Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair


Abstract
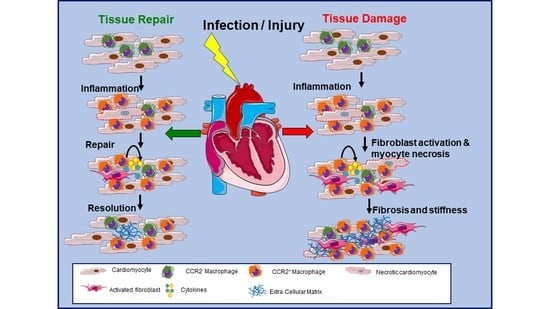
1. Introduction
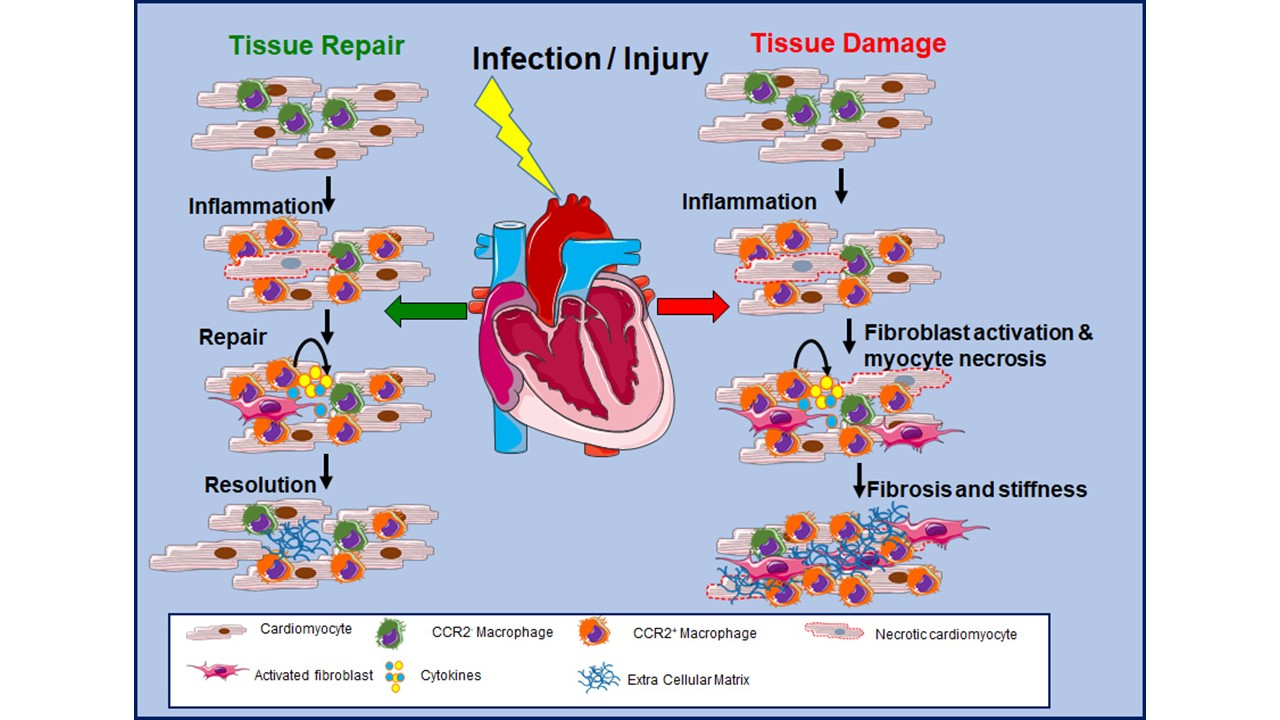
2. Cardiac Immune Cells
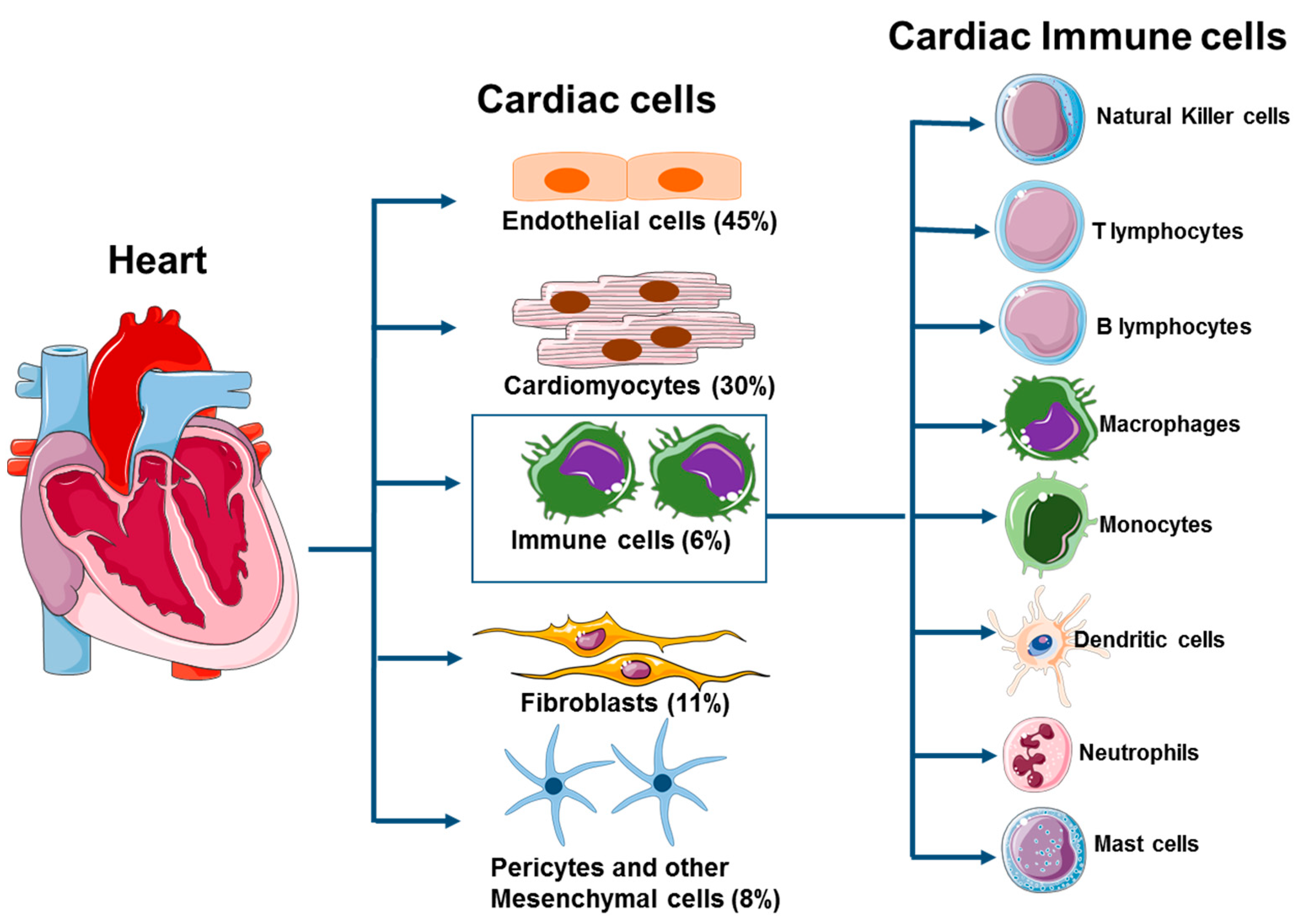
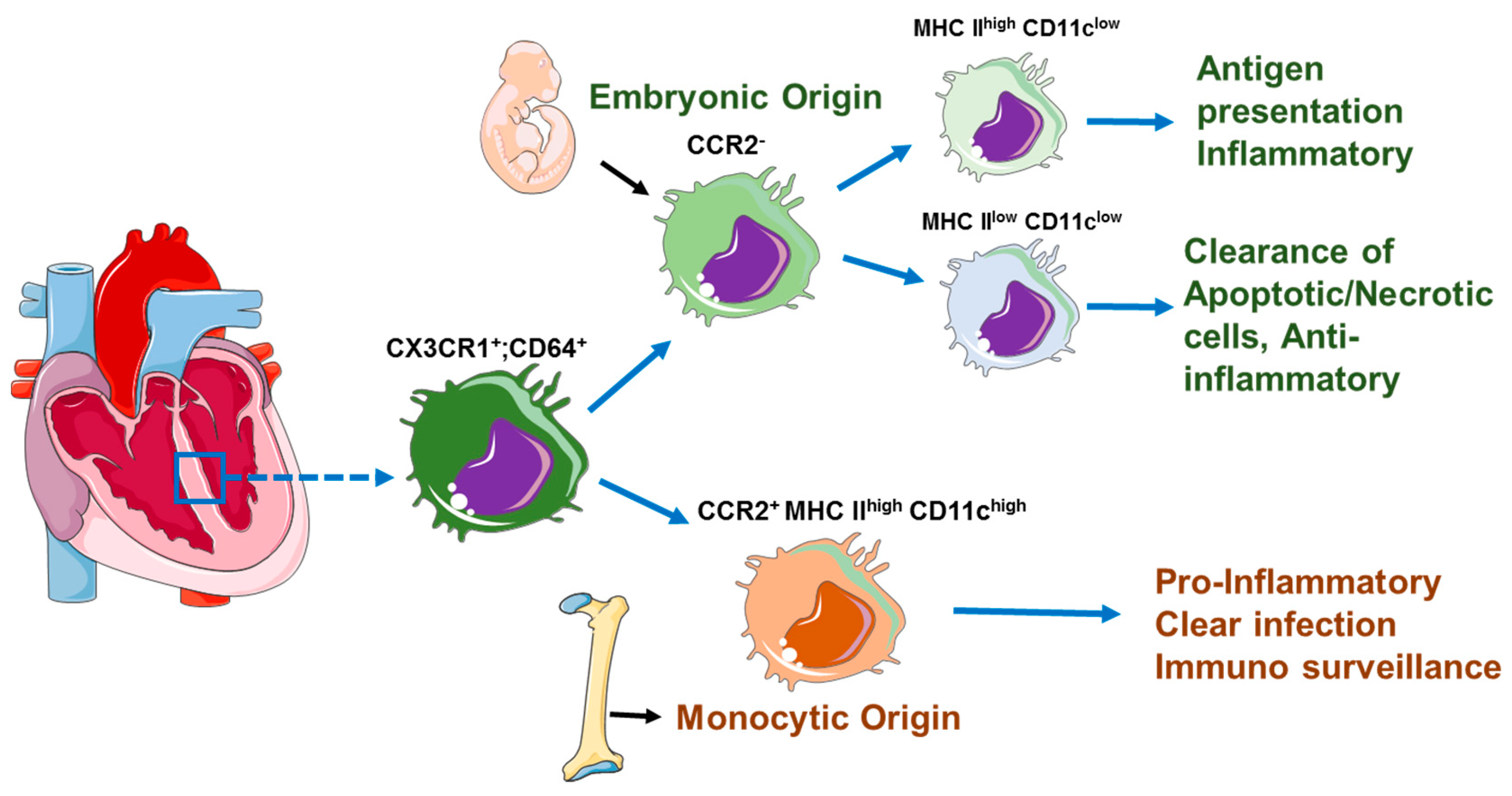
3. Cardiac-Resident Macrophages
4. Macrophage Role in Cardiac Inflammation and Cardiac Dysfunction
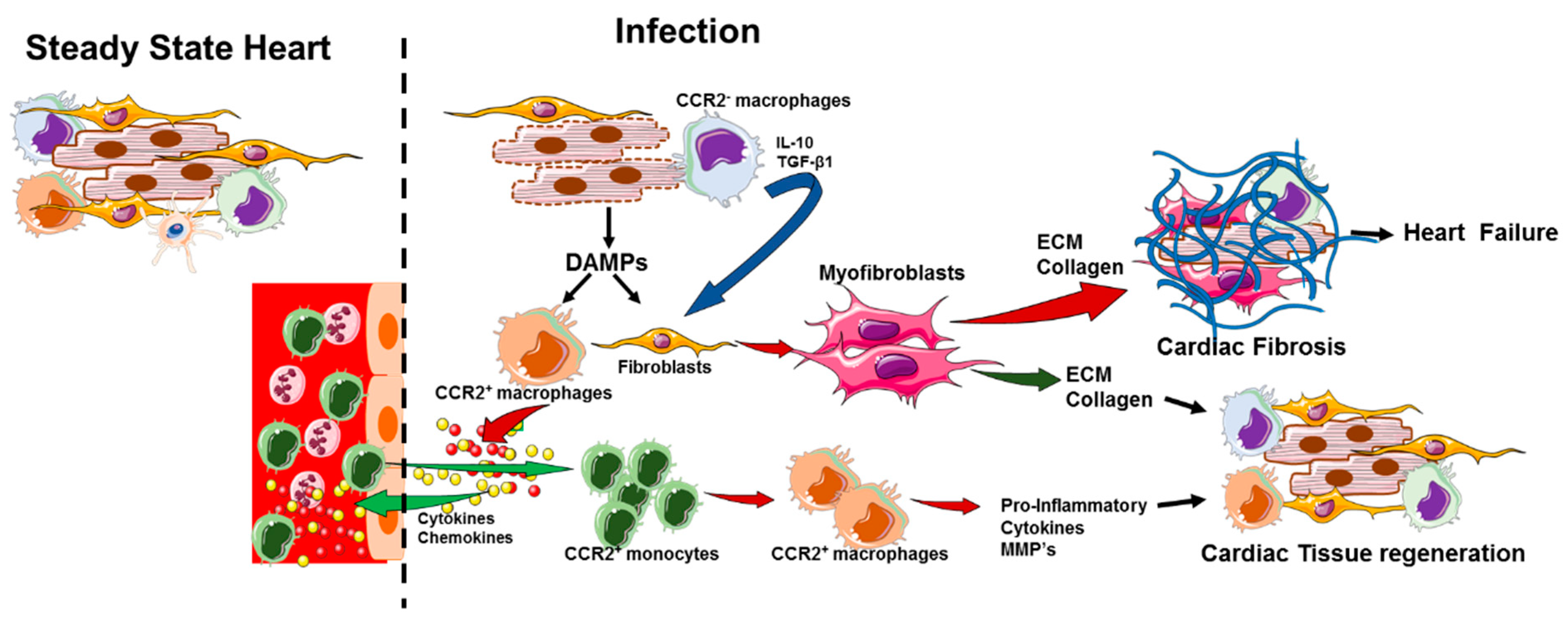
5. Activation of Cardiac Macrophages in Diseased Heart
6. Role of Cardiac-Resident Macrophages in Tissue Repair during Cardiac Injury
7. Role of Macrophages in Cardiac Remodeling in Hypertension and Diabetic Cardiomyopathy
8. Role of Macrophages on Mitochondrial Homeostasis and Collagen Secretion
9. Cardiac-Resident Macrophages and Cardiac Fibrosis
10. Future Direction of Cardiac Macrophages
11. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations Used in This Article
| HF | Heart failure |
| CVD | Cardiovascular diseases |
| CM | Cardiac macrophages |
| MI | Myocardial Infarction |
| DAMPs | Danger-Associated Molecular Patterns |
| PAMPs | Pathogen-Associated Molecular Patterns |
| Treg | T regulatory cell |
| cDC | Cardiac Dendritic cell |
| Nabs | Natural antibodies |
| CAR | Coxsackievirus and Adenovirus Receptor |
| DAF | Decay Accelerating Factor |
| MMP | Matrix metalloproteinase |
| HMGB1 | High mobility group box-1 |
| MERTK | Myeloid-epithelial-reproductive tyrosine kinase |
| EAT | epicardial adipose tissue |
References
- Gordon, S.; Martinez-Pomares, L. Physiological roles of macrophages. Pflugers Arch. 2017, 469, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Sam, F.; Nahrendorf, M. Monocyte and macrophage contributions to cardiac remodeling. J. Mol. Cell. Cardiol. 2016, 93, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Ramos, G.C.; van den Berg, A.; Nunes-Silva, V.; Weirather, J.; Peters, L.; Burkard, M.; Friedrich, M.; Pinnecker, J.; Abesser, M.; Heinze, K.G.; et al. Myocardial aging as a T-cell-mediated phenomenon. Proc. Natl. Acad. Sci. USA 2017, 114, E2420–E2429. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guerin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef]
- Weirather, J.; Hofmann, U.D.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef]
- Saxena, A.; Dobaczewski, M.; Rai, V.; Haque, Z.; Chen, W.; Li, N.; Frangogiannis, N.G. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1233–H1242. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Casares, X.; Hosseinzadeh, S.; Barbu, I.; Dick, S.A.; Macklin, J.A.; Wang, Y.; Momen, A.; Kantores, C.; Aronoff, L.; Farno, M.; et al. A CD103(+) Conventional Dendritic Cell Surveillance System Prevents Development of Overt Heart Failure during Subclinical Viral Myocarditis. Immunity 2017, 47, 974–989. [Google Scholar] [CrossRef] [PubMed]
- Bracamonte-Baran, W.; Chen, G.; Hou, X.; Talor, M.V.; Choi, H.S.; Davogustto, G.; Taegtmeyer, H.; Sung, J.; Hackam, D.J.; Nauen, D.; et al. Non-cytotoxic Cardiac Innate Lymphoid Cells Are a Resident and Quiescent Type 2-Commited Population. Front. Immunol. 2019, 10, 634. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.J.; Khan, M.; Mohsin, S. Healing the Broken Heart; The Immunomodulatory Effects of Stem Cell Therapy. Front. Immunol. 2020, 11, 639. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Maekawa, Y.; Anzai, T.; Yoshikawa, T.; Asakura, Y.; Takahashi, T.; Ishikawa, S.; Mitamura, H.; Ogawa, S. Prognostic significance of peripheral monocytosis after reperfused acute myocardial infarction:a possible role for left ventricular remodeling. J. Am. Coll. Cardiol. 2002, 39, 241–246. [Google Scholar] [CrossRef]
- Glezeva, N.; Voon, V.; Watson, C.; Horgan, S.; McDonald, K.; Ledwidge, M.; Baugh, J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: Evidence of M2 macrophage activation in disease pathogenesis. J. Card. Fail. 2015, 21, 167–177. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Gerhardt, L.M.; Tan, T.C.; Winter, C.; Holderried, T.A.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Dewald, O.; Xia, Y.; Ren, G.; Haudek, S.; Leucker, T.; Kraemer, D.; Taffet, G.; Rollins, B.J.; Entman, M.L. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation 2007, 115, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.N.; Fabre, J.W. Demonstration and characterization of Ia-positive dendritic cells in the interstitial connective tissues of rat heart and other tissues, but not brain. J. Exp. Med. 1981, 154, 347–361. [Google Scholar] [CrossRef]
- Van der Borght, K.; Lambrecht, B.N. Heart macrophages and dendritic cells in sickness and in health: A tale of a complicated marriage. Cell. Immunol. 2018, 330, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, A.; Risi, E.; Biganzoli, L. No pain, no gain... What we can learn from a trial reporting negative results. Ann. Oncol. 2017, 28, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Haak, S.; Sisirak, V.; Reizis, B. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 2013, 13, 566–577. [Google Scholar] [CrossRef]
- Van der Borght, K.; Scott, C.L.; Nindl, V.; Bouche, A.; Martens, L.; Sichien, D.; Van Moorleghem, J.; Vanheerswynghels, M.; De Prijck, S.; Saeys, Y.; et al. Myocardial Infarction Primes Autoreactive T Cells through Activation of Dendritic Cells. Cell Rep. 2017, 18, 3005–3017. [Google Scholar] [CrossRef]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Smith, S.C.; Allen, P.M. Expression of myosin-class II major histocompatibility complexes in the normal myocardium occurs before induction of autoimmune myocarditis. Proc. Natl. Acad. Sci. USA 1992, 89, 9131–9135. [Google Scholar] [CrossRef]
- Donermeyer, D.L.; Beisel, K.W.; Allen, P.M.; Smith, S.C. Myocarditis-inducing epitope of myosin binds constitutively and stably to I-Ak on antigen-presenting cells in the heart. J. Exp. Med. 1995, 182, 1291–1300. [Google Scholar] [CrossRef]
- Hofmann, U.; Beyersdorf, N.; Weirather, J.; Podolskaya, A.; Bauersachs, J.; Ertl, G.; Kerkau, T.; Frantz, S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 2012, 125, 1652–1663. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Xia, Y.; Bujak, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am. J. Pathol. 2010, 176, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Ogawa, M.; Suzuki, J.; Hirata, Y.; Nagai, R.; Isobe, M. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J. 2011, 52, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Day, Y.J.; Toufektsian, M.C.; Xu, Y.; Ramos, S.I.; Marshall, M.A.; French, B.A.; Linden, J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 2006, 114, 2056–2064. [Google Scholar] [CrossRef] [PubMed]
- Laroumanie, F.; Douin-Echinard, V.; Pozzo, J.; Lairez, O.; Tortosa, F.; Vinel, C.; Delage, C.; Calise, D.; Dutaur, M.; Parini, A.; et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 2014, 129, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Nevers, T.; Salvador, A.M.; Grodecki-Pena, A.; Knapp, A.; Velazquez, F.; Aronovitz, M.; Kapur, N.K.; Karas, R.H.; Blanton, R.M.; Alcaide, P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ. Heart Fail. 2015, 8, 776–787. [Google Scholar] [CrossRef] [PubMed]
- van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef]
- Sawyer, R.T.; Strausbauch, P.H.; Volkman, A. Resident macrophage proliferation in mice depleted of blood monocytes by strontium-89. Lab. Investig. 1982, 46, 165–170. [Google Scholar]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef]
- Jakubzick, C.; Gautier, E.L.; Gibbings, S.L.; Sojka, D.K.; Schlitzer, A.; Johnson, T.E.; Ivanov, S.; Duan, Q.; Bala, S.; Condon, T.; et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 2013, 39, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [PubMed]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef]
- Miyanishi, M.; Tada, K.; Koike, M.; Uchiyama, Y.; Kitamura, T.; Nagata, S. Identification of Tim4 as a phosphatidylserine receptor. Nature 2007, 450, 435–439. [Google Scholar] [CrossRef]
- Lim, H.Y.; Lim, S.Y.; Tan, C.K.; Thiam, C.H.; Goh, C.C.; Carbajo, D.; Chew, S.H.S.; See, P.; Chakarov, S.; Wang, X.N.; et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity 2018, 49, 326–341.e7. [Google Scholar] [CrossRef]
- Sagar, S.; Liu, P.P.; Cooper, L.T., Jr. Myocarditis. Lancet 2012, 379, 738–747. [Google Scholar] [CrossRef]
- Coura, J.R.; Borges-Pereira, J. Chagas disease: 100 years after its discovery. A systemic review. Acta Trop. 2010, 115, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Kindermann, I.; Barth, C.; Mahfoud, F.; Ukena, C.; Lenski, M.; Yilmaz, A.; Klingel, K.; Kandolf, R.; Sechtem, U.; Cooper, L.T.; et al. Update on myocarditis. J. Am. Coll. Cardiol. 2012, 59, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Savvatis, K.; Schultheiss, H.P.; Tschope, C. Immunomodulation and matrix metalloproteinases in viral myocarditis. J. Mol. Cell. Cardiol. 2010, 48, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.O.; Mann, B.; Gao, G.; Hankins, J.S.; Humann, J.; Giardina, J.; Faverio, P.; Restrepo, M.I.; Halade, G.V.; Mortensen, E.M.; et al. Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog. 2014, 10, e1004383. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.O.; Orihuela, C.J. Visualization of Streptococcus pneumoniae within Cardiac Microlesions and Subsequent Cardiac Remodeling. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gilley, R.P.; Gonzalez-Juarbe, N.; Shenoy, A.T.; Reyes, L.F.; Dube, P.H.; Restrepo, M.I.; Orihuela, C.J. Infiltrated Macrophages Die of Pneumolysin-Mediated Necroptosis following Pneumococcal Myocardial Invasion. Infect. Immun. 2016, 84, 1457–1469. [Google Scholar] [CrossRef]
- Alhamdi, Y.; Neill, D.R.; Abrams, S.T.; Malak, H.A.; Yahya, R.; Barrett-Jolley, R.; Wang, G.; Kadioglu, A.; Toh, C.H. Circulating Pneumolysin Is a Potent Inducer of Cardiac Injury during Pneumococcal Infection. PLoS Pathog. 2015, 11, e1004836. [Google Scholar] [CrossRef]
- Headley, C.A.; Gerberick, A.; Mehta, S.; Wu, Q.; Yu, L.; Fadda, P.; Khan, M.; Ganesan, L.P.; Turner, J.; Rajaram, M.V.S. Nontuberculous mycobacterium M. avium infection predisposes aged mice to cardiac abnormalities and inflammation. Aging Cell 2019, 18, e12926. [Google Scholar] [CrossRef]
- Makara, M.A.; Hoang, K.V.; Ganesan, L.P.; Crouser, E.D.; Gunn, J.S.; Turner, J.; Schlesinger, L.S.; Mohler, P.J.; Rajaram, M.V. Cardiac Electrical and Structural Changes During Bacterial Infection: An Instructive Model to Study Cardiac Dysfunction in Sepsis. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Esfandiarei, M.; McManus, B.M. Molecular biology and pathogenesis of viral myocarditis. Annu. Rev. Pathol. 2008, 3, 127–155. [Google Scholar] [CrossRef] [PubMed]
- Mena, I.; Perry, C.M.; Harkins, S.; Rodriguez, F.; Gebhard, J.; Whitton, J.L. The role of B lymphocytes in coxsackievirus B3 infection. Am. J. Pathol. 1999, 155, 1205–1215. [Google Scholar] [CrossRef]
- Cheung, C.; Luo, H.; Yanagawa, B.; Leong, H.S.; Samarasekera, D.; Lai, J.C.; Suarez, A.; Zhang, J.; McManus, B.M. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in coxsackievirus-induced myocarditis. Cardiovasc. Pathol. 2006, 15, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Spinale, F.G.; Coker, M.L.; Heung, L.J.; Bond, B.R.; Gunasinghe, H.R.; Etoh, T.; Goldberg, A.T.; Zellner, J.L.; Crumbley, A.J. A matrix metalloproteinase induction/activation system exists in the human left ventricular myocardium and is upregulated in heart failure. Circulation 2000, 102, 1944–1949. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. Jama 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Ranard, L.S.; Fried, J.A.; Abdalla, M.; Anstey, D.E.; Givens, R.C.; Kumaraiah, D.; Kodali, S.K.; Takeda, K.; Karmpaliotis, D.; Rabbani, L.E.; et al. Approach to Acute Cardiovascular Complications in COVID-19 Infection. Circ. Heart Fail. 2020, 13, e007220. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802–810. [Google Scholar] [CrossRef]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef]
- Long, B.; Brady, W.J.; Koyfman, A.; Gottlieb, M. Cardiovascular complications in COVID-19. Am. J. Emerg. Med. 2020, 38, 1504–1507. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhao, P.; Tang, D.; Zhu, T.; Han, R.; Zhan, C.; Liu, W.; Zeng, H.; Tao, Q.; Xia, L. Cardiac Involvement in Patients Recovered From COVID-2019 Identified Using Magnetic Resonance Imaging. JACC Cardiovasc. Imaging 2020, 13, 2330–2339. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.H.; Liu, Y.X.; Yuan, J.; Wang, F.X.; Wu, W.B.; Li, J.X.; Wang, L.F.; Gao, H.; Wang, Y.; Dong, C.F.; et al. First case of COVID-19 complicated with fulminant myocarditis: A case report and insights. Infection 2020, 48, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, H.; Li, M.; Zhang, L.; Xie, M. The prevalence, risk factors and outcome of cardiac dysfunction in hospitalized patients with COVID-19. Intensive Care Med. 2020, 46, 2096–2098. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Hou, K.; Xu, R.; Li, Z.; Fu, H.; Wen, L.; Xie, L.; Liu, H.; Selvanayagam, J.B.; Zhang, N.; et al. Clinical Characteristics and Risk Factors of Cardiac Involvement in COVID-19. J. Am. Heart Assoc. 2020, 9, e016807. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.P.; et al. Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020, 5, 1281–1285. [Google Scholar] [CrossRef]
- Dolhnikoff, M.; Ferreira Ferranti, J.; de Almeida Monteiro, R.A.; Duarte-Neto, A.N.; Soares Gomes-Gouvêa, M.; Viu Degaspare, N.; Figueiredo Delgado, A.; Montanari Fiorita, C.; Nunes Leal, G.; Rodrigues, R.M.; et al. SARS-CoV-2 in cardiac tissue of a child with COVID-19-related multisystem inflammatory syndrome. Lancet Child Adolesc. Health 2020, 4, 790–794. [Google Scholar] [CrossRef]
- Moriyama, N.; Lehtola, H.; Miyashita, H.; Piuhola, J.; Niemelä, M.; Laine, M. Hemodynamic comparison of transcatheter aortic valve replacement with the SAPIEN 3 Ultra versus SAPIEN 3: The HomoSAPIEN registry. Catheter. Cardiovasc. Interv. 2020. [Google Scholar] [CrossRef]
- Sharma, A.; Garcia, G.; Arumugaswami, V.; Svendsen, C.N. Human iPSC-Derived Cardiomyocytes are Susceptible to SARS-CoV-2 Infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pérez-Bermejo, J.A.; Kang, S.; Rockwood, S.J.; Simoneau, C.R.; Joy, D.A.; Ramadoss, G.N.; Silva, A.C.; Flanigan, W.R.; Li, H.; Nakamura, K.; et al. SARS-CoV-2 infection of human iPSC-derived cardiac cells predicts novel cytopathic features in hearts of COVID-19 patients. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e520. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.V. Resident Macrophages: Near and Dear to Your Heart. Cell 2017, 169, 376–377. [Google Scholar] [CrossRef]
- Esper, L.; Talvani, A.; Pimentel, P.; Teixeira, M.M.; Machado, F.S. Molecular mechanisms of myocarditis caused by Trypanosoma cruzi. Curr. Opin Infect. Dis. 2015, 28, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Hidron, A.; Vogenthaler, N.; Santos-Preciado, J.I.; Rodriguez-Morales, A.J.; Franco-Paredes, C.; Rassi, A., Jr. Cardiac involvement with parasitic infections. Clin. Microbiol. Rev. 2010, 23, 324–349. [Google Scholar] [CrossRef] [PubMed]
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Andrade, Z.A. Immunopathology of Chagas disease. Mem. Inst. Oswaldo Cruz 1999, 94 (Suppl. 1), 71–80. [Google Scholar] [CrossRef]
- Dias, E.; Laranja, F.S.; Miranda, A.; Nobrega, G. Chagas’ disease; a clinical, epidemiologic, and pathologic study. Circulation 1956, 14, 1035–1060. [Google Scholar] [CrossRef]
- Viotti, R.J.; Vigliano, C.; Laucella, S.; Lococo, B.; Petti, M.; Bertocchi, G.; Ruiz Vera, B.; Armenti, H. Value of echocardiography for diagnosis and prognosis of chronic Chagas disease cardiomyopathy without heart failure. Heart 2004, 90, 655–660. [Google Scholar] [CrossRef]
- Acquatella, H. Echocardiography in Chagas heart disease. Circulation 2007, 115, 1124–1131. [Google Scholar] [CrossRef]
- Salvador, F.; Trevino, B.; Sulleiro, E.; Pou, D.; Sanchez-Montalva, A.; Cabezos, J.; Soriano, A.; Serre, N.; Gomez, I.P.J.; Pahissa, A.; et al. Trypanosoma cruzi infection in a non-endemic country: Epidemiological and clinical profile. Clin. Microbiol. Infect. 2014, 20, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.M.; Rahimtoola, S.H. Chagas’ heart disease. Curr. Probl. Cardiol. 1995, 20, 825–924. [Google Scholar] [PubMed]
- Esper, L.; Utsch, L.; Soriani, F.M.; Brant, F.; Esteves Arantes, R.M.; Campos, C.F.; Pinho, V.; Souza, D.G.; Teixeira, M.M.; Tanowitz, H.B.; et al. Regulatory effects of IL-18 on cytokine profiles and development of myocarditis during Trypanosoma cruzi infection. Microbes Infect. 2014, 16, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsohn, I.A.; Coffman, R.L. Trypanosoma cruzi: IL-10, TNF, IFN-gamma, and IL-12 regulate innate and acquired immunity to infection. Exp. Parasitol. 1996, 84, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Martins, G.A.; Vieira, L.Q.; Cunha, F.Q.; Silva, J.S. Gamma interferon modulates CD95 (Fas) and CD95 ligand (Fas-L) expression and nitric oxide-induced apoptosis during the acute phase of Trypanosoma cruzi infection: A possible role in immune response control. Infect. Immun. 1999, 67, 3864–3871. [Google Scholar] [CrossRef]
- Talvani, A.; Ribeiro, C.S.; Aliberti, J.C.; Michailowsky, V.; Santos, P.V.; Murta, S.M.; Romanha, A.J.; Almeida, I.C.; Farber, J.; Lannes-Vieira, J.; et al. Kinetics of cytokine gene expression in experimental chagasic cardiomyopathy: Tissue parasitism and endogenous IFN-gamma as important determinants of chemokine mRNA expression during infection with Trypanosoma cruzi. Microbes Infect. 2000, 2, 851–866. [Google Scholar] [CrossRef]
- Machado, F.S.; Koyama, N.S.; Carregaro, V.; Ferreira, B.R.; Milanezi, C.M.; Teixeira, M.M.; Rossi, M.A.; Silva, J.S. CCR5 plays a critical role in the development of myocarditis and host protection in mice infected with Trypanosoma cruzi. J. Infect. Dis. 2005, 191, 627–636. [Google Scholar] [CrossRef]
- Machado, F.S.; Martins, G.A.; Aliberti, J.C.; Mestriner, F.L.; Cunha, F.Q.; Silva, J.S. Trypanosoma cruzi-infected cardiomyocytes produce chemokines and cytokines that trigger potent nitric oxide-dependent trypanocidal activity. Circulation 2000, 102, 3003–3008. [Google Scholar] [CrossRef]
- Wasi, F.; Shuter, J. Primary bacterial infection of the myocardium. Front. Biosci. 2003, 8, s228–s231. [Google Scholar] [CrossRef]
- Cheng, A.G.; Kim, H.K.; Burts, M.L.; Krausz, T.; Schneewind, O.; Missiakas, D.M. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 2009, 23, 3393–3404. [Google Scholar] [CrossRef]
- Martínez-Marcos, F.J.; Lomas-Cabezas, J.M.; Hidalgo-Tenorio, C.; de la Torre-Lima, J.; Plata-Ciézar, A.; Reguera-Iglesias, J.M.; Ruiz-Morales, J.; Márquez-Solero, M.; Gálvez-Acebal, J.; de Alarcón-González, A. [Enterococcal endocarditis: A multicenter study of 76 cases]. Enferm. Infecc. Microbiol. Clin. 2009, 27, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Sy, R.W.; Kritharides, L. Health care exposure and age in infective endocarditis: Results of a contemporary population-based profile of 1536 patients in Australia. Eur. Heart J. 2010, 31, 1890–1897. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.R.; Olaison, L.; Anderson, D.J.; Hoen, B.; Miro, J.M.; Eykyn, S.; Abrutyn, E.; Fowler, V.G., Jr.; Habib, G.; Selton-Suty, C.; et al. Enterococcal endocarditis: 107 cases from the international collaboration on endocarditis merged database. Am. J. Med. 2005, 118, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Moreillon, P.; Que, Y.A. Infective endocarditis. Lancet 2004, 363, 139–149. [Google Scholar] [CrossRef]
- Rice, L.B.; Calderwood, S.B.; Eliopoulos, G.M.; Farber, B.F.; Karchmer, A.W. Enterococcal endocarditis: A comparison of prosthetic and native valve disease. Rev. Infect. Dis. 1991, 13, 1–7. [Google Scholar] [CrossRef]
- Di Salvo, G.; Thuny, F.; Rosenberg, V.; Pergola, V.; Belliard, O.; Derumeaux, G.; Cohen, A.; Iarussi, D.; Giorgi, R.; Casalta, J.P.; et al. Endocarditis in the elderly: Clinical, echocardiographic, and prognostic features. Eur. Heart J. 2003, 24, 1576–1583. [Google Scholar] [CrossRef]
- Braun, J.S.; Sublett, J.E.; Freyer, D.; Mitchell, T.J.; Cleveland, J.L.; Tuomanen, E.I.; Weber, J.R. Pneumococcal pneumolysin and H(2)O(2) mediate brain cell apoptosis during meningitis. J. Clin. Investig. 2002, 109, 19–27. [Google Scholar] [CrossRef]
- Collier, R.J. Diphtheria toxin: Mode of action and structure. Bacteriol. Rev. 1975, 39, 54–85. [Google Scholar] [CrossRef]
- Hadfield, T.L.; McEvoy, P.; Polotsky, Y.; Tzinserling, V.A.; Yakovlev, A.A. The pathology of diphtheria. J. Infect. Dis. 2000, 181 (Suppl. 1), S116–S120. [Google Scholar] [CrossRef]
- Lakkireddy, D.R.; Kondur, A.K.; Chediak, E.J.; Nair, C.K.; Khan, I.A. Cardiac troponin I release in non-ischemic reversible myocardial injury from acute diphtheric myocarditis. Int. J. Cardiol. 2005, 98, 351–354. [Google Scholar] [CrossRef]
- Cahill, T.J.; Prendergast, B.D. Infective endocarditis. Lancet 2016, 387, 882–893. [Google Scholar] [CrossRef]
- Doctor, N.S.; Shah, A.B.; Coplan, N.; Kronzon, I. Acute Pericarditis. Prog. Cardiovasc. Dis. 2017, 59, 349–359. [Google Scholar] [CrossRef]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef]
- Tan, Y.; Li, X.; Prabhu, S.D.; Brittian, K.R.; Chen, Q.; Yin, X.; McClain, C.J.; Zhou, Z.; Cai, L. Angiotensin II plays a critical role in alcohol-induced cardiac nitrative damage, cell death, remodeling, and cardiomyopathy in a protein kinase C/nicotinamide adenine dinucleotide phosphate oxidase-dependent manner. J. Am. Coll. Cardiol. 2012, 59, 1477–1486. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Zhang, X.; Mosser, D.M. Macrophage activation by endogenous danger signals. J. Pathol. 2008, 214, 161–178. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Plüddemann, A.; Gordon, S. Macrophage pattern recognition receptors in immunity, homeostasis and self tolerance. Adv. Exp. Med. Biol. 2009, 653, 1–14. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef]
- Mann, D.L.; Topkara, V.K.; Evans, S.; Barger, P.M. Innate immunity in the adult mammalian heart: For whom the cell tolls. Trans. Am. Clin. Clim. Assoc. 2010, 121, 34–50; discussion 50–51. [Google Scholar]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef]
- Nishimura, M.; Naito, S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol. Pharm. Bull. 2005, 28, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Kelly, R.A.; Bourcier, T. Role of TLR-2 in the activation of nuclear factor kappaB by oxidative stress in cardiac myocytes. J. Biol. Chem 2001, 276, 5197–5203. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Kobzik, L.; Kim, Y.D.; Fukazawa, R.; Medzhitov, R.; Lee, R.T.; Kelly, R.A. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J. Clin. Investig. 1999, 104, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, M.; Imanishi, T.; Ozaki, Y.; Satogami, K.; Masuno, T.; Wada, T.; Nakatani, Y.; Ishibashi, K.; Komukai, K.; Tanimoto, T.; et al. Differential expression of Toll-like receptor 4 and human monocyte subsets in acute myocardial infarction. Atherosclerosis 2012, 221, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; Smeets, M.B.; O’Neill, L.A.; Keogh, B.; McGuirk, P.; Timmers, L.; Tersteeg, C.; Hoefer, I.E.; Doevendans, P.A.; Pasterkamp, G.; et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation 2010, 121, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Neu, N.; Rose, N.R.; Beisel, K.W.; Herskowitz, A.; Gurri-Glass, G.; Craig, S.W. Cardiac myosin induces myocarditis in genetically predisposed mice. J. Immunol. 1987, 139, 3630–3636. [Google Scholar]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Haubner, B.J.; Adamowicz-Brice, M.; Khadayate, S.; Tiefenthaler, V.; Metzler, B.; Aitman, T.; Penninger, J.M. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging (Albany NY) 2012, 4, 966–977. [Google Scholar] [CrossRef]
- Dick, S.A.; Zaman, R.; Epelman, S. Using High-Dimensional Approaches to Probe Monocytes and Macrophages in Cardiovascular Disease. Front. Immunol. 2019, 10, 2146. [Google Scholar] [CrossRef] [PubMed]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.M.; Wong, A.L.; Bezuhly, M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair 2015, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- King, K.R.; Aguirre, A.D.; Ye, Y.X.; Sun, Y.; Roh, J.D.; Ng, R.P., Jr.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef]
- Anzai, A.; Choi, J.L.; He, S.; Fenn, A.M.; Nairz, M.; Rattik, S.; McAlpine, C.S.; Mindur, J.E.; Chan, C.T.; Iwamoto, Y.; et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J. Exp. Med. 2017, 214, 3293–3310. [Google Scholar] [CrossRef]
- Mitchell, A.J.; Roediger, B.; Weninger, W. Monocyte homeostasis and the plasticity of inflammatory monocytes. Cell. Immunol. 2014, 291, 22–31. [Google Scholar] [CrossRef]
- Wan, E.; Yeap, X.Y.; Dehn, S.; Terry, R.; Novak, M.; Zhang, S.; Iwata, S.; Han, X.; Homma, S.; Drosatos, K.; et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 2013, 113, 1004–1012. [Google Scholar] [CrossRef]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ. Res. 2016, 119, 414–417. [Google Scholar] [CrossRef] [PubMed]
- DeBerge, M.; Shah, S.J.; Wilsbacher, L.; Thorp, E.B. Macrophages in Heart Failure with Reduced versus Preserved Ejection Fraction. Trends Mol. Med. 2019, 25, 328–340. [Google Scholar] [CrossRef]
- Mouton, A.J.; Li, X.; Hall, M.E.; Hall, J.E. Obesity, Hypertension, and Cardiac Dysfunction: Novel Roles of Immunometabolism in Macrophage Activation and Inflammation. Circ. Res. 2020, 126, 789–806. [Google Scholar] [CrossRef]
- Lopatin, Y. Metabolic Therapy in Heart Failure. Card. Fail. Rev. 2015, 1, 112–117. [Google Scholar] [CrossRef]
- Berthiaume, J.M.; Young, M.E.; Chen, X.; McElfresh, T.A.; Yu, X.; Chandler, M.P. Normalizing the metabolic phenotype after myocardial infarction: Impact of subchronic high fat feeding. J. Mol. Cell. Cardiol. 2012, 53, 125–133. [Google Scholar] [CrossRef][Green Version]
- Cavalera, M.; Wang, J.; Frangogiannis, N.G. Obesity, metabolic dysfunction, and cardiac fibrosis: Pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl. Res. 2014, 164, 323–335. [Google Scholar] [CrossRef]
- Dodd, M.S.; Ball, D.R.; Schroeder, M.A.; Le Page, L.M.; Atherton, H.J.; Heather, L.C.; Seymour, A.M.; Ashrafian, H.; Watkins, H.; Clarke, K.; et al. In vivo alterations in cardiac metabolism and function in the spontaneously hypertensive rat heart. Cardiovasc. Res. 2012, 95, 69–76. [Google Scholar] [CrossRef]
- Lauterbach, M.A.; Wunderlich, F.T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflugers Arch. 2017, 469, 385–396. [Google Scholar] [CrossRef]
- Boutens, L.; Hooiveld, G.J.; Dhingra, S.; Cramer, R.A.; Netea, M.G.; Stienstra, R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia 2018, 61, 942–953. [Google Scholar] [CrossRef]
- Groh, L.; Keating, S.T.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. Monocyte and macrophage immunometabolism in atherosclerosis. Semin ImmunoPathol. 2018, 40, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, G.I.; Langley, K.G.; Berglund, N.A.; Kammoun, H.L.; Reibe, S.; Estevez, E.; Weir, J.; Mellett, N.A.; Pernes, G.; Conway, J.R.W.; et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018, 27, 1096–1110.e1095. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Zhang, L.F.; Ding, P.S.; Liu, Y.J.; Wu, X.; Zhou, J.N. Microglial activation mediates host neuronal survival induced by neural stem cells. J. Cell. Mol. Med. 2014, 18, 1300–1312. [Google Scholar] [CrossRef]
- Wicks, K.; Torbica, T.; Mace, K.A. Myeloid cell dysfunction and the pathogenesis of the diabetic chronic wound. Semin Immunol. 2014, 26, 341–353. [Google Scholar] [CrossRef]
- Kanter, J.E.; Kramer, F.; Barnhart, S.; Averill, M.M.; Vivekanandan-Giri, A.; Vickery, T.; Li, L.O.; Becker, L.; Yuan, W.; Chait, A.; et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc. Natl. Acad. Sci. USA 2012, 109, E715–E724. [Google Scholar] [CrossRef]
- Pavlou, S.; Lindsay, J.; Ingram, R.; Xu, H.; Chen, M. Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol. 2018, 19, 24. [Google Scholar] [CrossRef]
- Jin, X.; Yao, T.; Zhou, Z.; Zhu, J.; Zhang, S.; Hu, W.; Shen, C. Advanced Glycation End Products Enhance Macrophages Polarization into M1 Phenotype through Activating RAGE/NF-κB Pathway. BioMed Res. Int. 2015, 2015, 732450. [Google Scholar] [CrossRef]
- Mishra, P.K.; Ying, W.; Nandi, S.S.; Bandyopadhyay, G.K.; Patel, K.K.; Mahata, S.K. Diabetic Cardiomyopathy: An Immunometabolic Perspective. Front. Endocrinol. 2017, 8, 72. [Google Scholar] [CrossRef]
- Watanabe, R.; Hilhorst, M.; Zhang, H.; Zeisbrich, M.; Berry, G.J.; Wallis, B.B.; Harrison, D.G.; Giacomini, J.C.; Goronzy, J.J.; Weyand, C.M. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, Fat Mass and Immune System: Role for Leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef] [PubMed]
- Kain, V.; Ingle, K.A.; Kachman, M.; Baum, H.; Shanmugam, G.; Rajasekaran, N.S.; Young, M.E.; Halade, G.V. Excess ω-6 fatty acids influx in aging drives metabolic dysregulation, electrocardiographic alterations, and low-grade chronic inflammation. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H160–h169. [Google Scholar] [CrossRef]
- Namgaladze, D.; Brüne, B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim. Biophys. Acta 2016, 1861, 1796–1807. [Google Scholar] [CrossRef]
- Ben-Mordechai, T.; Holbova, R.; Landa-Rouben, N.; Harel-Adar, T.; Feinberg, M.S.; Abd Elrahman, I.; Blum, G.; Epstein, F.H.; Silman, Z.; Cohen, S.; et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J. Am. Coll. Cardiol. 2013, 62, 1890–1901. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef]
- Liao, X.; Shen, Y.; Zhang, R.; Sugi, K.; Vasudevan, N.T.; Alaiti, M.A.; Sweet, D.R.; Zhou, L.; Qing, Y.; Gerson, S.L.; et al. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2018, 115, E4661–e4669. [Google Scholar] [CrossRef]
- Horckmans, M.; Bianchini, M.; Santovito, D.; Megens, R.T.A.; Springael, J.Y.; Negri, I.; Vacca, M.; Di Eusanio, M.; Moschetta, A.; Weber, C.; et al. Pericardial Adipose Tissue Regulates Granulopoiesis, Fibrosis, and Cardiac Function After Myocardial Infarction. Circulation 2018, 137, 948–960. [Google Scholar] [CrossRef]
- Aldiss, P.; Davies, G.; Woods, R.; Budge, H.; Sacks, H.S.; Symonds, M.E. ‘Browning’ the cardiac and peri-vascular adipose tissues to modulate cardiovascular risk. Int. J. Cardiol. 2017, 228, 265–274. [Google Scholar] [CrossRef]
- Vianello, E.; Marrocco-Trischitta Massimiliano, M.; Dozio, E.; Bandera, F.; Tacchini, L.; Canciani, E.; Dellavia, C.; Schmitz, G.; Lorenzo, M.; Corsi Romanelli Massimiliano, M. Correlational study on altered epicardial adipose tissue as a stratification risk factor for valve disease progression through IL-13 signaling. J. Mol. Cell. Cardiol. 2019, 132, 210–218. [Google Scholar] [CrossRef]
- Guzzardi, M.A.; Iozzo, P. Brain functional imaging in obese and diabetic patients. Acta Diabetol. 2019, 56, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Gruzdeva, O.; Uchasova, E.; Dyleva, Y.; Borodkina, D.; Akbasheva, O.; Belik, E.; Karetnikova, V.; Brel, N.; Kokov, A.; Kashtalap, V.; et al. Relationships between epicardial adipose tissue thickness and adipo-fibrokine indicator profiles post-myocardial infarction. Cardiovasc. Diabetol. 2018, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; DeLeon-Pennell, K.Y.; Rivera Gonzalez, O.J.; Flynn, E.R.; Freeman, T.C.; Saucerman, J.J.; Garrett, M.R.; Ma, Y.; Harmancey, R.; Lindsey, M.L. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 2018, 113, 26. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Savoia, C.; Sada, L.; Zezza, L.; Pucci, L.; Lauri, F.M.; Befani, A.; Alonzo, A.; Volpe, M. Vascular inflammation and endothelial dysfunction in experimental hypertension. Int. J. Hypertens. 2011, 2011, 281240. [Google Scholar] [CrossRef]
- Andreadou, I.; Cabrera-Fuentes, H.A.; Devaux, Y.; Frangogiannis, N.G.; Frantz, S.; Guzik, T.; Liehn, E.A.; Gomes, C.P.C.; Schulz, R.; Hausenloy, D.J. Immune cells as targets for cardioprotection: New players and novel therapeutic opportunities. Cardiovasc. Res. 2019, 115, 1117–1130. [Google Scholar] [CrossRef]
- Chen, B.; Frangogiannis, N.G. The Role of Macrophages in Nonischemic Heart Failure. JACC Basic Transl. Sci. 2018, 3, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; Ma, Y.; Rivera Gonzalez, O.J.; Daseke, M.J., 2nd; Flynn, E.R.; Freeman, T.C.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 2019, 114, 6. [Google Scholar] [CrossRef]
- Lewis, A.J.M.; Miller, J.J.; Lau, A.Z.; Curtis, M.K.; Rider, O.J.; Choudhury, R.P.; Neubauer, S.; Cunningham, C.H.; Carr, C.A.; Tyler, D.J. Noninvasive Immunometabolic Cardiac Inflammation Imaging Using Hyperpolarized Magnetic Resonance. Circ. Res. 2018, 122, 1084–1093. [Google Scholar] [CrossRef]
- Zhang, S.; Weinberg, S.; DeBerge, M.; Gainullina, A.; Schipma, M.; Kinchen, J.M.; Ben-Sahra, I.; Gius, D.R.; Yvan-Charvet, L.; Chandel, N.S.; et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019, 29, 443–456.e445. [Google Scholar] [CrossRef]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Muñoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Phagocytosis: An Immunobiologic Process. Immunity 2016, 44, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 2019, 363. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K. Monocyte and macrophage heterogeneity in the heart. Circ. Res. 2013, 112, 1624–1633. [Google Scholar] [CrossRef]
- Tong, M.; Sadoshima, J. Mitochondrial autophagy in cardiomyopathy. Curr. Opin. Genet. Dev. 2016, 38, 8–15. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; Díaz-García, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e123. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef]
- Aurora, A.B.; Porrello, E.R.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef]
- Godwin, J.W.; Pinto, A.R.; Rosenthal, N.A. Macrophages are required for adult salamander limb regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 9415–9420. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.L.; Marín-Juez, R.; Moura, P.L.; Kuenne, C.; Lai, J.K.H.; Tsedeke, A.T.; Guenther, S.; Looso, M.; Stainier, D.Y. Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Simões, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020, 11, 600. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The immune system and cardiac repair. Pharmacol. Res. 2008, 58, 88–111. [Google Scholar] [CrossRef]
- Huynh, M.L.; Fadok, V.A.; Henson, P.M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Investig. 2002, 109, 41–50. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog. Mol. Biol. Transl. Sci. 2017, 147, 75–100. [Google Scholar] [CrossRef]
- Westermann, D.; Savvatis, K.; Lindner, D.; Zietsch, C.; Becher, P.M.; Hammer, E.; Heimesaat, M.M.; Bereswill, S.; Volker, U.; Escher, F.; et al. Reduced degradation of the chemokine MCP-3 by matrix metalloproteinase-2 exacerbates myocardial inflammation in experimental viral cardiomyopathy. Circulation 2011, 124, 2082–2093. [Google Scholar] [CrossRef]
- Rifkin, D.B.; Mazzieri, R.; Munger, J.S.; Noguera, I.; Sung, J. Proteolytic control of growth factor availability. APMIS 1999, 107, 80–85. [Google Scholar] [CrossRef]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Alger, R.; Berkowitz, L.L.; Bergeron, B.; Buskett, D. Computer networking in an ambulatory health care setting. J. Med. Pr. Manag. 1999, 14, 303–307. [Google Scholar]
- Serini, G.; Bochaton-Piallat, M.L.; Ropraz, P.; Geinoz, A.; Borsi, L.; Zardi, L.; Gabbiani, G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol. 1998, 142, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Willems, I.E.; Havenith, M.G.; De Mey, J.G.; Daemen, M.J. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am. J. Pathol. 1994, 145, 868–875. [Google Scholar]
- Blyszczuk, P.; Muller-Edenborn, B.; Valenta, T.; Osto, E.; Stellato, M.; Behnke, S.; Glatz, K.; Basler, K.; Luscher, T.F.; Distler, O.; et al. Transforming growth factor-beta-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur. Heart J. 2017, 38, 1413–1425. [Google Scholar] [CrossRef]
- Leslie, K.O.; Taatjes, D.J.; Schwarz, J.; vonTurkovich, M.; Low, R.B. Cardiac myofibroblasts express alpha smooth muscle actin during right ventricular pressure overload in the rabbit. Am. J. Pathol. 1991, 139, 207–216. [Google Scholar]
- Law, B.A.; Levick, S.P.; Carver, W.E. Alterations in cardiac structure and function in a murine model of chronic alcohol consumption. Microsc. Microanal. 2012, 18, 453–461. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Chaturvedi, R.R.; Herron, T.; Simmons, R.; Shore, D.; Kumar, P.; Sethia, B.; Chua, F.; Vassiliadis, E.; Kentish, J.C. Passive stiffness of myocardium from congenital heart disease and implications for diastole. Circulation 2010, 121, 979–988. [Google Scholar] [CrossRef]
- Espira, L.; Czubryt, M.P. Emerging concepts in cardiac matrix biology. Can. J. Physiol. Pharmacol. 2009, 87, 996–1008. [Google Scholar] [CrossRef]
- de Bakker, J.M.; van Capelle, F.J.; Janse, M.J.; Tasseron, S.; Vermeulen, J.T.; de Jonge, N.; Lahpor, J.R. Fractionated electrograms in dilated cardiomyopathy: Origin and relation to abnormal conduction. J. Am. Coll. Cardiol. 1996, 27, 1071–1078. [Google Scholar] [CrossRef]
- Spach, M.S.; Boineau, J.P. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: A major mechanism of structural heart disease arrhythmias. Pacing Clin. Electrophysiol. 1997, 20, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Kai, H.; Mori, T.; Tokuda, K.; Takayama, N.; Tahara, N.; Takemiya, K.; Kudo, H.; Sugi, Y.; Fukui, D.; Yasukawa, H.; et al. Pressure overload-induced transient oxidative stress mediates perivascular inflammation and cardiac fibrosis through angiotensin II. Hypertens. Res. 2006, 29, 711–718. [Google Scholar] [CrossRef] [PubMed]
- van den Borne, S.W.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol. 2010, 7, 30–37. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD45 + Cells | Markers | Steady State #/mg | Day 1–3 MI #/mg | 7 Day MI #/mg | References |
|---|---|---|---|---|---|
| Macrophage and DC | CD11b+ F4/80+ MHC II high/lowCD64+ Ly6G− | 567 | 1000 | 2000 | [7,8] |
| Total Monocytes | CD11b+ F4/80− CD64− Ly6G− | 80 | 50,000 | 40,000 | unpublished data, [8] |
| Classical monocytes | CCR2high Ly6Chigh CD11clow CX3CR1low | 25 | 37,500 | 10,000 | unpublished data, [8] |
| Non-Classical monocytes | CCR2high Ly6Clow CD11chighCX3CR1high | 50 | 12,500 | 30,000 | unpublished data, [8] |
| T Cells | CD11b− Ly6G− CD3ε+ | 41 | 210 | 110 | [7,9] |
| T Regs | CD4+ CD25+ Foxp3+ | 60 | 90 | 175 | [10] |
| B Cells | CD11b− Ly6G− B220+ | 160 | 800 | 100 | [9] |
| Neutrophils | CD11b+ Ly6G+ | 18 | 100,000 | 5000 | [7,8,9] |
| Cardiac Macrophage Populations | Markers | Origin | Transcriptional Analysis (Cluster Defining Genes) | Function |
|---|---|---|---|---|
| Leid et al., and Bajpai et al., [43,45] | ||||
| C-C chemokine receptor type 2 (CCR2)− | CD64+, MERTK+, CX3CR1high | Embryonic | Cx3cr1, Lyve, Emr1, CD207, Ccl12, Igf1, Pdgfc, Hbegf, Cyr61 | Pro-angiogenic and myogenic, coronary development, and remodeling |
| CCR2+ | CD64+, MERTK+, CX3CR1interm | Monocyte | Ly6C, CXCR2, Sel1, Irf5, Nr4a1 | Inflammatory, Type I Interferon response |
| Epelman et al., [6] | ||||
| CCR2− MHCIIhigh | CD64+, MERTK+, CX3CR1high, CD206interm | Embryonic | MHCII genes, Cd74, Slc11a1, March1, Fcgr2b, Fcgr3, Stab1, Ccl12 | Antigen Presentation, Inflammatory |
| CCR2− MHCIIlow | CD64+, MERTK+, CX3CR1interm, CD206high | Embryonic | Fcna, Lyve1, Igf1, Slco2b1, Vsig4, C1qb, Tnk2, Cd164, Snx8, Rab34 | Homeostasis, Clearance of necrotic and apoptotic cells |
| R3 CCR2+ MHCIIhigh | CD64+, MERTK+, CX3CR1high, CD206high | Monocyte | MHCII genes, Cd74, Nlrp3, Nod2, Ptgs2, IL1b, Il18, Mefv, Tnsf14, Rgs1 | Antigen Presentation, Pro-inflammatory, IL-1β secretion |
| Dick et al., [46] Steady-State Populations | ||||
| Timid4 Cluster (Equivalent to CCR2− MHCIIlow) | CCR2−, TIMD4+, LYVE1+, Igf1, MHCIIlow | Embryonic | Timd4, Lyve1, Folr1, Mrc1, Nrp1, Igfbp4, Wwp1, Igf1, Fxyd2, Maf, Gas6 | Homeostasis, regenerative functions (endocytosis, lysosome function) |
| MHCII Cluster (Equivalent to CCR2− MHCIIhigh) | CCR2−, TIMD4−, LYVE1−, MHCIIhigh | Embryonic and Monocyte | MHCII genes, Cd74, Cx3cr1, Axl, Rgs1, Ccl4. Cxcl2, CCl3, Tmsb10, Hexb | Antigen Presentation, Chemokine Response |
| Isg Cluster | CCR2+, TIMD4−, LYVE1−, Irf7, Isg20, Ifit1, MHCIIlow | Monocyte | Irf7, Isg20, Ifit1, C1qa, Stat1, Gbp2, Isg15, Mx1, Xaf1, Ifit2, Ifit3, Oas3, Usp18 | Inflammatory, Type I Interferon Response |
| CCR2 Cluster (Equivalent to CCR2+ MHCIIhigh) | CCR2+, TIMD4−, LYVE1−, MHCIIhigh | Monocyte | MHCII genes, Ccr2, Msrb1, Gstp1, Anxa2, Cox5a, Mif, Lgals3, Rac2, Clec12a | Antigen Presentation, Pro-inflammatory, IL-12 and IFN-γ responses |
| Dick et al. [46] Populations after MI | ||||
| HIf1α Cluster | Hif1α | Monocytic | Il1b, S100a11, Cdk2ap2, Msrb1, Adssl1, Tmsb10, Hif1a, Cyp4f18, Plbd1 | Response to Hypoxia |
| cDC2 Cluster | CD209a | Monocytic | Ifitm3, CD209a Samsn1, Tmsb10, Rasgrp2, Nr4a1, Il17ra, Gngt2, Adgre5, Samhd1 | Conventional DC cluster |
| Cluster 6 | Ifitm3, | Monocytic | Vegfa, Arg1, Slc7a11, F10, Timp1, Ass1, Cxcl3, Fn1, Clec4e | Extracellular Matrix interactions |
| Cluster 8 | Cd72, IL1b | Monocytic | Cd72, F11r, Sh2d1b1, Napsa, Tmem119, Il1b | Osteoclast Differentiation, Chemokine signaling |
| Cluster 9 | Fcrls, Ccl8 | Monocytic | Fcrls, Ccl8 | PI-3K signaling, ECM-receptor interactions |
| Cluster 10 | Clec4d, Lrg1 | Monocytic | Saa3, Lrg1, Prtn3, Arg1, Fn1, Gda, C4b, Clec4d, Thbs1, Wfdc17, Fam20c | Degradative pathways |
| Cluster 11 | Cd63, Cd9 | Monocytic | Gpnmb, Syngr1, Ctsd, Lgals3, Trem2, Ms4a7, Cd63, Fabp5, Clec4d, Cd9 etc. | Lysosome and Glycosaminoglycan degradation, Glutathione metabolism |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2021, 10, 51. https://doi.org/10.3390/cells10010051
Lafuse WP, Wozniak DJ, Rajaram MVS. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells. 2021; 10(1):51. https://doi.org/10.3390/cells10010051
Chicago/Turabian StyleLafuse, William P., Daniel J. Wozniak, and Murugesan V. S. Rajaram. 2021. "Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair" Cells 10, no. 1: 51. https://doi.org/10.3390/cells10010051
APA StyleLafuse, W. P., Wozniak, D. J., & Rajaram, M. V. S. (2021). Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells, 10(1), 51. https://doi.org/10.3390/cells10010051