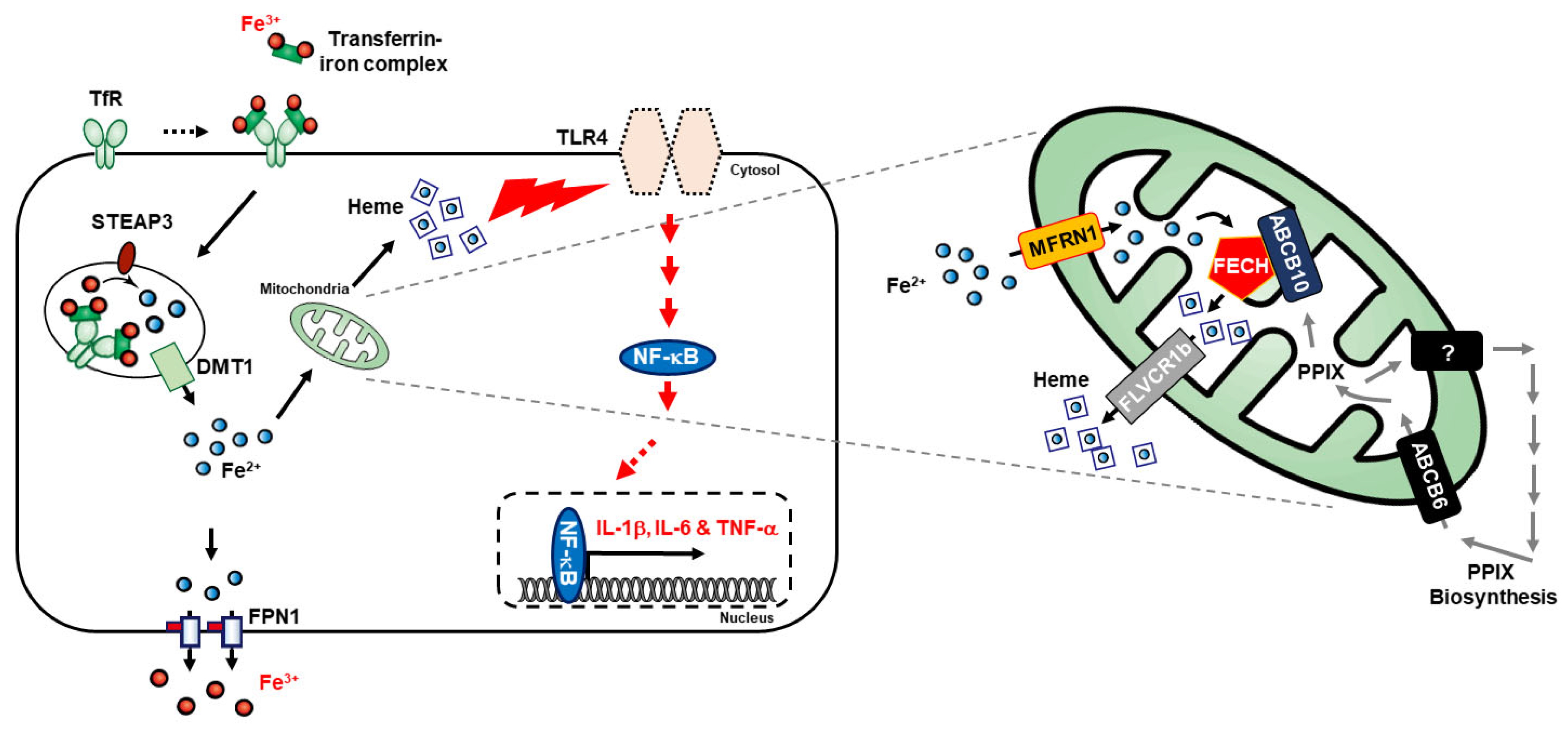
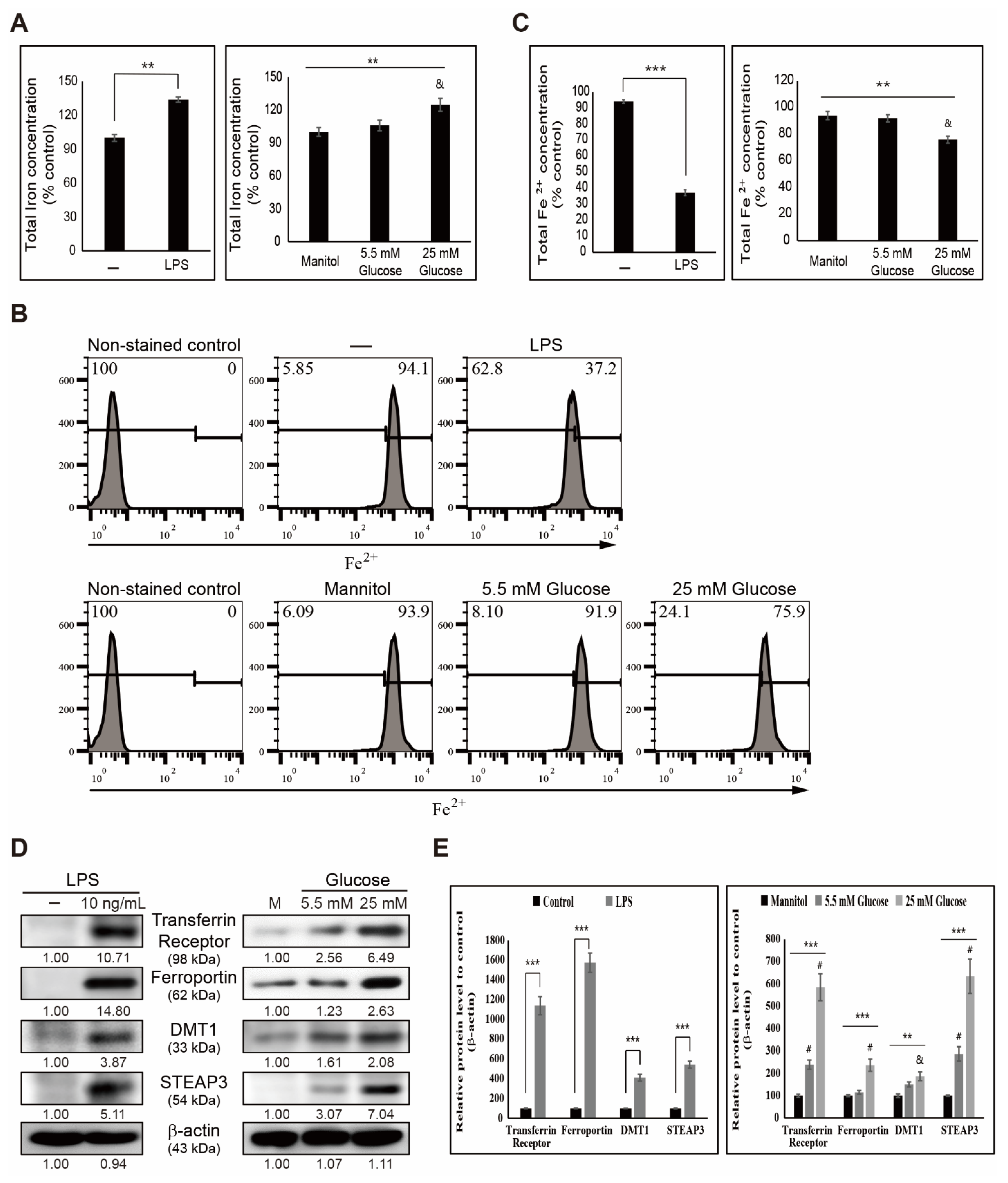
3.1. LPS- or Glucose-Induced Inflammation Elevates Iron Metabolism
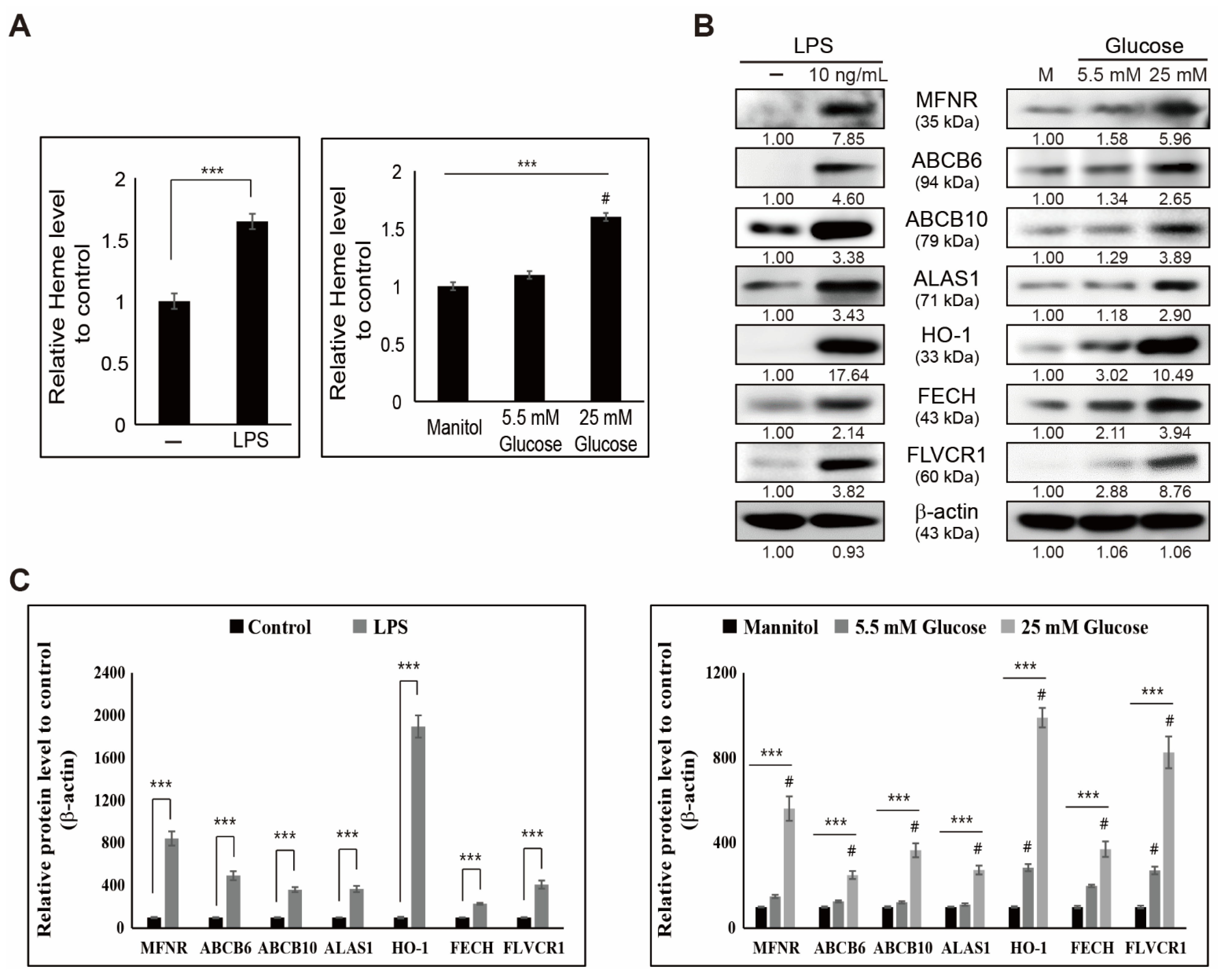
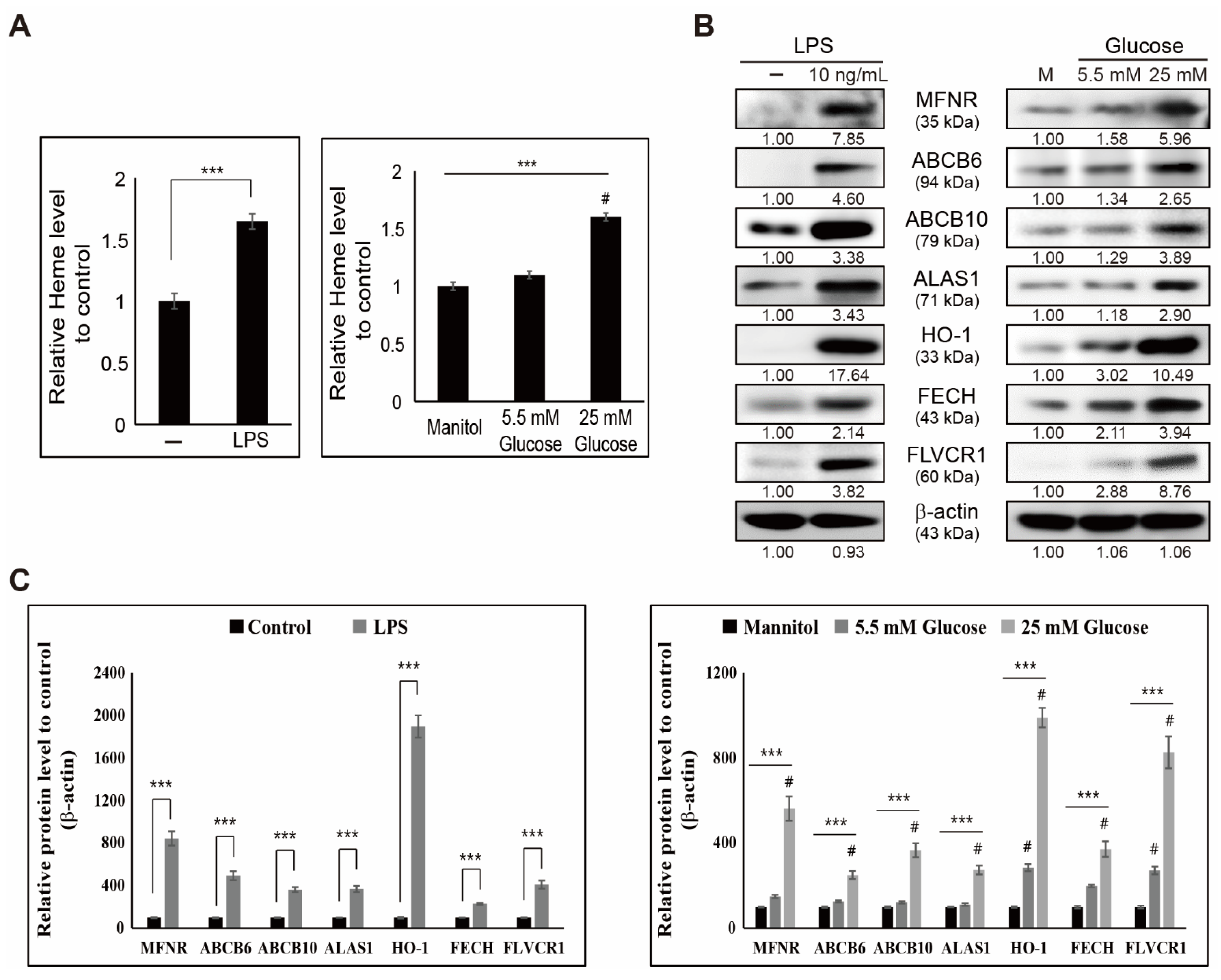
3.2. LPS- or Glucose-Induced Inflammation Enhances Heme Biosynthesis
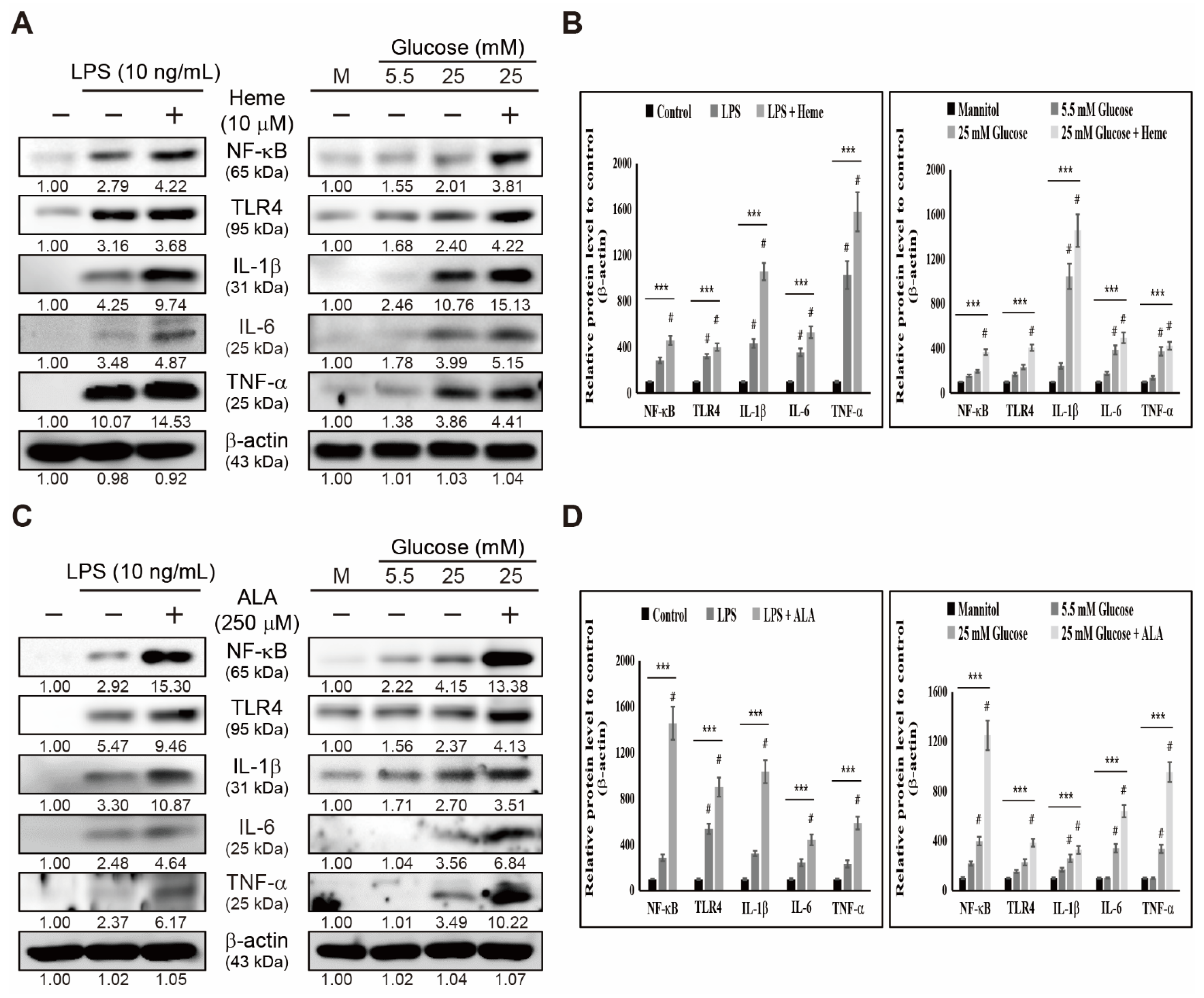
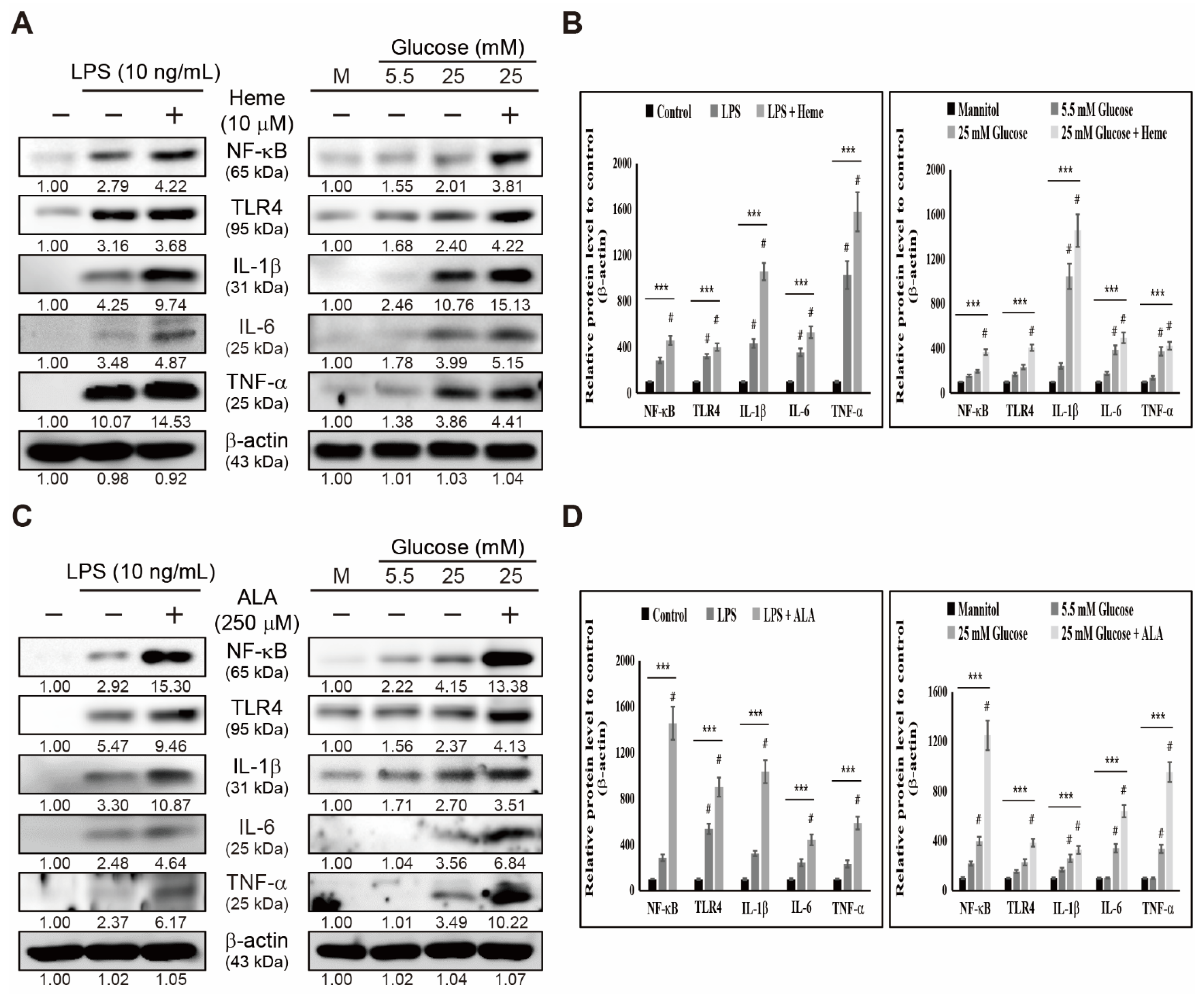
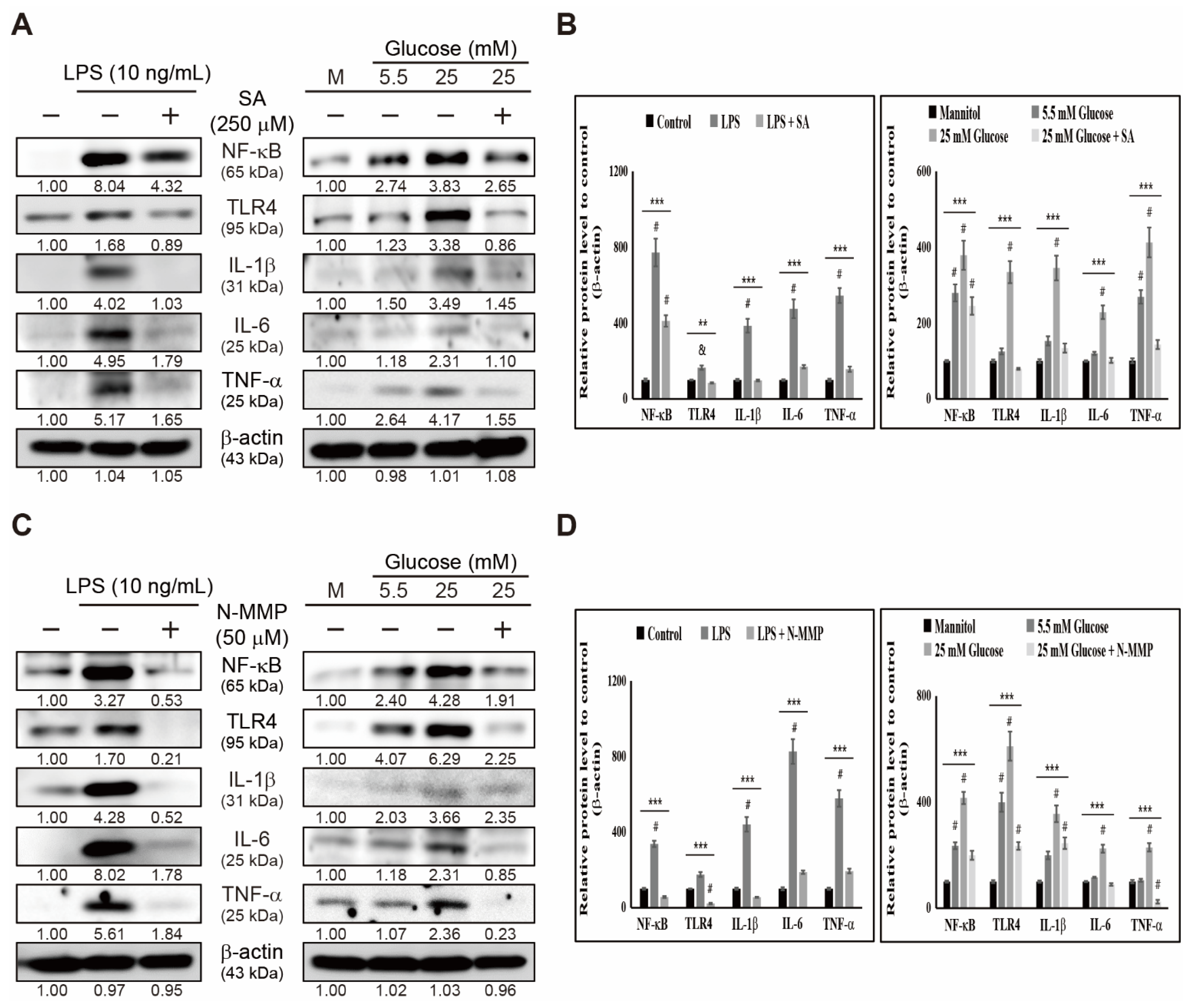
3.3. Heme Biosynthesis Induces Inflammatory Responses through TLR4/NF-κB Signaling in THP-1 Cells
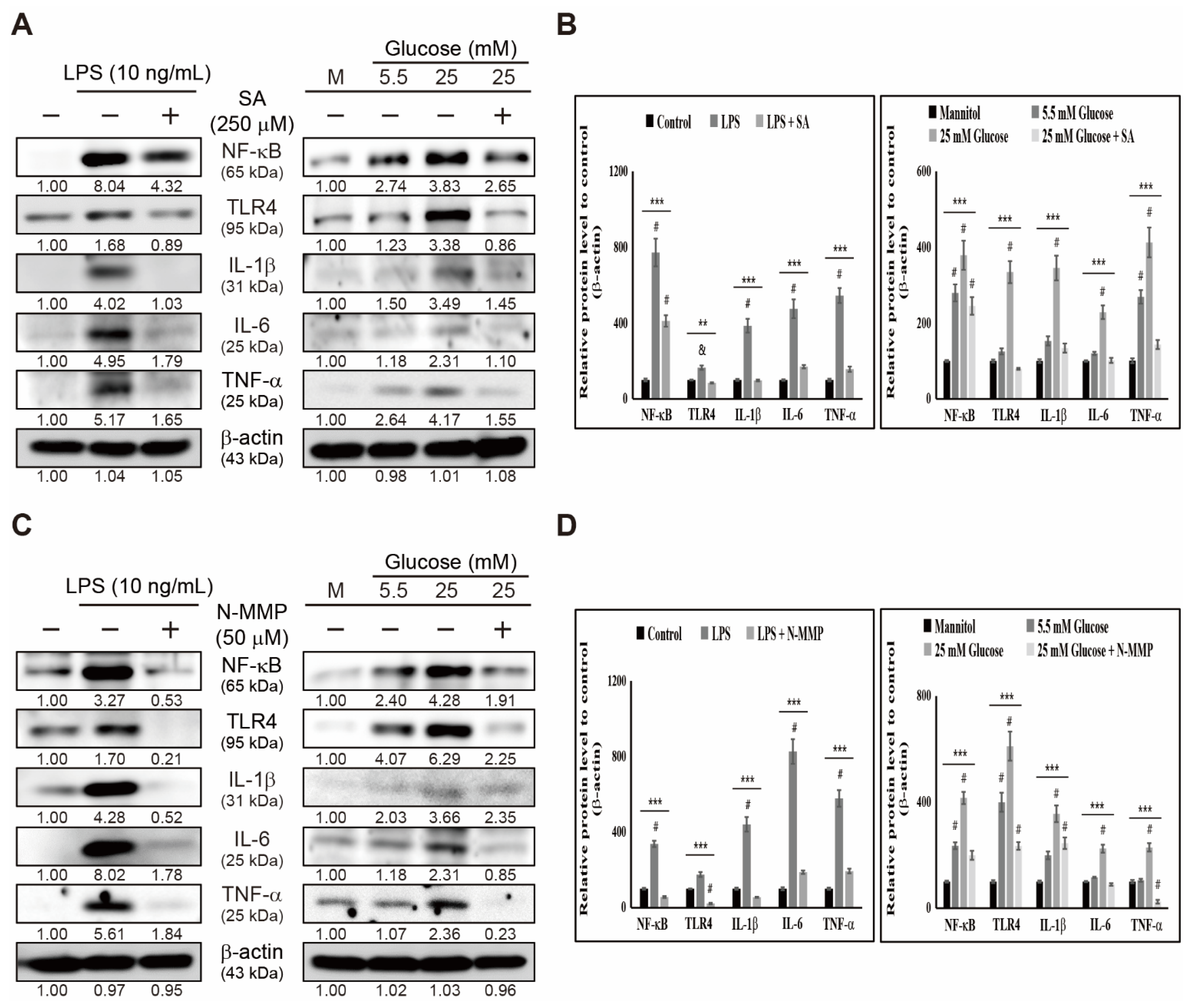
3.4. Inhibition of Heme Biosynthesis Causes Inflammatory Response Suppression in THP-1 Cells
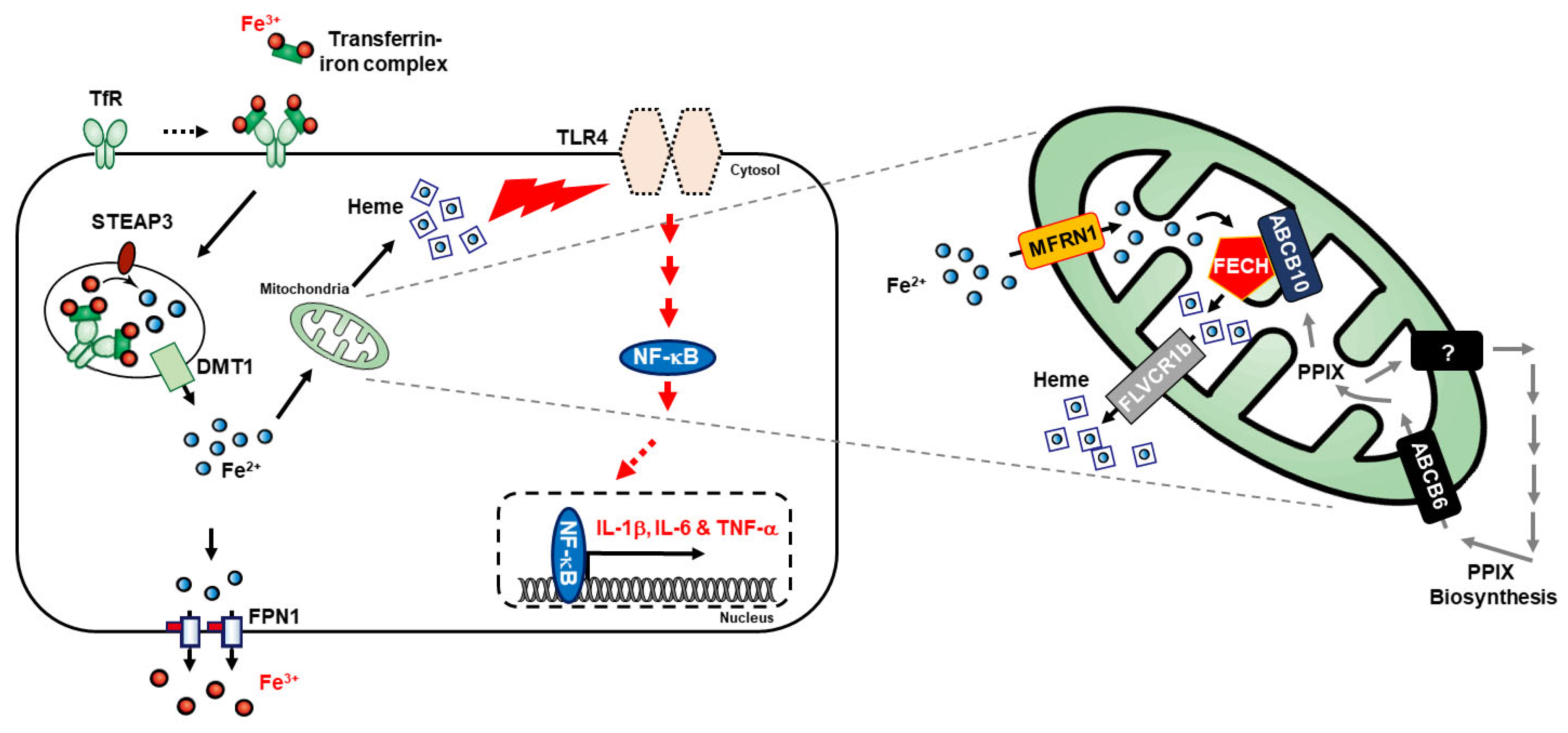
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feng, H.; Gu, J.; Gou, F.; Huang, W.; Gao, C.; Chen, G.; Long, Y.; Zhou, X.; Yang, M.; Liu, S.; et al. High glucose and lipopolysaccharide prime NLRP3 inflammasome via ROS/TXNIP pathway in mesangial cells. J. Diabetes Res. 2016, 2016, 6973175. [Google Scholar] [CrossRef] [Green Version]
- Libby, P. Inflammatory mechanisms: The molecular basis of inflammation and disease. Nutr. Rev. 2007, 65, S140–S146. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uematsu, S.; Akira, S. The role of toll-like receptors in immune disorders. Expert Opin. Biol. Ther. 2006, 6, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.S.; Sp, N.; Kang, D.Y.; Rugamba, A.; Kim, I.H.; Bae, S.W.; Liu, Q.; Jang, K.J.; Yang, Y.M. Sulfur Compounds inhibit high glucose-induced inflammation by regulating NF-kappaB signaling in human monocytes. Molecules 2020, 25, 2342. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C.; Liu, Z.G. A special issue on NF-kappaB signaling and function. Cell Res. 2011, 21, 1–2. [Google Scholar] [CrossRef]
- Ramana, K.V.; Friedrich, B.; Srivastava, S.; Bhatnagar, A.; Srivastava, S.K. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes 2004, 53, 2910–2920. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms linking inflammation to insulin resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef]
- Kang, D.Y.; Sp, N.; Jo, E.S.; Rugamba, A.; Kim, H.D.; Kim, I.H.; Park, J.C.; Bae, S.W.; Jang, K.J.; Yang, Y.M. Non-toxic sulfur inhibits LPS-induced inflammation by regulating TLR-4 and JAK2/STAT3 through IL-6 signaling. Mol. Med. Rep. 2021, 24, 1–8. [Google Scholar] [CrossRef]
- Yucel, G.; Zhao, Z.; El-Battrawy, I.; Lan, H.; Lang, S.; Li, X.; Buljubasic, F.; Zimmermann, W.H.; Cyganek, L.; Utikal, J.; et al. Lipopolysaccharides induced inflammatory responses and electrophysiological dysfunctions in human-induced pluripotent stem cell derived cardiomyocytes. Sci. Rep. 2017, 7, 2935. [Google Scholar] [CrossRef]
- Piktel, E.; Wnorowska, U.; Ciesluk, M.; Deptula, P.; Pogoda, K.; Misztalewska-Turkowicz, I.; Paprocka, P.; Niemirowicz-Laskowska, K.; Wilczewska, A.Z.; Janmey, P.A.; et al. Inhibition of inflammatory response in human keratinocytes by magnetic nanoparticles functionalized with PBP10 peptide derived from the PIP2-binding site of human plasma gelsolin. J. Nanobiotechnol. 2019, 17, 22. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-like receptors and innate immunity. J. Mol. Med. 2006, 84, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Sp, N.; Kang, D.Y.; Kim, H.D.; Rugamba, A.; Jo, E.S.; Park, J.C.; Bae, S.W.; Lee, J.M.; Jang, K.J. Natural sulfurs inhibit LPS-induced inflammatory responses through NF-kappaB signaling in CCD-986Sk skin fibroblasts. Life 2021, 11, 427. [Google Scholar] [CrossRef]
- Tsilidis, K.K.; Kasimis, J.C.; Lopez, D.S.; Ntzani, E.E.; Ioannidis, J.P. Type 2 diabetes and cancer: Umbrella review of meta-analyses of observational studies. BMJ 2015, 350, g7607. [Google Scholar] [CrossRef] [Green Version]
- Tsilidis, K.K.; Allen, N.E.; Appleby, P.N.; Rohrmann, S.; Nothlings, U.; Arriola, L.; Gunter, M.J.; Chajes, V.; Rinaldi, S.; Romieu, I.; et al. Diabetes mellitus and risk of prostate cancer in the European Prospective Investigation into Cancer and Nutrition. Int. J. Cancer 2015, 136, 372–381. [Google Scholar] [CrossRef]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The diabetes mellitus-atherosclerosis connection: The role of lipid and glucose metabolism and chronic inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruderman, N.B.; Haudenschild, C. Diabetes as an atherogenic factor. Prog. Cardiovasc. Dis. 1984, 26, 373–412. [Google Scholar] [CrossRef]
- Mazzone, T.; Chait, A.; Plutzky, J. Cardiovascular disease risk in type 2 diabetes mellitus: Insights from mechanistic studies. Lancet 2008, 371, 1800–1809. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelissen, A.; Guo, L.; Sakamoto, A.; Virmani, R.; Finn, A.V. New insights into the role of iron in inflammation and atherosclerosis. EBioMedicine 2019, 47, 598–606. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Harrington, L.; Trebicka, E.; Shi, H.N.; Kagan, J.C.; Hong, C.C.; Lin, H.Y.; Babitt, J.L.; Cherayil, B.J. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J. Clin. Investig. 2009, 119, 3322–3328. [Google Scholar] [CrossRef] [Green Version]
- Ponka, P. Cell biology of heme. Am. J. Med. Sci. 1999, 318, 241–256. [Google Scholar] [CrossRef]
- Wagener, F.A.; Volk, H.D.; Willis, D.; Abraham, N.G.; Soares, M.P.; Adema, G.J.; Figdor, C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev. 2003, 55, 551–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A.; Vercellotti, G.M.; Grande, J.P.; Miyoshi, H.; Paya, C.V.; Manivel, J.C.; Haggard, J.J.; Croatt, A.J.; Payne, W.D.; Alam, J. Heme protein-induced chronic renal inflammation: Suppressive effect of induced heme oxygenase-1. Kidney Int. 2001, 59, 106–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagener, F.A.; Eggert, A.; Boerman, O.C.; Oyen, W.J.; Verhofstad, A.; Abraham, N.G.; Adema, G.; van Kooyk, Y.; de Witte, T.; Figdor, C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001, 98, 1802–1811. [Google Scholar] [CrossRef] [PubMed]
- Wagener, F.A.; Feldman, E.; de Witte, T.; Abraham, N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc. Soc. Exp. Biol. Med. 1997, 216, 456–463. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Dasu, M.R.; Devaraj, S.; Jialal, I. High glucose induces IL-1beta expression in human monocytes: Mechanistic insights. Am. J. Physiol Endocrinol Metab 2007, 293, E337–E346. [Google Scholar] [CrossRef] [Green Version]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Olson, C.M.; Hedrick, M.N.; Izadi, H.; Bates, T.C.; Olivera, E.R.; Anguita, J. p38 mitogen-activated protein kinase controls NF-kappaB transcriptional activation and tumor necrosis factor alpha production through RelA phosphorylation mediated by mitogen- and stress-activated protein kinase 1 in response to Borrelia burgdorferi antigens. Infect. Immun. 2007, 75, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, A.C.; Almeida, J.I.; Lima, I.S.; Kapitao, A.S.; Gozzelino, R. Iron metabolism and the inflammatory response. IUBMB Life 2017, 69, 442–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konijn, A.M. Iron metabolism in inflammation. Baillieres Clin. Haematol. 1994, 7, 829–849. [Google Scholar] [CrossRef]
- Zhang, M.W.; Yang, G.; Zhou, Y.F.; Qian, C.; Mu, M.D.; Ke, Y.; Qian, Z.M. Regulating ferroportin-1 and transferrin receptor-1 expression: A novel function of hydrogen sulfide. J. Cell Physiol. 2019, 234, 3158–3169. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O.; Van der Westhuizen, J.; Vulpe, C.D.; Anderson, G.J.; Simpson, R.J.; McKie, A.T. Molecular and functional roles of duodenal cytochrome B (Dcytb) in iron metabolism. Blood Cells Mol. Dis. 2002, 29, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.D.; Romano, M.A.; Su, M.A.; Garrick, L.M.; Garrick, M.D.; Andrews, N.C. Nramp2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp2 in endosomal iron transport. Proc. Natl. Acad. Sci. USA 1998, 95, 1148–1153. [Google Scholar] [CrossRef] [Green Version]
- Byrne, S.L.; Krishnamurthy, D.; Wessling-Resnick, M. Pharmacology of iron transport. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 17–36. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.R.; Lane, D.J.; Becker, E.M.; Huang, M.L.; Whitnall, M.; Suryo Rahmanto, Y.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Mercurio, S.; Tolosano, E. Heme and erythropoieis: More than a structural role. Haematologica 2014, 99, 973–983. [Google Scholar] [CrossRef]
- Chen, W.; Paradkar, P.N.; Li, L.; Pierce, E.L.; Langer, N.B.; Takahashi-Makise, N.; Hyde, B.B.; Shirihai, O.S.; Ward, D.M.; Kaplan, J.; et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 16263–16268. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Investig. 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [Green Version]
- Malik, Z.; Bessler, H.; Djaldetti, M. The role of hemin in the regulation of heme synthesis by fetal mouse liver erythroblasts in culture. Exp. Hematol. 1979, 7, 183–188. [Google Scholar]
- Ponka, P.; Schulman, H.M. Regulation of heme synthesis in erythroid cells: Hemin inhibits transferrin iron utilization but not protoporphyrin synthesis. Blood 1985, 65, 850–857. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, K.T.; Shang, M.; Wu, R.; Chang, H.C.; Khechaduri, A.; Sato, T.; Kamide, C.; Liu, T.; Naga Prasad, S.V.; Ardehali, H. Increased heme levels in the heart lead to exacerbated ischemic injury. J. Am. Heart Assoc. 2015, 4, e002272. [Google Scholar] [CrossRef] [Green Version]
- Ebert, P.S.; Hess, R.A.; Frykholm, B.C.; Tschudy, D.P. Succinylacetone, a potent inhibitor of heme biosynthesis: Effect on cell growth, heme content and delta-aminolevulinic acid dehydratase activity of malignant murine erythroleukemia cells. Biochem Biophys. Res. Commun. 1979, 88, 1382–1390. [Google Scholar] [CrossRef]
- Beale, S.I.; Chen, N.C. N-methyl mesoporphyrin IX inhibits phycocyanin, but not chlorophyll synthesis in cyanidium caldarium. Plant. Physiol. 1983, 71, 263–268. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, D.Y.; Sp, N.; Jo, E.S.; Lee, J.-M.; Jang, K.-J. New Insights into the Pivotal Role of Iron/Heme Metabolism in TLR4/NF-κB Signaling-Mediated Inflammatory Responses in Human Monocytes. Cells 2021, 10, 2549. https://doi.org/10.3390/cells10102549
Kang DY, Sp N, Jo ES, Lee J-M, Jang K-J. New Insights into the Pivotal Role of Iron/Heme Metabolism in TLR4/NF-κB Signaling-Mediated Inflammatory Responses in Human Monocytes. Cells. 2021; 10(10):2549. https://doi.org/10.3390/cells10102549
Chicago/Turabian StyleKang, Dong Young, Nipin Sp, Eun Seong Jo, Jin-Moo Lee, and Kyoung-Jin Jang. 2021. "New Insights into the Pivotal Role of Iron/Heme Metabolism in TLR4/NF-κB Signaling-Mediated Inflammatory Responses in Human Monocytes" Cells 10, no. 10: 2549. https://doi.org/10.3390/cells10102549
APA StyleKang, D. Y., Sp, N., Jo, E. S., Lee, J.-M., & Jang, K.-J. (2021). New Insights into the Pivotal Role of Iron/Heme Metabolism in TLR4/NF-κB Signaling-Mediated Inflammatory Responses in Human Monocytes. Cells, 10(10), 2549. https://doi.org/10.3390/cells10102549