Comparing Biochemical and Raman Microscopy Analyses of Starch, Lipids, Polyphosphate, and Guanine Pools during the Cell Cycle of Desmodesmus quadricauda

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Organism
2.2. Algae Cultivation and Synchronization of Cultures
2.3. 4′,6-diamidine-2′-phenylindole Dihydrochloride (DAPI) Staining
2.4. Cell Size and Number
2.5. Assessment of Commitment Points and Cell Division Curves
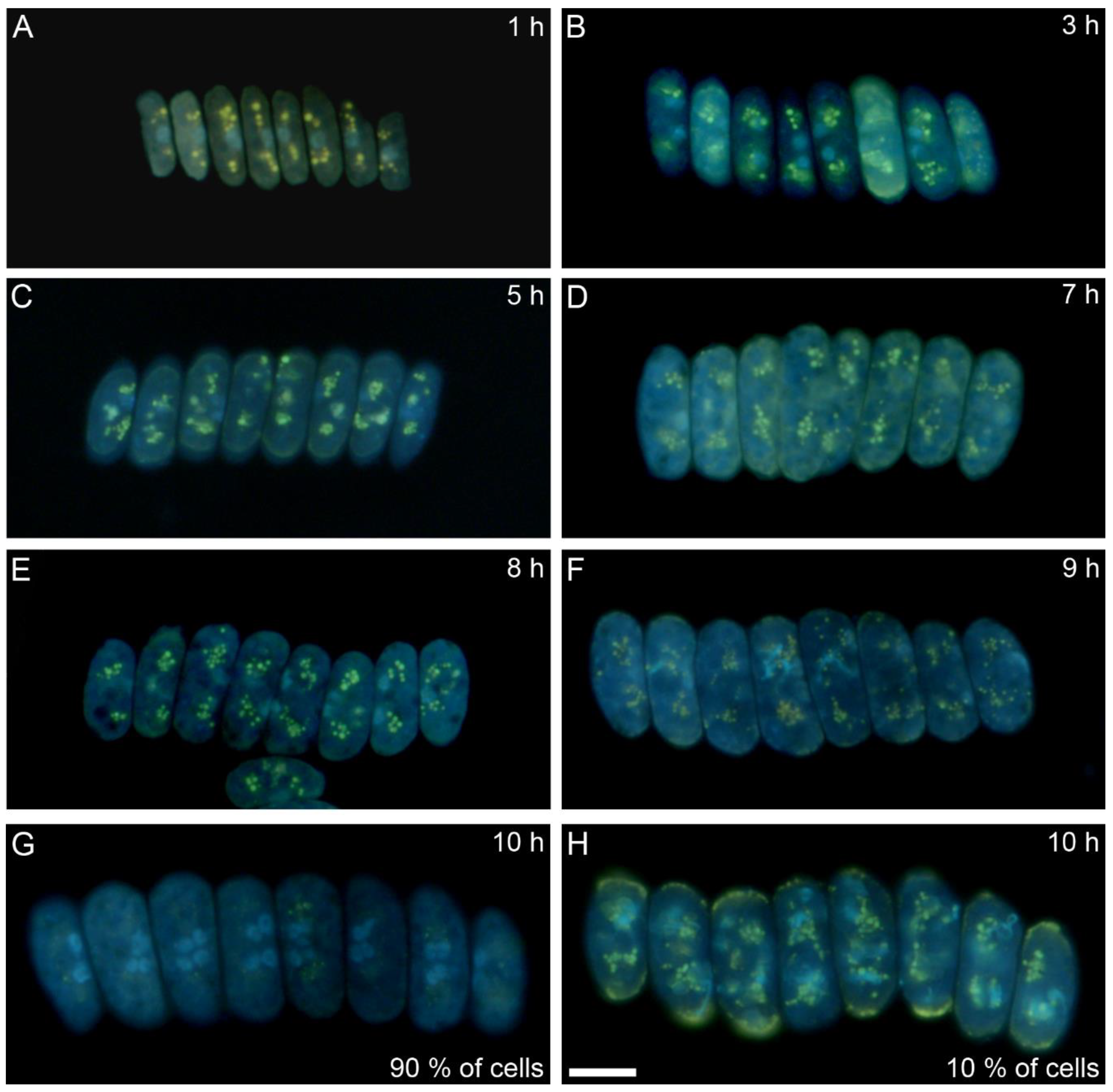
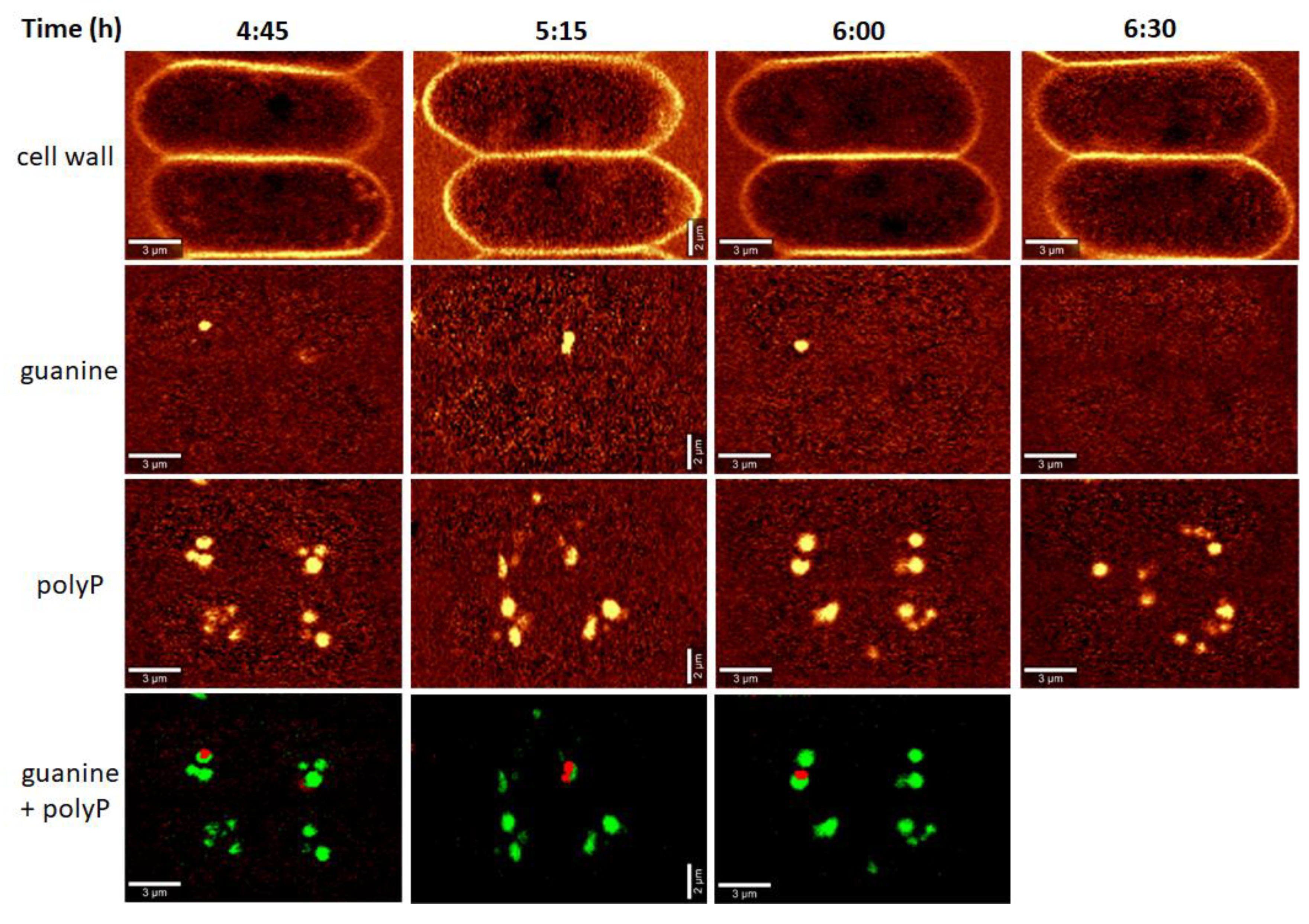
2.6. Polyphosphate Visualization
2.7. Biochemical Analyses
2.7.1. Estimation of Bulk RNA, DNA, and Protein
2.7.2. Starch Assay
2.7.3. Total Phosphate Assay
2.7.4. Polyphosphate Assays
2.8. Raman Analyses
2.8.1. Sample Preparation
2.8.2. Raman Measurement
2.8.3. Data Treatment
3. Results
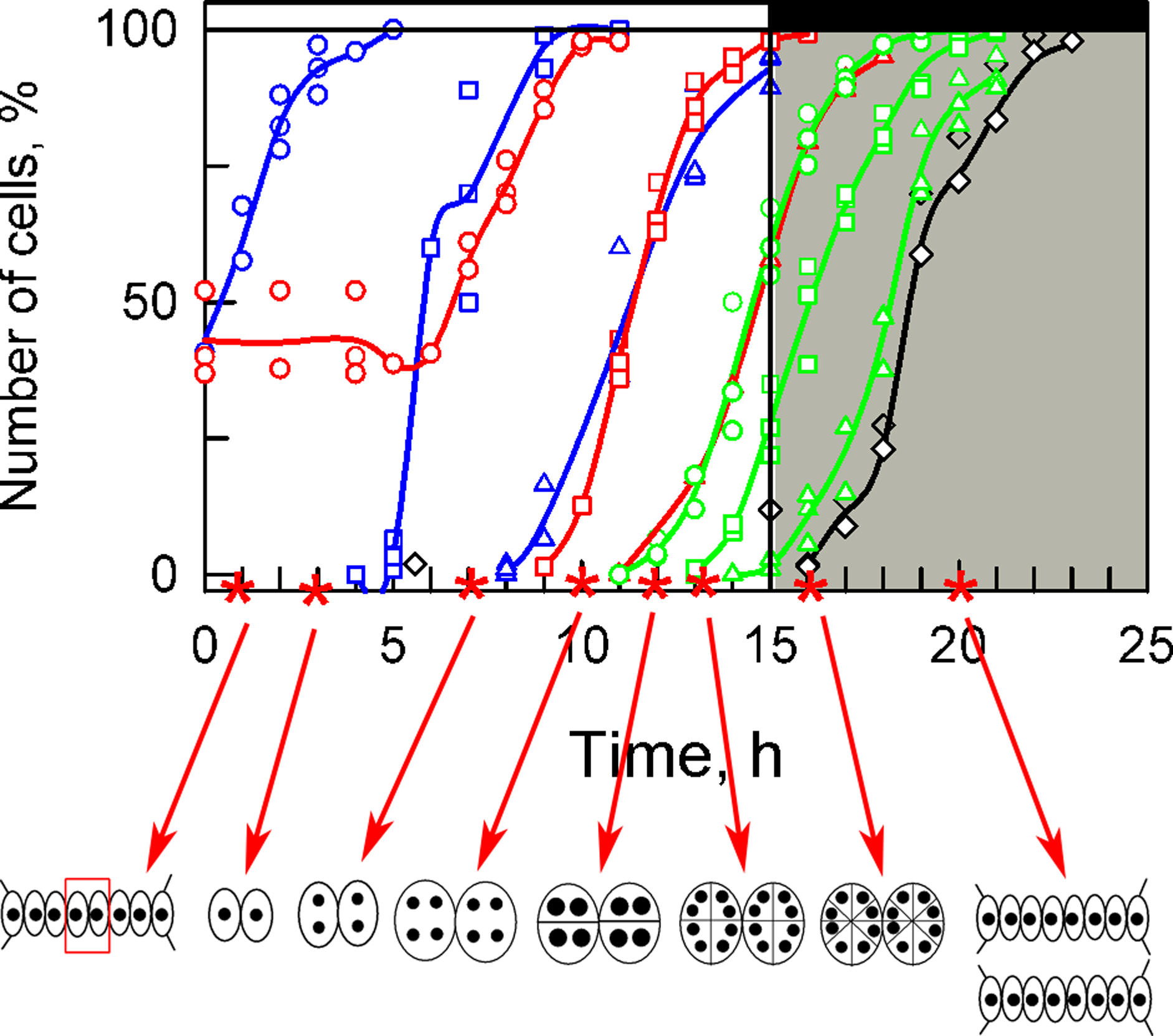
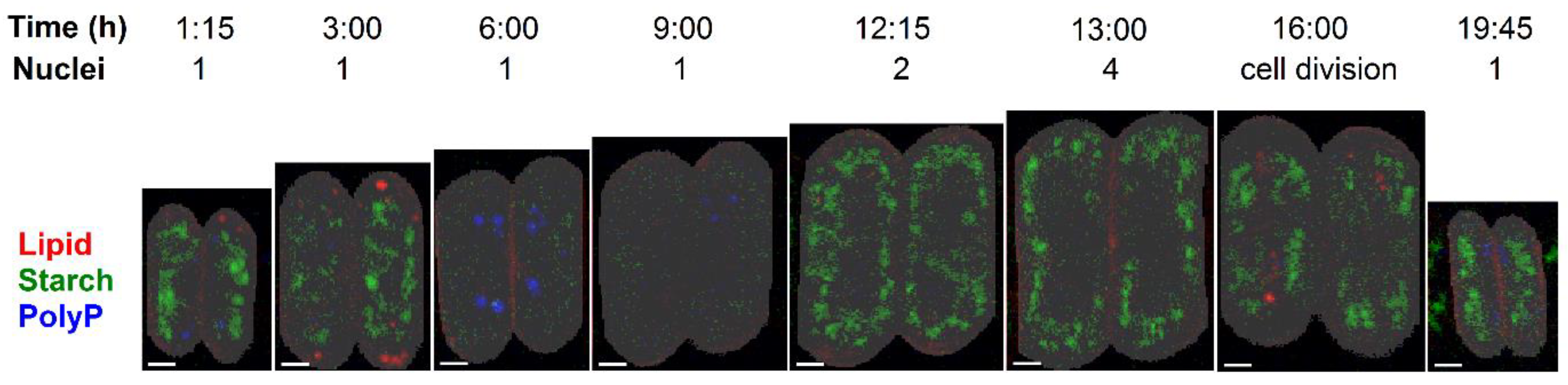
3.1. Cell Cycle Characteristics

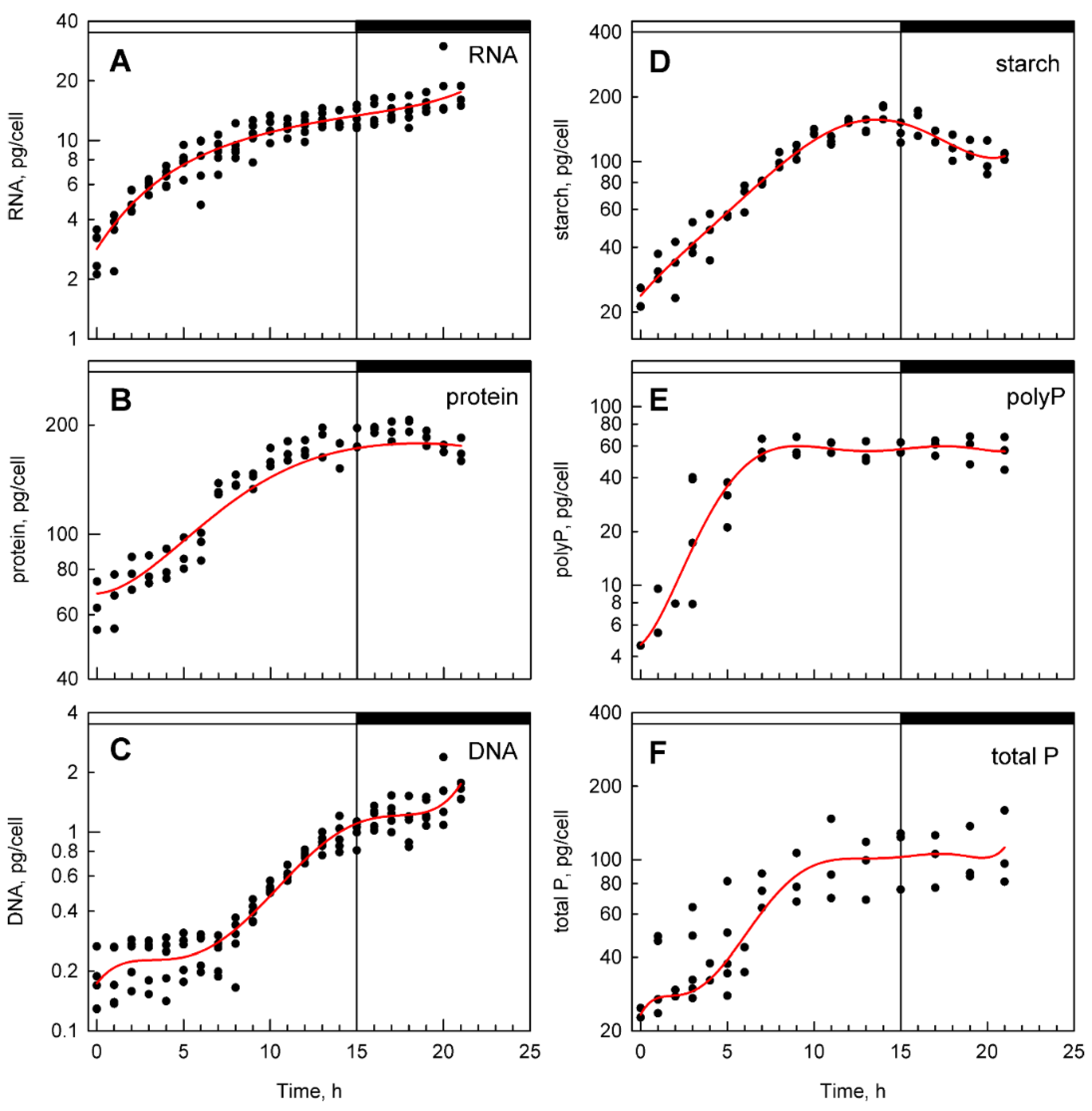
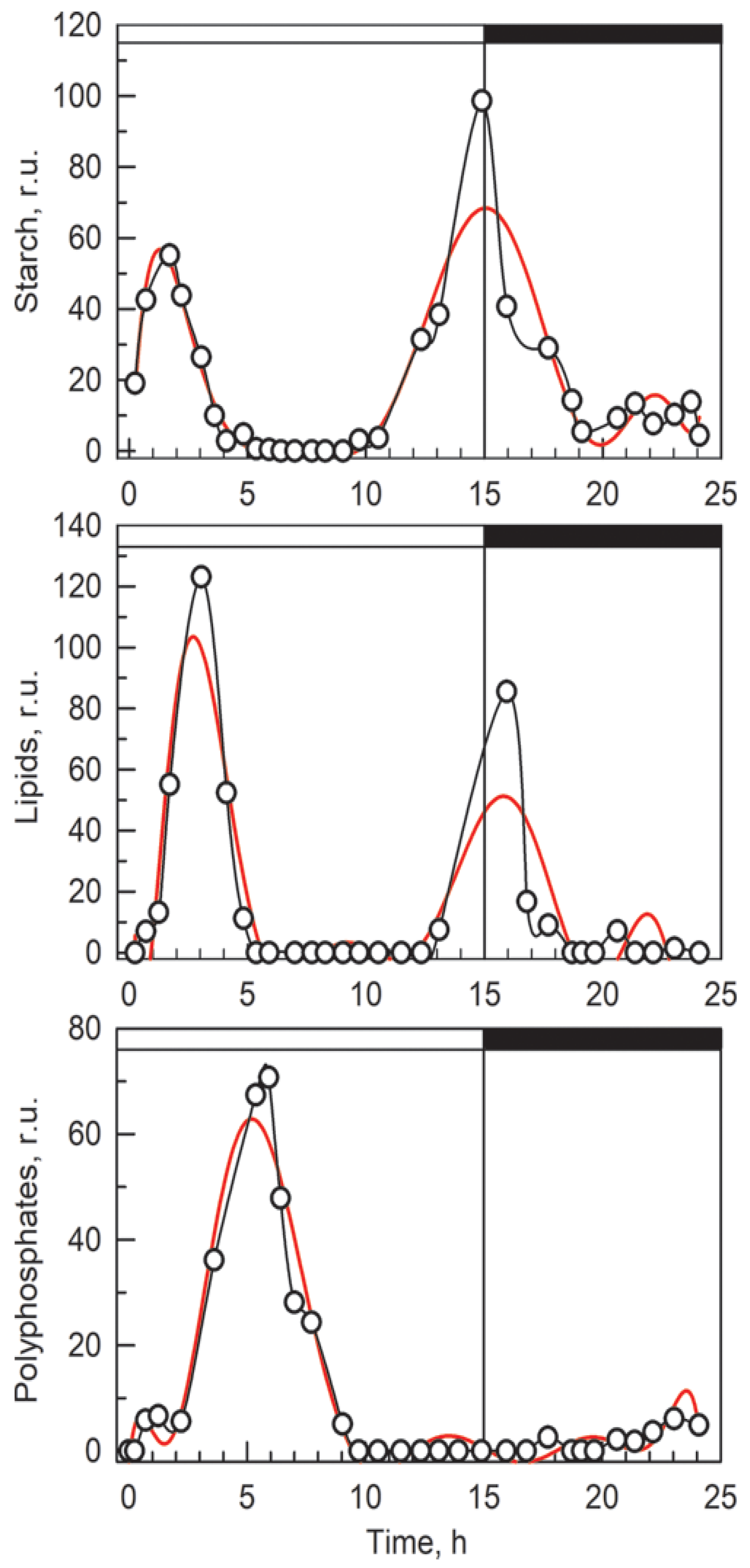
3.2. Bulk Analysis of Cell Composition
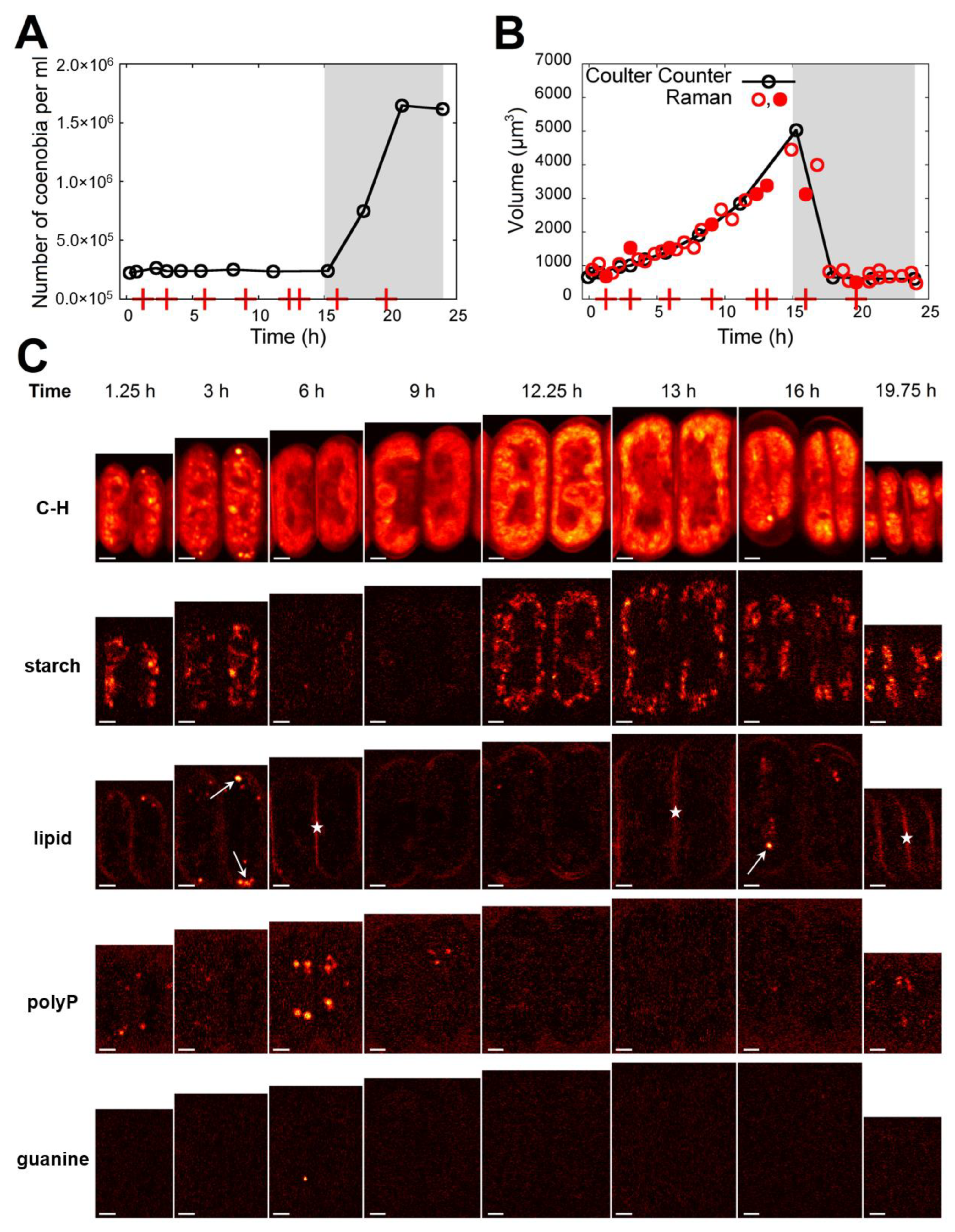
3.3. Raman Estimates of Cell Volume and Chemical Composition
3.4. Comparison of Raman and Bulk Biochemical Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zachleder, V.; Bišová, K.; Vítová, M. The cell cycle of microalgae. In The Physiology of Microalgae; Borowitzka, M.A., Beardall, J., Raven, J.A., Eds.; Springer: Dordrecht, The Netherlands, 2016; Volume 6, pp. 3–46. [Google Scholar]
- Moudříková, Š.; Nedbal, L.; Solovchenko, A.; Mojzeš, P. Raman microscopy shows that nitrogen-rich cellular inclusions in microalgae are microcrystalline guanine. Algal Res. 2017, 23, 216–222. [Google Scholar] [CrossRef]
- Mojzeš, P.; Gao, L.; Ismagulova, T.; Pilátová, J.; Moudříková, Š.; Gorelova, O.; Solovchenko, A.; Nedbal, L.; Salih, A. Guanine, a high-capacity and rapid-turnover nitrogen reserve in microalgal cells. Proc. Natl. Acad. Sci. USA 2020, 117, 32722–32730. [Google Scholar] [CrossRef]
- Šetlík, I.; Zachleder, V. The multiple fission cell reproductive patterns in algae. In The Microbial Cell Cycle; Nurse, P., Streiblová, E., Eds.; CRC Press Inc.: Boca Raton, FL, USA, 1984; pp. 253–279. [Google Scholar]
- Zachleder, V.; van den Ende, H. Cell cycle events in the green alga Chlamydomonas eugametos and their control by environmental factors. J. Cell Sci. 1992, 102, 469–474. [Google Scholar]
- Donnan, L.; John, P.C.L. Cell cycle control by timer and sizer in Chlamydomonas. Nature 1983, 304, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Šetlík, I.; Berková, E.; Doucha, J.; Kubín, S.; Vendlová, J.; Zachleder, V. The coupling of synthetic and reproduction processes in Scenedesmus quadricauda. Arch. Hydrobiol. Algol. Stud. 1972, 7, 172–217. [Google Scholar]
- Zachleder, V.; Šetlík, I. Distinct controls of DNA-replication and of nuclear division in the cell-cycles of the chlorococcal alga Scenedesmus quadricauda. J. Cell Sci. 1988, 91, 531–539. [Google Scholar]
- Zachleder, V.; Šetlík, I. Timing of events in overlapping cell reproductive sequences and their mutual interactions in the alga Scenedesmus quadricauda. J. Cell Sci. 1990, 97, 631–638. [Google Scholar]
- Zachleder, V. The course of reproductive events in the chloroplast cycle of the chlorococcal alga Scenedesmus quadricauda as revealed by using inhibitors of DNA replication. Plant Cell Physiol. 1997, 38, 56. [Google Scholar]
- Tukaj, Z.; Kubínová, A.; Zachleder, V. Effect of irradiance on growth and reproductive processes during the cell cycle in Scenedesmus armatus (Chlorophyta). J. Phycol. 1996, 32, 624–631. [Google Scholar] [CrossRef]
- Zachleder, V.; Doucha, J.; Berková, E.; Šetlík, I. The effect of synchronizing dark period on populations of Scenedesmus quadricauda. Biol Plant. 1975, 17, 416–433. [Google Scholar] [CrossRef]
- Zachleder, V.; Bišová, K.; Vítová, M.; Kubín, Š.; Hendrychová, J. Variety of cell cycle patterns in the alga Scenedesmus quadricauda (Chlorophyta) as revealed by application of illumination regimes and inhibitors. Eur. J. Phycol. 2002, 37, 361–371. [Google Scholar] [CrossRef]
- Vítová, M.; Zachleder, V. Points of commitment to reproductive events as a tool for analysis of the cell cycle in synchronous cultures of algae. Folia Microbiol. 2005, 50, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Stessman, D.; Hart, J.H.; Dong, H.L.; Wang, Y.J.; Wright, D.A.; Nikolau, B.J.; Spalding, M.H.; Halverson, L.J. High-throughput fluorescence-activated cell sorting for lipid hyperaccumulating Chlamydomonas reinhardtii mutants. Plant Biotechnol. J. 2014, 12, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Terashima, M.; Freeman, E.S.; Jinkerson, R.E.; Jonikas, M.C. A fluorescence-activated cell sorting-based strategy for rapid isolation of high-lipid Chlamydomonas mutants. Plant J. 2014, 81, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Vítová, M.; Hendrychová, J.; Cepák, V.; Zachleder, V. Visualization of DNA-containing structures in various species of Chlorophyta, Rhodophyta and Cyanophyta using SYBR green I dye. Folia Microbiol. 2005, 50, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Dieing, T.; Hollricher, O.; Toporski, J. Confocal Raman Microscopy; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar] [CrossRef]
- Butler, H.J.; Ashton, L.; Bird, B.; Cinque, G.; Curtis, K.; Dorney, J.; Esmonde-White, K.; Fullwood, N.J.; Gardner, B.; Martin-Hirsch, P.L.; et al. Using Raman spectroscopy to characterize biological materials. Nat. Protoc. 2016, 11, 664–687. [Google Scholar] [CrossRef]
- Koch, M.; Zagermann, S.; Kniggendorf, A.K.; Meinhardt-Wollweber, M.; Roth, B. Violaxanthin cycle kinetics analysed in vivo with resonance Raman spectroscopy. J. Raman Spectrosc. 2017, 48, 686–691. [Google Scholar] [CrossRef]
- Jehlička, J.; Edwards, H.G.M.; Orenc, A. Raman spectroscopy of microbial pigments. Appl. Environ. Microbiol. 2014, 80, 3286–3295. [Google Scholar] [CrossRef]
- Li, K.; Cheng, J.; Ye, Q.; He, Y.; Zhou, J.H.; Cen, K.F. In vivo kinetics of lipids and astaxanthin evolution in Haematococcus pluvialis mutant under 15% CO2 using Raman microspectroscopy. Bioresour. Technol. 2017, 244, 1439–1444. [Google Scholar] [CrossRef]
- Meksiarun, P.; Spegazzini, N.; Matsui, H.; Nakajima, K.; Matsuda, Y.; Sato, H. In vivo study of lipid accumulation in the microalgae marine diatom Thalassiosira pseudonana using Raman spectroscopy. Appl. Spectrosc. 2015, 69, 45–51. [Google Scholar] [CrossRef]
- Samek, O.; Jonáš, A.; Pilát, Z.; Zemánek, P.; Nedbal, L.; Tříska, J.; Kotas, P.; Trtílek, M. Raman microspectroscopy of individual algal cells: Sensing unsaturation of storage lipids in vivo. Sensors 2010, 10, 8635–8651. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Volponi, J.V.; Oliver, A.E.; Parikh, A.N.; Simmons, B.A.; Singh, S. In vivo lipidomics using single-cell Raman spectroscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 3809–3814. [Google Scholar] [CrossRef] [PubMed]
- Chiu, L.-D.; Ho, S.-H.; Shimada, R.; Ren, N.-Q.; Ozawa, T. Rapid in vivo lipid/carbohydrate quantification of single microalgal cell by Raman spectral imaging to reveal salinity-induced starch-to-lipid shift. Biotechnol. Biofuels 2017, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Beal, C.M.; Cai, W.W.; Ruoff, R.S.; Terentjev, E.M. Micro-Raman spectroscopy of algae: Composition analysis and fluorescence background behavior. Biotechnol. Bioenergy 2010, 105, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Moudříková, Š.; Mojzeš, P.; Zachleder, V.; Pfaff, C.; Behrendt, D.; Nedbal, L. Raman and fluorescence microscopy sensing energy-transducing and energy-storing structures in microalgae. Algal Res. 2016, 16, 224–232. [Google Scholar] [CrossRef]
- Zachleder, V.; Šetlík, I. Effect of irradiance on the course of RNA synthesis in the cell cycle of Scenedesmus quadricauda. Biol. Plant. 1982, 24, 341–353. [Google Scholar] [CrossRef]
- Hlavová, M.; Vítová, M.; Bišová, K. Synchronization of green algae by light and dark regimes for cell cycle and cell division studies. In Plant Cell Division; Caillaud, M.-C., Ed.; Springer Science: New York, NY, USA; Berlin/Heidelberg, Germany; Dordrecht, The Netherlands; London, UK, 2016; pp. 3–16. [Google Scholar]
- Zachleder, V.; Cepák, V. Visualization of DNA containing structures by fluorochrome DAPI in those algal cells which are not freely permeable to the dye. Arch. Hydrobiol. Algol. Stud. 1987, 47, 157–168. [Google Scholar]
- Ota, S.; Yoshihara, M.; Yamazaki, T.; Takeshita, T.; Hirata, A.; Konomi, M.; Oshima, K.; Hattori, M.; Bišová, K.; Zachleder, V.; et al. Deciphering the relationship among phosphate dynamics, electron-dense body and lipid accumulation in the green alga Parachlorella kessleri. Sci. Rep. 2016, 6, 25731. [Google Scholar] [CrossRef]
- Wanka, F. Die Bestimmung der Nucleinsäuren in Chlorella pyrenoidosa. Planta 1962, 58, 594–619. [Google Scholar] [CrossRef]
- Lukavský, J.; Tetík, K.; Vendlová, J. Extraction of nucleic acid from the alga Scenedesmus Quadricauda. Arch. Hydrobiol. Algol. Stud. 1973, 9, 416–426. [Google Scholar]
- Decallonne, J.R.; Weyns, C.J. A shortened procedure of the diphenylamine reaction for measurement of deoxyribonucleic acid by using light activation. Anal. Biochem. 1976, 74, 448–456. [Google Scholar] [CrossRef]
- Zachleder, V. Optimization of nucleic acids assay in green and blue-green algae: Extraction procedures and the light-activated reaction for DNA. Arch. Hydrobiol. Algol. Stud. 1984, 36, 313–328. [Google Scholar] [CrossRef]
- Miller, G.L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 1959, 31, 426–428. [Google Scholar] [CrossRef]
- Everall, N.J. Confocal Raman microscopy: Performance, pitfalls, and best practice. Appl. Spectrosc. 2009, 63, 245A–262A. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.W.; Bateman, D.; Hauberg, S.; Wehbring, R. GNU Octave Version 4.0.0 Manual: A High-Level Interactive Language for Numerical Computations; CreateSpace Independent Publishing Platform: Boston, MA, USA, 2015. [Google Scholar]
- Palacký, J.; Mojzeš, P.; Bok, J. SVD-based method for intensity normalization, background correction and solvent subtraction in Raman spectroscopy exploiting the properties of water stretching vibrations. J. Raman Spectrosc. 2011, 42, 1528–1539. [Google Scholar] [CrossRef]
- Zachleder, V.; Schläfli, O.; Boschetti, A. Growth-controlled oscillation in activity of histone H1 kinase during the cell cycle of Chlamydomonas reinhardtii (Chlorophyta). J. Phycol. 1997, 33, 673–681. [Google Scholar] [CrossRef]
- Bišová, K.; Zachleder, V. Cell-cycle regulation in green algae dividing by multiple fission. J. Exp. Bot. 2014, 65, 2585–2602. [Google Scholar] [CrossRef]
- Miyachi, S.; Tamiya, H. Distribution and turnover of phosphate compounds in growing Chlorella cells. Plant Cell Physiol. 1961, 2, 405–414. [Google Scholar]
- Miyachi, S.; Tamiya, H. Some observations on the phosphorus metabolism in growing Chlorella cells. Biochim. Biophys. Acta 1961, 46, 200–202. [Google Scholar] [CrossRef]
- Miyachi, S.; Kanai, R.; Mihara, S.; Miyachi, S.; Aoki, S. Metabolic roles of inorganic polyphosphates in Chlorella cells. Biochim. Biophys. Acta 1964, 93, 625–634. [Google Scholar] [CrossRef]
- Juppner, J.; Mubeen, U.; Leisse, A.; Caldana, C.; Wiszniewski, A.; Steinhauser, D.; Giavalisco, P. The target of rapamycin kinase affects biomass accumulation and cell cycle progression by altering carbon/nitrogen balance in synchronized Chlamydomonas reinhardtii cells. Plant J. 2018, 93, 355–376. [Google Scholar] [CrossRef] [PubMed]
- Vítová, M.; Bišová, K.; Umysová, D.; Hlavová, M.; Kawano, S.; Zachleder, V.; Čížková, M. Chlamydomonas reinhardtii: Duration of its cell cycle and phases at growth rates affected by light intensity. Planta 2011, 233, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; He, Y.; Cui, Y.; Wang, T.; Wang, Y.; Li, Y.; Huang, W.E.; Xu, J. Raman spectroscopy provides a rapid, non-invasive method for quantitation of starch in live, unicellular microalgae. Biotechnol. J. 2014, 9, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Ando, M.; Mukai, S.; Osada, K.; Yoshino, T.; Hamaguchi, H.; Tanaka, T. In vivo live cell imaging for the quantitative monitoring of lipids by using raman microspectroscopy. Anal. Chem. 2014, 86, 8224–8230. [Google Scholar] [CrossRef] [PubMed]
- Moudříková, Š.; Sadowsky, A.; Metzger, S.; Nedbal, L.; Mettler-Altmann, T.; Mojzeš, P. Quantification of polyphosphate in microalgae by Raman microscopy and by a reference enzymatic assay. Anal. Chem. 2017, 89, 12006–12013. [Google Scholar] [CrossRef]
- Solovchenko, A.; Khozin-Goldberg, I.; Selyakh, I.; Semenova, L.; Ismagulova, T.; Lukyanov, A.; Mamedov, I.; Vinogradova, E.; Karpova, O.; Konyukhov, I.; et al. Phosphorus starvation and luxury uptake in green microalgae revisited. Algal Res. 2019, 43, 101651. [Google Scholar] [CrossRef]
- Barcytė, D.; Pilátová, J.; Mojzeš, P.; Nedbalová, L. The arctic Cylindrocystis (Zygnematophyceae, Streptophyta) green algae are genetically and morphologically diverse and exhibit effective accumulation of polyphosphate. J. Phycol. 2020, 56, 217–232. [Google Scholar] [CrossRef]
- Siebers, N.; Hofmann, D.; Schiedung, H.; Landsrath, A.; Ackermann, B.; Gao, L.; Mojzeš, P.; Jablonowski, N.D.; Nedbal, L.; Amelung, W. Towards phosphorus recycling for agriculture by algae: Soil incubation and rhizotron studies using 33P-labeled microalgal biomass. Algal Res. 2019, 43, 101634. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moudříková, Š.; Ivanov, I.N.; Vítová, M.; Nedbal, L.; Zachleder, V.; Mojzeš, P.; Bišová, K. Comparing Biochemical and Raman Microscopy Analyses of Starch, Lipids, Polyphosphate, and Guanine Pools during the Cell Cycle of Desmodesmus quadricauda. Cells 2021, 10, 62. https://doi.org/10.3390/cells10010062
Moudříková Š, Ivanov IN, Vítová M, Nedbal L, Zachleder V, Mojzeš P, Bišová K. Comparing Biochemical and Raman Microscopy Analyses of Starch, Lipids, Polyphosphate, and Guanine Pools during the Cell Cycle of Desmodesmus quadricauda. Cells. 2021; 10(1):62. https://doi.org/10.3390/cells10010062
Chicago/Turabian StyleMoudříková, Šárka, Ivan Nedyalkov Ivanov, Milada Vítová, Ladislav Nedbal, Vilém Zachleder, Peter Mojzeš, and Kateřina Bišová. 2021. "Comparing Biochemical and Raman Microscopy Analyses of Starch, Lipids, Polyphosphate, and Guanine Pools during the Cell Cycle of Desmodesmus quadricauda" Cells 10, no. 1: 62. https://doi.org/10.3390/cells10010062
APA StyleMoudříková, Š., Ivanov, I. N., Vítová, M., Nedbal, L., Zachleder, V., Mojzeš, P., & Bišová, K. (2021). Comparing Biochemical and Raman Microscopy Analyses of Starch, Lipids, Polyphosphate, and Guanine Pools during the Cell Cycle of Desmodesmus quadricauda. Cells, 10(1), 62. https://doi.org/10.3390/cells10010062