Tumor Suppressors Having Oncogenic Functions: The Double Agents

Abstract

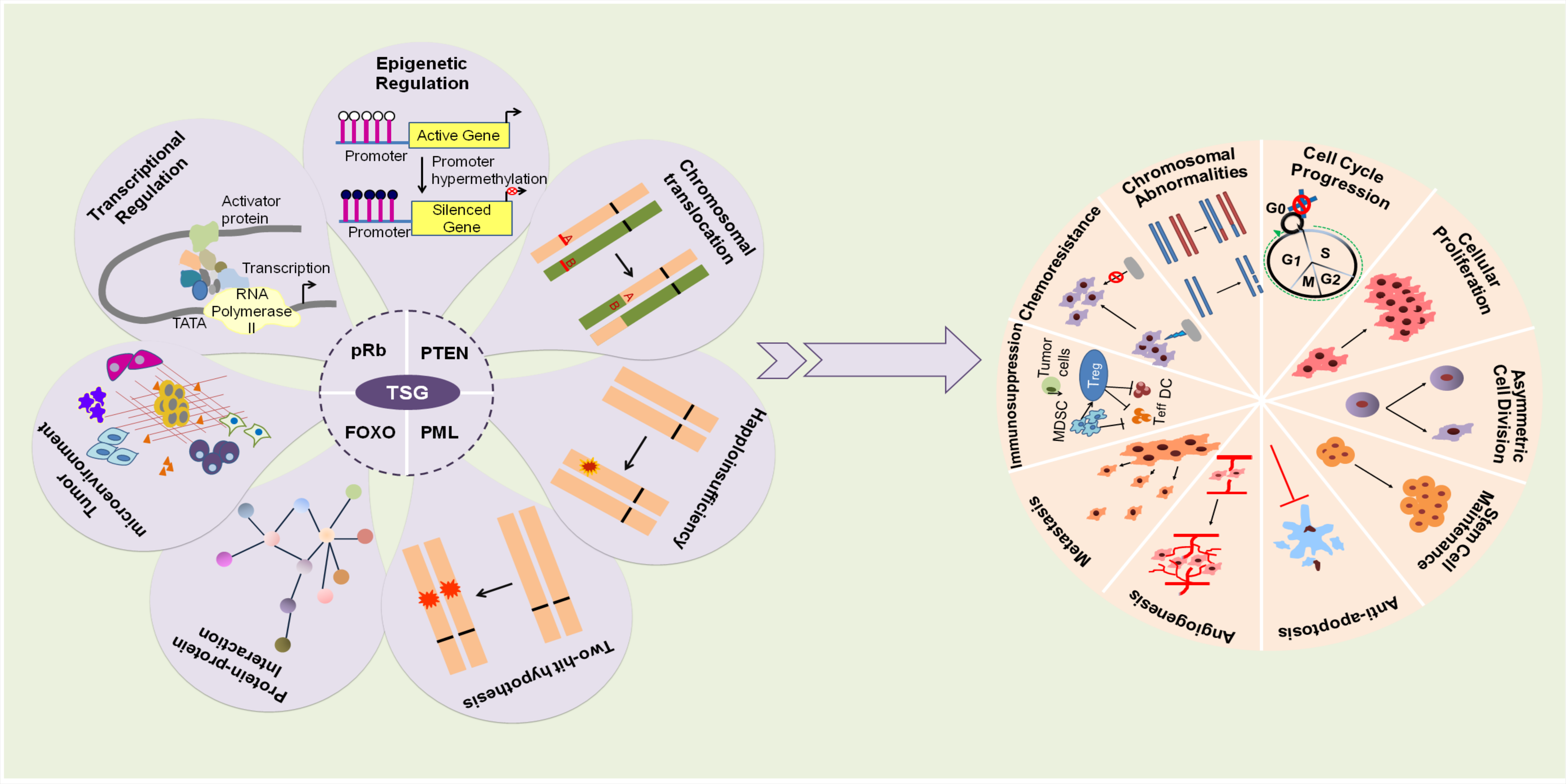
1. Introduction
2. TSG Mutations and Cancer
3. Retinoblastoma (RB1)
3.1. pRb and Angiogenesis
3.2. pRb and Cell Cycle
3.3. Anti-Apoptotic Role of pRb
3.4. pRb and E2F Signaling
3.5. pRb and Ras Families
4. PTEN
4.1. Haploinsufficiency of PTEN
4.2. Epigenetic Control of PTEN
4.3. PTEN and PI3K-Akt/mTOR Pathway
4.4. PTEN and p53 Interplay
4.5. PTEN’s Dual Lipid and Protein Phosphatase Activity on Proliferation
4.6. Immunosuppressive Function of PTEN in Tumor Microenvironment (TME)
4.7. Oncogenic Role of PTEN in Leukemia
5. FOXO Family
5.1. FOXO’s Chromosomal Translocation in Cancer
5.2. Oncogenic Role of Nuclear FOXO
5.3. Foxo3a and p53
5.4. FOXO and Estrogen Receptor Alpha (ERα)
5.5. Interactions with Other Oncogenes
5.6. TME and FOXOs
5.7. Epigenetics and Maintaining Stem Cells Pool
5.8. Involvement in Stress Resistance and Longevity
6. PML
6.1. Oncogenic Role of PML through PML-NB Interactions
6.2. Stem Cell Maintenance
6.3. Epigenetics, Chromatin Association, and Transcriptional Control
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Harris, H.; Miller, O.J.; Klein, G.; Worst, P.; Tachibana, T. Suppression of Malignancy by Cell Fusion. Nature 1969, 223, 363–368. [Google Scholar] [CrossRef]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Haber, D.; Harlow, E. Tumour-suppressor genes: Evolving definitions in the genomic age. Nat. Genet. 1997, 16, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Shi, Q.; Wang, W. Double agents: Genes with both oncogenic and tumor-suppressor functions. Oncogenesis 2018, 7, 25. [Google Scholar] [CrossRef]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef]
- Paige, A.J.W. Redefining tumour suppressor genes: Exceptions to the two-hit hypothesis. Cell. Mol. Life Sci. CMLS 2003, 60, 2147–2163. [Google Scholar] [CrossRef]
- Inoue, K.; Fry, E.A. Haploinsufficient tumor suppressor genes. Adv. Med. Biol. 2017, 118, 83–122. [Google Scholar]
- Izatt, L.; Greenman, J.; Hodgson, S.; Ellis, D.; Watts, S.; Scott, G.; Jacobs, C.; Liebmann, R.; Zvelebil, M.J.; Mathew, C.; et al. Identification of germline missense mutations and rare allelic variants in the ATM gene in early-onset breast cancer. Genes Chromosomes Cancer 1999, 26, 286–294. [Google Scholar] [CrossRef]
- Scott, S.P.; Bendix, R.; Chen, P.; Clark, R.; Dörk, T.; Lavin, M.F. Missense mutations but not allelic variants alter the function of ATM by dominant interference in patients with breast cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 925–930. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Harlow, E. The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2002, 2, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Stirzaker, C.; Millar, D.S.; Paul, C.L.; Warnecke, P.M.; Harrison, J.; Vincent, P.C.; Frommer, M.; Clark, S.J. Extensive DNA methylation spanning the Rb promoter in retinoblastoma tumors. Cancer Res. 1997, 57, 2229–2237. [Google Scholar] [PubMed]
- Ferres-Marco, D.; Gutiérrez-García, I.; Vallejo, D.M.; Bolivar, J.; Gutiérrez-Aviñó, F.J.; Dominguez, M. Epigenetic silencers and Notch collaborate to promote malignant tumours by Rb silencing. Nature 2006, 439, 430–436. [Google Scholar] [CrossRef]
- Sdek, P.; Ying, H.; Chang, D.L.F.; Qiu, W.; Zheng, H.; Touitou, R.; Allday, M.J.; Xiao, Z.-X.J. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol. Cell 2005, 20, 699–708. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ghosh, M.K. HAUSP, a novel deubiquitinase for Rb-MDM2 the critical regulator. FEBS J. 2014, 281, 3061–3078. [Google Scholar] [CrossRef]
- Gope, R.; Gope, M.L. Abundance and state of phosphorylation of the retinoblastoma susceptibility gene product in human colon cancer. Mol. Cell. Biochem. 1992, 110, 123–133. [Google Scholar] [CrossRef]
- Gope, M.L.; Chun, M.; Gope, R. Comparative study of the expression of Rb and p53 genes in human colorectal cancers, colon carcinoma cell lines and synchronized human fibroblasts. Mol. Cell. Biochem. 1991, 107, 55–63. [Google Scholar] [CrossRef]
- Wegiel, B.; Bjartell, A.; Ekberg, J.; Gadaleanu, V.; Brunhoff, C.; Persson, J.L. A role for cyclin A1 in mediating the autocrine expression of vascular endothelial growth factor in prostate cancer. Oncogene 2005, 24, 6385–6393. [Google Scholar] [CrossRef] [PubMed]
- Budde, A.; Schneiderhan-Marra, N.; Petersen, G.; Brüne, B. Retinoblastoma susceptibility gene product pRB activates hypoxia-inducible factor-1 (HIF-1). Oncogene 2005, 24, 1802–1808. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lasorella, A.; Rothschild, G.; Yokota, Y.; Russell, R.G.; Iavarone, A. Id2 Mediates Tumor Initiation, Proliferation, and Angiogenesis in Rb Mutant Mice. Mol. Cell. Biol. 2005, 25, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shepherd, A.T.; Eason, D.D.; Wei, S.; Diaz, J.I.; Djeu, J.Y.; Wu, G.D.; Blanck, G. Retinoblastoma Protein Expression Leads to Reduced Oct-1 DNA Binding Activity and Enhances Interleukin-8 Expression. Cell Growth Differ. 1999, 10, 457–465. [Google Scholar] [PubMed]
- Vidal, A.; Zacharoulis, S.; Guo, W.; Shaffer, D.; Giancotti, F.; Bramley, A.H.; De La Hoz, C.; Jensen, K.K.; Kato, D.; Macdonald, D.D.; et al. p130Rb2 and p27kip1 cooperate to control mobilization of angiogenic progenitors from the bone marrow. Proc. Natl. Acad. Sci. USA 2005, 102, 6890–6895. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.; Helin, K. The E2F transcription factors: Key regulators of cell proliferation. Biochim. Biophys. Acta 2000, 1470, M1–M12. [Google Scholar] [CrossRef]
- Jares, P.; Campo, E.; Pinyol, M.; Bosch, F.; Miquel, R.; Fernandez, P.L.; Sanchez-Beato, M.; Soler, F.; Perez-Losada, A.; Nayach, I.; et al. Expression of retinoblastoma gene product (pRb) in mantle cell lymphomas. Correlation with cyclin D1 (PRAD1/CCND1) mRNA levels and proliferative activity. Am. J. Pathol. 1996, 148, 1591–1600. [Google Scholar]
- Soletti, R.C.; Biasoli, D.; Rodrigues, N.A.L.V.; Delou, J.M.A.; Maciel, R.; Chagas, V.L.; Martins, R.A.P.; Rehen, S.; Borges, H.L. Inhibition of pRB Pathway Differentially Modulates Apoptosis in Esophageal Cancer Cells. Transl. Oncol. 2017, 10, 726–733. [Google Scholar] [CrossRef]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3. [Google Scholar] [CrossRef]
- Buggins, A.G.; Milojkovic, D.; Arno, M.J.; Lea, N.C.; Mufti, G.J.; Thomas, N.S.; Hirst, W.J. Microenvironment produced by acute myeloid leukemia cells prevents T cell activation and proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways. J. Immunol. Baltim Md 1950 2001, 167, 6021–6030. [Google Scholar]
- Ezhevsky, S.A.; Ho, A.; Becker-Hapak, M.; Davis, P.K.; Dowdy, S.F. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol. Cell. Biol. 2001, 21, 4773–4784. [Google Scholar] [CrossRef] [PubMed]
- Chau, B.N.; Wang, J.Y.J. Coordinated regulation of life and death by RB. Nat. Rev. Cancer 2003, 3, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Indovina, P.; Pentimalli, F.; Casini, N.; Vocca, I.; Giordano, A. RB1 dual role in proliferation and apoptosis: Cell fate control and implications for cancer therapy. Oncotarget 2015, 6, 17873–17890. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.; Yan, P.S.; Cechvala, M.; Huang, T.; Farnham, P.J. Identification of novel pRb binding sites using CpG microarrays suggests that E2F recruits pRb to specific genomic sites during S phase. Oncogene 2003, 22, 1445–1460. [Google Scholar] [CrossRef]
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell. Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef]
- Bernards, R. Cancer: Entangled pathways. Nature 2008, 455, 479–480. [Google Scholar] [CrossRef]
- Wu, Z.; Zheng, S.; Li, Z.; Tan, J.; Yu, Q. E2F1 suppresses Wnt/β-catenin activity through transactivation of β-catenin interacting protein ICAT. Oncogene 2011, 30, 3979–3984. [Google Scholar] [CrossRef]
- Xie, W.; Jin, L.; Mei, Y.; Wu, M. E2F1 represses beta-catenin/TCF activity by direct up-regulation of Siah1. J. Cell. Mol. Med. 2009, 13, 1719–1727. [Google Scholar] [CrossRef]
- Jho, E.; Zhang, T.; Domon, C.; Joo, C.-K.; Freund, J.-N.; Costantini, F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 2002, 22, 1172–1183. [Google Scholar] [CrossRef]
- Morris, E.J.; Ji, J.-Y.; Yang, F.; Di Stefano, L.; Herr, A.; Moon, N.-S.; Kwon, E.-J.; Haigis, K.M.; Näär, A.M.; Dyson, N.J. E2F1 represses β-catenin transcription and is antagonized by both pRB and CDK8. Nature 2008, 455, 552–556. [Google Scholar] [CrossRef]
- Clemo, N.; Arhel, N.; Barnes, J.; Baker, J.; Moorghen, M.; Packham, G.; Paraskeva, C.; Williams, A.C. The role of the retinoblastoma protein (Rb) in the nuclear localization of BAG-1: Implications for colorectal tumour cell survival. Biochem. Soc. Trans. 2005, 33, 676–678. [Google Scholar] [CrossRef]
- Collard, T.J.; Urban, B.C.; Patsos, H.A.; Hague, A.; Townsend, P.A.; Paraskeva, C.; Williams, A.C. The retinoblastoma protein (Rb) as an anti-apoptotic factor: Expression of Rb is required for the anti-apoptotic function of BAG-1 protein in colorectal tumour cells. Cell Death Dis. 2012, 3, e408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Y.; Pfeiffer, H.K.; Mellert, H.S.; Stanek, T.J.; Sussman, R.T.; Kumari, A.; Yu, D.; Rigoutsos, I.; Thomas-Tikhonenko, A.; Seidel, H.E.; et al. Inhibition of the Single Downstream Target BAG1 Activates the Latent Apoptotic Potential of MYC. Mol. Cell. Biol. 2011, 31, 5037–5045. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma gene and gene product. Cancer Surv. 1992, 12, 43–57. [Google Scholar] [PubMed]
- Adjei, A.A. Blocking Oncogenic Ras Signaling for Cancer Therapy. JNCI J. Natl. Cancer Inst. 2001, 93, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Ladha, M.H.; McMahon, C.; Ewen, M.E. The retinoblastoma protein is linked to the activation of Ras. Mol. Cell. Biol. 1999, 19, 7724–7732. [Google Scholar] [CrossRef]
- Williams, J.P.; Stewart, T.; Li, B.; Mulloy, R.; Dimova, D.; Classon, M. The Retinoblastoma Protein Is Required for Ras-Induced Oncogenic Transformation. Mol. Cell. Biol. 2006, 26, 1170–1182. [Google Scholar] [CrossRef]
- Walter, D.M.; Yates, T.J.; Ruiz-Torres, M.; Kim-Kiselak, C.; Gudiel, A.A.; Deshpande, C.; Wang, W.Z.; Cicchini, M.; Stokes, K.L.; Tobias, J.W.; et al. RB constrains lineage fidelity and multiple stages of tumour progression and metastasis. Nature 2019, 569, 423–427. [Google Scholar] [CrossRef]
- Lee, E.Y.; Hu, N.; Yuan, S.S.; Cox, L.A.; Bradley, A.; Lee, W.H.; Herrup, K. Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev. 1994, 8, 2008–2021. [Google Scholar] [CrossRef]
- Jiang, Z.; Zacksenhaus, E.; Gallie, B.; Phillips, R.A. The retinoblastoma gene family is differentially expressed during embryogenesis. Oncogene 1997, 14, 1789–1797. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jiang, Z.; Zacksenhaus, E. Activation of retinoblastoma protein in mammary gland leads to ductal growth suppression, precocious differentiation, and adenocarcinoma. J. Cell Biol. 2002, 156, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Das, N.; Sarkar, M.; Chatterjee, U.; Chatterjee, S.; Ghosh, M.K. Exosome-mediated delivery of the intrinsic C-terminus domain of PTEN protects it from proteasomal degradation and ablates tumorigenesis. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.U.; Schriml, L.M.; Dean, M. Mutational spectra of PTEN/MMAC1 gene: A tumor suppressor with lipid phosphatase activity. J. Natl. Cancer Inst. 1999, 91, 1922–1932. [Google Scholar] [CrossRef]
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef]
- Kwon, C.-H.; Zhao, D.; Chen, J.; Alcantara, S.; Li, Y.; Burns, D.K.; Mason, R.P.; Lee, E.Y.-H.P.; Wu, H.; Parada, L.F. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008, 68, 3286–3294. [Google Scholar] [CrossRef]
- Kwabi-Addo, B.; Giri, D.; Schmidt, K.; Podsypanina, K.; Parsons, R.; Greenberg, N.; Ittmann, M. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc. Natl. Acad. Sci. USA 2001, 98, 11563–11568. [Google Scholar] [CrossRef]
- Phin, S.; Moore, M.W.; Cotter, P.D. Genomic Rearrangements of PTEN in Prostate Cancer. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef]
- Alvarez-Nuñez, F.; Bussaglia, E.; Mauricio, D.; Ybarra, J.; Vilar, M.; Lerma, E.; De Leiva, A.; Matias-Guiu, X. PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid. Off. J. Am. Thyroid. Assoc. 2006, 16, 17–23. [Google Scholar] [CrossRef]
- García, J.M.; Silva, J.; Peña, C.; Garcia, V.; Rodríguez, R.; Cruz, M.A.; Cantos, B.; Provencio, M.; España, P.; Bonilla, F. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer 2004, 41, 117–124. [Google Scholar] [CrossRef]
- Wang, L.; Wang, W.-L.; Zhang, Y.; Guo, S.-P.; Zhang, J.; Li, Q.-L. Epigenetic and genetic alterations of PTEN in hepatocellular carcinoma. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2007, 37, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-P.; Gimm, O.; Hampel, H.; Niemann, T.; Walker, M.J.; Eng, C. Epigenetic PTEN Silencing in Malignant Melanomas without PTEN Mutation. Am. J. Pathol. 2000, 157, 1123–1128. [Google Scholar] [CrossRef]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Udaka, K.; Yokoyama, A. Imatinib causes epigenetic alterations of PTEN gene via upregulation of DNA methyltransferases and polycomb group proteins. Blood Cancer J. 2011, 1, e48. [Google Scholar] [CrossRef]
- Montiel-Duarte, C.; Cordeu, L.; Agirre, X.; Román-Gómez, J.; Jiménez-Velasco, A.; José-Eneriz, E.S.; Garate, L.; Andreu, E.J.; Calasanz, M.J.; Heiniger, A.; et al. Resistance to Imatinib Mesylate-induced apoptosis in acute lymphoblastic leukemia is associated with PTEN down-regulation due to promoter hypermethylation. Leuk. Res. 2008, 32, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jeong, H.; Kong, N.; Yang, Y.; Carroll, J.; Luo, H.R.; Silberstein, L.E.; YupoMa; Chai, L. Stem Cell Factor SALL4 Represses the Transcriptions of PTEN and SALL1 through an Epigenetic Repressor Complex. PLoS ONE 2009, 4, e5577. [Google Scholar] [CrossRef]
- Yoshimi, A.; Goyama, S.; Watanabe-Okochi, N.; Yoshiki, Y.; Nannya, Y.; Nitta, E.; Arai, S.; Sato, T.; Shimabe, M.; Nakagawa, M.; et al. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood 2011, 117, 3617–3628. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Fan, C.; Gillis, A.; Feng, X.; Sanatani, M.; Hotte, S.; Kapoor, A.; Tang, D. Co-existence of high levels of the PTEN protein with enhanced Akt activation in renal cell carcinoma. Biochim. Biophys. Acta BBA Mol. Basis Dis 2007, 1772, 1134–1142. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Stemke-Hale, K.; Sahin, A.; Liu, S.; Barrera, J.A.; Burgues, O.; Lluch, A.M.; Chen, H.; Hortobagyi, G.N.; et al. PI3K Pathway Mutations and PTEN Levels in Primary and Metastatic Breast Cancer. Mol. Cancer Ther. 2011, 10, 1093–1101. [Google Scholar] [CrossRef]
- Joshi, S.; Singh, A.R.; Zulcic, M.; Durden, D.L. A macrophage-dominant PI3K isoform controls hypoxia-induced HIF1α and HIF2α stability and tumor growth, angiogenesis, and metastasis. Mol. Cancer Res. MCR 2014, 12, 1520–1531. [Google Scholar] [CrossRef]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B.; et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000, 14, 391–396. [Google Scholar]
- Petrella, B.L.; Brinckerhoff, C.E. PTEN suppression of YY1 induces HIF-2 activity in von-Hippel-Lindau-null renal-cell carcinoma. Cancer Biol. Ther. 2009, 8, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.; Li, J.; Liaw, D.; Hennessy, I.; Oliner, J.; Christians, F.; Parsons, R. PTEN Expression Causes Feedback Upregulation of Insulin Receptor Substrate 2. Mol. Cell. Biol. 2001, 21, 3947–3958. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.C.J.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Pandolfi, P.P. PTEN and p53: Who will get the upper hand? Cancer Cell 2003, 3, 97–99. [Google Scholar] [CrossRef]
- Freeman, D.J.; Li, A.G.; Wei, G.; Li, H.-H.; Kertesz, N.; Lesche, R.; Whale, A.D.; Martinez-Diaz, H.; Rozengurt, N.; Cardiff, R.D.; et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell 2003, 3, 117–130. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef]
- Li, Y.; Guessous, F.; Kwon, S.; Kumar, M.; Ibidapo, O.; Fuller, L.; Johnson, E.; Lal, B.; Hussaini, I.; Bao, Y.; et al. PTEN has tumor promoting properties in the setting of gain-of-function p53 mutations. Cancer Res. 2008, 68. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, Y.; Tang, Y.; Butler, N.; Kim, J.; Guessous, F.; Schiff, D.; Mandell, J.; Abounader, R. A Novel PTEN/Mutant p53/c-Myc/Bcl-XL Axis Mediates Context-Dependent Oncogenic Effects of PTEN with Implications for Cancer Prognosis and Therapy. Neoplasia N. Y. 2013, 15, 952–965. [Google Scholar] [CrossRef]
- Deb, T.B.; Barndt, R.J.; Zuo, A.H.; Sengupta, S.; Coticchia, C.M.; Johnson, M.D. PTEN-mediated ERK1/2 inhibition and paradoxical cellular proliferation following Pnck overexpression. Cell Cycle Georget. Tex 2014, 13, 961–973. [Google Scholar] [CrossRef]
- Deb, T.B.; Zuo, A.H.; Wang, Y.; Barndt, R.J.; Cheema, A.K.; Sengupta, S.; Coticchia, C.M.; Johnson, M.D. Pnck induces ligand-independent EGFR degradation by probable perturbation of the Hsp90 chaperone complex. Am. J. Physiol. Cell Physiol. 2011, 300, C1139–C1154. [Google Scholar] [CrossRef]
- Zimmermann, S.; Moelling, K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Moelling, K.; Schad, K.; Bosse, M.; Zimmermann, S.; Schweneker, M. Regulation of Raf-Akt Cross-talk. J. Biol. Chem. 2002, 277, 31099–31106. [Google Scholar] [CrossRef] [PubMed]
- Lima-Fernandes, E.; Enslen, H.; Camand, E.; Kotelevets, L.; Boularan, C.; Achour, L.; Benmerah, A.; Gibson, L.C.D.; Baillie, G.S.; Pitcher, J.A.; et al. Distinct functional outputs of PTEN signalling are controlled by dynamic association with β-arrestins. EMBO J. 2011, 30, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.D.; Shinde, R.; McGaha, T.L.; Huang, L.; Holmgaard, R.B.; Wolchok, J.D.; Mautino, M.R.; Celis, E.; Sharpe, A.H.; Francisco, L.M.; et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci. Adv. 2015, 1, e1500845. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Tay, J.; Ton, S.; Agrawal, S.; Gupta, S. Increased Reactivity of Dendritic Cells from Aged Subjects to Self-Antigen, the Human DNA. J. Immunol. 2009, 182, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kang, T.H.; Noh, K.H.; Kim, S.-H.; Lee, Y.-H.; Kim, K.W.; Bae, H.C.; Ahn, Y.-H.; Choi, E.Y.; Kim, J.-S.; et al. Enhancement of DC vaccine potency by activating the PI3K/AKT pathway with a small interfering RNA targeting PTEN. Immunol. Lett. 2010, 134, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Paganelli, F.; Fazio, A.; Bazzichetto, C.; Conciatori, F.; McCubrey, J.A. The Key Roles of PTEN in T-Cell Acute Lymphoblastic Leukemia Development, Progression, and Therapeutic Response. Cancers 2019, 11, 629. [Google Scholar] [CrossRef]
- Shojaee, S.; Garcia, C.; Wu, H.; Muschen, M. The Tumor Suppressor PTEN Is Required to Prevent Cellular Senescence and Cell Cycle Arrest In B Cell Lineage and Chronic Myeloid Leukemia. Blood 2010, 116, 513. [Google Scholar] [CrossRef]
- Shojaee, S.; Chan, L.N.; Buchner, M.; Cazzaniga, V.; Cosgun, K.N.; Geng, H.; Qiu, Y.H.; von Minden, M.D.; Ernst, T.; Hochhaus, A.; et al. PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat. Med. 2016, 22, 379–387. [Google Scholar] [CrossRef]
- Keenan, R.A.; De Riva, A.; Corleis, B.; Hepburn, L.; Licence, S.; Winkler, T.H.; Mårtensson, I.-L. Censoring of autoreactive B cell development by the pre-B cell receptor. Science 2008, 321, 696–699. [Google Scholar] [CrossRef]
- Gomes, A.M.; Soares, M.V.D.; Ribeiro, P.; Caldas, J.; Póvoa, V.; Martins, L.R.; Melão, A.; Serra-Caetano, A.; de Sousa, A.B.; Lacerda, J.F.; et al. Adult B-cell acute lymphoblastic leukemia cells display decreased PTEN activity and constitutive hyperactivation of PI3K/Akt pathway despite high PTEN protein levels. Haematologica 2014, 99, 1062–1068. [Google Scholar] [CrossRef]
- Barata, J.T. The impact of PTEN regulation by CK2 on PI3K-dependent signaling and leukemia cell survival. Adv. Enzyme. Regul. 2011, 51, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Pulido, R. The Tumor Suppressor PTEN Is Phosphorylated by the Protein Kinase CK2 at Its C Terminus IMPLICATIONS FOR PTEN STABILITY TO PROTEASOME-MEDIATED DEGRADATION. J. Biol. Chem. 2001, 276, 993–998. [Google Scholar] [CrossRef]
- Santamaría, C.M.; Chillón, M.C.; García-Sanz, R.; Pérez, C.; Caballero, M.D.; Ramos, F.; de Coca, A.G.; Alonso, J.M.; Giraldo, P.; Bernal, T.; et al. High FOXO3a expression is associated with a poorer prognosis in AML with normal cytogenetics. Leuk. Res. 2009, 33, 1706–1709. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, J.-I.; Tian, T.; Wang, Y.; Hori, Y.; Honma, K.; Wada, N.; Morii, E. Expression of FoxO3a in clinical cases of malignant lymphoma. Pathol. Res. Pract. 2013, 209, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.-S.; Kondo, E.; Miyake, T.; Shibata, M.; Takashima, T.; Liu, Y.-X.; Hayashi, K.; Akagi, T.; Yoshino, T. Expression and Intracellular Localization of FKHRL1 in Mammary Gland Neoplasms. Acta Med. Okayama 2004, 58, 9. [Google Scholar]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J.; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef]
- Barr, F.G. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 2001, 20, 5736–5746. [Google Scholar] [CrossRef]
- Davis, R.J.; D’Cruz, C.M.; Lovell, M.A.; Biegel, J.A.; Barr, F.G. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994, 54, 2869–2872. [Google Scholar]
- Parry, P.; Wei, Y.; Evans, G. Cloning and characterization of the t(X;11) breakpoint from a leukemic cell line identify a new member of the forkhead gene family. Genes Chromosomes Cancer 1994, 11, 79–84. [Google Scholar] [CrossRef]
- Borkhardt, A.; Repp, R.; Haas, O.A.; Leis, T.; Harbott, J.; Kreuder, J.; Hammermann, J.; Henn, T.; Lampert, F. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11)(q13;q23). Oncogene 1997, 14, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J.; Viars, C.S.; Czekaya, S.; Caveneeabc, W.K.; Arden, K.C. Cloning and characterization of three human forkhead genes that comprise an FKHR-like gene subfamily. Genomics 1998, 47, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Hillion, J.; Le Coniat, M.; Jonveaux, P.; Berger, R.; Bernard, O.A. AF6q21, a novel partner of the MLL gene in t(6;11)(q21;q23), defines a forkhead transcriptional factor subfamily. Blood 1997, 90, 3714–3719. [Google Scholar] [CrossRef] [PubMed]
- Hui, R.C.-Y.; Gomes, A.R.; Constantinidou, D.; Costa, J.R.; Karadedou, C.T.; Fernandez de Mattos, S.; Wymann, M.P.; Brosens, J.J.; Schulze, A.; Lam, E.W.-F. The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Mol. Cell. Biol. 2008, 28, 5886–5898. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Lin, A.; Piao, H.-L.; Zhuang, L.; Sarbassov, D.D.; Ma, L.; Gan, B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K-AKT pathway. Cancer Res. 2014, 74, 1682–1693. [Google Scholar] [CrossRef]
- Yang, L.; Xie, S.; Jamaluddin, M.S.; Altuwaijri, S.; Ni, J.; Kim, E.; Chen, Y.-T.; Hu, Y.-C.; Wang, L.; Chuang, K.-H.; et al. Induction of Androgen Receptor Expression by Phosphatidylinositol 3-Kinase/Akt Downstream Substrate, FOXO3a, and Their Roles in Apoptosis of LNCaP Prostate Cancer Cells. J. Biol. Chem. 2005, 280, 33558–33565. [Google Scholar] [CrossRef]
- Hornsveld, M.; Dansen, T.B.; Derksen, P.W.; Burgering, B.M.T. Re-evaluating the role of FOXOs in cancer. Semin. Cancer Biol. 2018, 50, 90–100. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef]
- Chen, J.; Gomes, A.R.; Monteiro, L.J.; Wong, S.Y.; Wu, L.H.; Ng, T.-T.; Karadedou, C.T.; Millour, J.; Ip, Y.-C.; Cheung, Y.N.; et al. Constitutively Nuclear FOXO3a Localization Predicts Poor Survival and Promotes Akt Phosphorylation in Breast Cancer. PLoS ONE 2010, 5, e12293. [Google Scholar] [CrossRef]
- Storz, P.; Döppler, H.; Copland, J.A.; Simpson, K.J.; Toker, A. FOXO3a Promotes Tumor Cell Invasion through the Induction of Matrix Metalloproteinases. Mol. Cell. Biol. 2009, 29, 4906–4917. [Google Scholar] [CrossRef] [PubMed]
- Marlow, L.A.; von Roemeling, C.A.; Cooper, S.J.; Zhang, Y.; Rohl, S.D.; Arora, S.; Gonzales, I.M.; Azorsa, D.O.; Reddi, H.V.; Tun, H.W.; et al. Foxo3a drives proliferation in anaplastic thyroid carcinoma through transcriptional regulation of cyclin A1: A paradigm shift that impacts current therapeutic strategies. J. Cell Sci. 2012, 125, 4253–4263. [Google Scholar] [CrossRef] [PubMed]
- Link, W. Nuclear accumulation of β-catenin and forkhead box O3a in colon cancer: Dangerous liaison. World J. Biol. Chem. 2012, 3, 175. [Google Scholar] [CrossRef] [PubMed]
- You, H. Crosstalk between p53 and FOXO Transcription Factors. Cell Cycle 2004, 4, 37–38. [Google Scholar] [CrossRef]
- Zhao, H.H.; Herrera, R.E.; Coronado-Heinsohn, E.; Yang, M.C.; Ludes-Meyers, J.H.; Seybold-Tilson, K.J.; Nawaz, Z.; Yee, D.; Barr, F.G.; Diab, S.G.; et al. Forkhead Homologue in Rhabdomyosarcoma Functions as a Bifunctional Nuclear Receptor-interacting Protein with Both Coactivator and Corepressor Functions. J. Biol. Chem. 2001, 276, 27907–27912. [Google Scholar] [CrossRef]
- Schuur, E.R.; Loktev, A.V.; Sharma, M.; Sun, Z.; Roth, R.A.; Weigel, R.J. Ligand-dependent Interaction of Estrogen Receptor-α with Members of the Forkhead Transcription Factor Family. J. Biol. Chem. 2001, 276, 33554–33560. [Google Scholar] [CrossRef]
- Guo, S.; Sonenshein, G.E. Forkhead Box Transcription Factor FOXO3a Regulates Estrogen Receptor Alpha Expression and Is Repressed by the Her-2/neu/Phosphatidylinositol 3-Kinase/Akt Signaling Pathway. Mol. Cell. Biol. 2004, 24, 8681–8690. [Google Scholar] [CrossRef]
- Sisci, D.; Maris, P.; Cesario, M.G.; Anselmo, W.; Coroniti, R.; Trombino, G.E.; Romeo, F.; Ferraro, A.; Lanzino, M.; Aquila, S.; et al. The estrogen receptor α is the key regulator of the bifunctional role of FoxO3a transcription factor in breast cancer motility and invasiveness. Cell Cycle Georget. Tex. 2013, 12, 3405–3420. [Google Scholar] [CrossRef]
- Belguise, K.; Guo, S.; Sonenshein, G.E. Activation of FOXO3a by the green tea polyphenol epigallocatechin-3-gallate induces estrogen receptor alpha expression reversing invasive phenotype of breast cancer cells. Cancer Res. 2007, 67, 5763–5770. [Google Scholar] [CrossRef]
- Madureira, P.A.; Varshochi, R.; Constantinidou, D.; Francis, R.E.; Coombes, R.C.; Yao, K.-M.; Lam, E.W.-F. The Forkhead Box M1 Protein Regulates the Transcription of the Estrogen Receptor α in Breast Cancer Cells. J. Biol. Chem. 2006, 281, 25167–25176. [Google Scholar] [CrossRef]
- Zou, Y.; Tsai, W.-B.; Cheng, C.-J.; Hsu, C.; Chung, Y.M.; Li, P.-C.; Lin, S.-H.; Hu, M.C. Forkhead box transcription factor FOXO3a suppresses estrogen-dependent breast cancer cell proliferation and tumorigenesis. Breast Cancer Res. 2008, 10. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Feld, F.; Krüger, K.-D.; Bahrenberg, G.; Roth, R.A.; Joost, H.-G.; Heinrich, P.C.; Behrmann, I.; Barthel, A. Akt Modulates STAT3-mediated Gene Expression through a FKHR (FOXO1a)-dependent Mechanism. J. Biol. Chem. 2003, 278, 5242–5249. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Marcos-Villar, L.; Gallego, P.; Arroyo, J.; Da Costa, M.; Pomeranz, K.M.; Lam, E.W.-F.; Rivas, C. Latent protein LANA2 from Kaposi’s sarcoma-associated herpesvirus interacts with 14-3-3 proteins and inhibits FOXO3a transcription factor. J. Virol. 2007, 81, 1511–1516. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chandramohan, V.; Mineva, N.D.; Burke, B.; Jeay, S.; Wu, M.; Shen, J.; Yang, W.; Hann, S.R.; Sonenshein, G.E. c-Myc represses FOXO3a-mediated transcription of the gene encoding the p27(Kip1) cyclin dependent kinase inhibitor. J. Cell. Biochem. 2008, 104, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Le, H.-V.; Shen, L.; Anderson, S.A.; Massagué, J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H.; Chen, Y.; Fan, L.; Fang, J. Forkhead Transcription Factor FOXO3a Protein Activates Nuclear Factor κB through B-cell lymphoma/leukemia 10 (BCL10) Protein and Promotes Tumor Cell Survival in Serum Deprivation. J. Biol. Chem. 2012, 287, 17737–17745. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, F.; Hughes, T.; Yu, J. FOXOs in Cancer Immunity: Knowns and Unknowns. Semin. Cancer Biol. 2018, 50, 53–64. [Google Scholar] [CrossRef]
- Jensen, K.S.; Binderup, T.; Jensen, K.T.; Therkelsen, I.; Borup, R.; Nilsson, E.; Multhaupt, H.; Bouchard, C.; Quistorff, B.; Kjaer, A.; et al. FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 2011, 30, 4554–4570. [Google Scholar] [CrossRef]
- Ferber, E.C.; Peck, B.; Delpuech, O.; Bell, G.; East, P.; Schulze, A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968–979. [Google Scholar] [CrossRef]
- Peck, B.; Ferber, E.C.; Schulze, A. Antagonism between FOXO and MYC Regulates Cellular Powerhouse. Front. Oncol 2013, 3. [Google Scholar] [CrossRef]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a Is Essential for Maintenance of the Hematopoietic Stem Cell Pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Pollina, E.A.; Brunet, A. Epigenetic regulation of aging stem cells. Oncogene 2011, 30, 3105–3126. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-β–FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Ptasinska, A.; Chen, X.; Shrestha, M.; Assi, S.A.; Chin, P.S.; Imperato, M.R.; Aronow, B.J.; Zhang, J.; Weirauch, M.T.; et al. A FOXO1-induced oncogenic network defines the AML1-ETO preleukemic program. Blood 2017, 130, 1213–1222. [Google Scholar] [CrossRef]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs Are Critical Mediators of Hematopoietic Stem Cell Resistance to Physiologic Oxidative Stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef]
- Liu, H.; Song, Y.; Qiu, H.; Liu, Y.; Luo, K.; Yi, Y.; Jiang, G.; Lu, M.; Zhang, Z.; Yin, J.; et al. Downregulation of FOXO3a by DNMT1 promotes breast cancer stem cell properties and tumorigenesis. Cell Death Differ. 2020, 27, 966–983. [Google Scholar] [CrossRef]
- Kumazoe, M.; Takai, M.; Hiroi, S.; Takeuchi, C.; Kadomatsu, M.; Nojiri, T.; Onda, H.; Bae, J.; Huang, Y.; Takamatsu, K.; et al. The FOXO3/PGC-1β signaling axis is essential for cancer stem cell properties of pancreatic ductal adenocarcinoma. J. Biol. Chem. 2017, 292, 10813–10823. [Google Scholar] [CrossRef]
- Matkar, S.; Sharma, P.; Gao, S.; Gurung, B.; Katona, B.W.; Liao, J.; Muhammad, A.B.; Kong, X.-C.; Wang, L.; Jin, G.; et al. An Epigenetic Pathway Regulates Sensitivity of Breast Cancer Cells to HER2 Inhibition via FOXO/c-Myc Axis. Cancer Cell 2015, 28, 472–485. [Google Scholar] [CrossRef]
- Essers, M.A.G.; Weijzen, S.; de Vries-Smits, A.M.M.; Saarloos, I.; de Ruiter, N.D.; Bos, J.L. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004, 23, 4802–4812. [Google Scholar] [CrossRef]
- Das, N.; Datta, N.; Chatterjee, U.; Ghosh, M.K. Estrogen receptor alpha transcriptionally activates casein kinase 2 alpha: A pivotal regulator of promyelocytic leukaemia protein (PML) and AKT in oncogenesis. Cell Signal. 2016, 28, 675–687. [Google Scholar] [CrossRef]
- Chatterjee, A.; Chatterjee, U.; Ghosh, M.K. Activation of protein kinase CK2 attenuates FOXO3a functioning in a PML-dependent manner: Implications in human prostate cancer. Cell Death Dis. 2013, 4, e543. [Google Scholar] [CrossRef] [PubMed]
- Datta, N.; Islam, S.; Chatterjee, U.; Chatterjee, S.; Panda, C.K.; Ghosh, M.K. Promyelocytic Leukemia (PML) gene regulation: Implication towards curbing oncogenesis. Cell Death Dis. 2019, 10, 656. [Google Scholar] [CrossRef] [PubMed]
- Bellodi, C.; Kindle, K.; Bernassola, F.; Cossarizza, A.; Dinsdale, D.; Melino, G.; Salomoni, P. A cytoplasmic PML mutant inhibits p53 function. Cell Cycle Georget. Tex. 2006, 5, 2688–2692. [Google Scholar] [CrossRef]
- Bellodi, C.; Kindle, K.; Bernassola, F.; Dinsdale, D.; Cossarizza, A.; Melino, G.; Heery, D.; Salomoni, P. Cytoplasmic function of mutant promyelocytic leukemia (PML) and PML-retinoic acid receptor-alpha. J. Biol. Chem. 2006, 281, 14465–14473. [Google Scholar] [CrossRef]
- Lin, H.-K.; Bergmann, S.; Pandolfi, P.P. Cytoplasmic PML function in TGF-β signalling. Nature 2004, 431, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Buczek, M.E.; Miles, A.K.; Green, W.; Johnson, C.; Boocock, D.J.; Pockley, A.G.; Rees, R.C.; Hulman, G.; van Schalkwyk, G.; Parkinson, R.; et al. Cytoplasmic PML promotes TGF-β-associated epithelial–mesenchymal transition and invasion in prostate cancer. Oncogene 2016, 35, 3465–3475. [Google Scholar] [CrossRef]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.-M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002, 21, 2383–2396. [Google Scholar] [CrossRef]
- Renner, F.; Moreno, R.; Schmitz, M.L. SUMOylation-Dependent Localization of IKKɛ in PML Nuclear Bodies Is Essential for Protection against DNA-Damage-Triggered Cell Death. Mol. Cell 2010, 37, 503–515. [Google Scholar] [CrossRef]
- Wimmer, P.; Berscheminski, J.; Blanchette, P.; Groitl, P.; Branton, P.E.; Hay, R.T.; Dobner, T.; Schreiner, S. PML isoforms IV and V contribute to adenovirus-mediated oncogenic transformation by functionally inhibiting the tumor-suppressor p53. Oncogene 2016, 35, 69–82. [Google Scholar] [CrossRef]
- Carracedo, A.; Ito, K.; Pandolfi, P.P. The nuclear bodies inside out: PML conquers the cytoplasm. Curr. Opin. Cell Biol. 2011, 23, 360–366. [Google Scholar] [CrossRef]
- Iwanami, A.; Gini, B.; Zanca, C.; Matsutani, T.; Assuncao, A.; Nael, A.; Dang, J.; Yang, H.; Zhu, S.; Kohyama, J.; et al. PML mediates glioblastoma resistance to mammalian target of rapamycin (mTOR)-targeted therapies. Proc. Natl. Acad. Sci. USA 2013, 110, 4339–4344. [Google Scholar] [CrossRef] [PubMed]
- de Thé, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejean, A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Di Croce, L.; Raker, V.A.; Corsaro, M.; Fazi, F.; Fanelli, M.; Faretta, M.; Fuks, F.; Lo Coco, F.; Kouzarides, T.; Nervi, C.; et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 2002, 295, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Nasr, R.; de Thé, H. Eradication of acute promyelocytic leukemia-initiating cells by PML/RARA-targeting. Int. J. Hematol. 2010, 91, 742–747. [Google Scholar] [CrossRef]
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008, 453, 1072–1078. [Google Scholar] [CrossRef]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef]
- Zhou, W.; Cheng, L.; Shi, Y.; Ke, S.Q.; Huang, Z.; Fang, X.; Chu, C.; Xie, Q.; Bian, X.; Rich, J.N.; et al. Arsenic trioxide disrupts glioma stem cells via promoting PML degradation to inhibit tumor growth. Oncotarget 2015, 6, 37300–37315. [Google Scholar] [CrossRef]
- Tang, H.; Jin, Y.; Jin, S.; Tan, Z.; Peng, Z.; Kuang, Y. Arsenite inhibits the function of CD133+ CD13+ liver cancer stem cells by reducing PML and Oct4 protein expression. Tumour. Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 14103–14115. [Google Scholar] [CrossRef]
- Sun, J.; Fu, S.; Zhong, W.; Huang, H. PML overexpression inhibits proliferation and promotes the osteogenic differentiation of human mesenchymal stem cells. Oncol. Rep. 2013, 30, 2785–2794. [Google Scholar] [CrossRef]
- Guarnerio, J.; Mendez, L.M.; Asada, N.; Menon, A.V.; Fung, J.; Berry, K.; Frenette, P.S.; Ito, K.; Pandolfi, P.P. A non-cell-autonomous role for Pml in the maintenance of leukemia from the niche. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Aoto, T.; Saitoh, N.; Ichimura, T.; Niwa, H.; Nakao, M. Nuclear and chromatin reorganization in the MHC-Oct3/4 locus at developmental phases of embryonic stem cell differentiation. Dev. Biol. 2006, 298, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Hu, X.; Gupta, P.; Lin, Y.-P.; Gil Ha, S.G.; Wei, L.-N. SUMOylation of Tr2 orphan receptor involves Pml and fine-tunes Oct4 expression in stem cells. Nat. Struct. Mol. Biol. 2007, 14, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Park, S.W.; Farooqui, M.; Wei, L.-N. Orphan nuclear receptor TR2, a mediator of preadipocyte proliferation, is differentially regulated by RA through exchange of coactivator PCAF with corepressor RIP140 on a platform molecule GRIP1. Nucleic Acids Res. 2007, 35, 2269–2282. [Google Scholar] [CrossRef] [PubMed]
- Hadjimichael, C.; Chanoumidou, K.; Nikolaou, C.; Klonizakis, A.; Theodosi, G.-I.; Makatounakis, T.; Papamatheakis, J.; Kretsovali, A. Promyelocytic Leukemia Protein Is an Essential Regulator of Stem Cell Pluripotency and Somatic Cell Reprogramming. Stem Cell Rep. 2017, 8, 1366–1378. [Google Scholar] [CrossRef]
- Torok, D.; Ching, R.W.; Bazett-Jones, D.P. PML nuclear bodies as sites of epigenetic regulation. Front. Biosci Landmark Ed. 2009, 14, 1325–1336. [Google Scholar] [CrossRef]
- Isogai, Y.; Tjian, R. Targeting genes and transcription factors to segregated nuclear compartments. Curr. Opin. Cell Biol. 2003, 15, 296–303. [Google Scholar] [CrossRef]
- Wang, J.; Shiels, C.; Sasieni, P.; Wu, P.J.; Islam, S.A.; Freemont, P.S.; Sheer, D. Promyelocytic leukemia nuclear bodies associate with transcriptionally active genomic regions. J. Cell Biol. 2004, 164, 515–526. [Google Scholar] [CrossRef]
- Vallian, S.; Gäken, J.A.; Gingold, E.B.; Kouzarides, T.; Chang, K.-S.; Farzaneh, F. Modulation of Fos-mediated AP-1 transcription by the promyelocytic leukemia protein. Oncogene 1998, 16. Available online: http://search.ebscohost.com/login.aspx?direct=true&profile=ehost&scope=site&authtype=crawler&jrnl=09509232&AN=8910781&h=n4%2BCVENu7WmMK27aXwaC4KTM%2Fw1NmRuOuQFrAcAhJOpUyNG9SLXjg96d5hFmjxqyQm1vkv%2BDc22rExPvRpW7rw%3D%3D&crl=c (accessed on 4 September 2017). [CrossRef][Green Version]
- Salomoni, P. The promyelocytic leukemia protein PML regulates c-Jun function in response to DNA damage. Blood 2005, 105, 3686–3690. [Google Scholar] [CrossRef][Green Version]
- Vallian, S.; Chin, K.V.; Chang, K.S. The promyelocytic leukemia protein interacts with Sp1 and inhibits its transactivation of the epidermal growth factor receptor promoter. Mol. Cell. Biol. 1998, 18, 7147–7156. [Google Scholar] [CrossRef][Green Version]
- Li, J.; Zou, W.-X.; Chang, K.-S. Inhibition of Sp1 functions by its sequestration into PML nuclear bodies. PLoS ONE 2014, 9, e94450. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, Z.-X.; Zhao, R.-X.; Ding, T.; Tran, T.T.; Zhang, W.; Pandolfi, P.P.; Chang, K.-S. Promyelocytic Leukemia Protein 4 Induces Apoptosis by Inhibition of Survivin Expression. J. Biol. Chem. 2004, 279, 1838–1844. [Google Scholar] [CrossRef] [PubMed]
- Shtutman, M.; Zhurinsky, J.; Oren, M.; Levina, E.; Ben-Ze’ev, A. PML is a target gene of beta-catenin and plakoglobin, and coactivates beta-catenin-mediated transcription. Cancer Res. 2002, 62, 5947–5954. [Google Scholar] [PubMed]
- Ponente, M.; Campanini, L.; Cuttano, R.; Piunti, A.; Delledonne, G.A.; Coltella, N.; Valsecchi, R.; Villa, A.; Cavallaro, U.; Pattini, L.; et al. PML promotes metastasis of triple-negative breast cancer through transcriptional regulation of HIF1A target genes. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.D.; Searleman, A.C.; Swamidass, S.J.; Griffith, O.L.; Bose, R. Statistically identifying tumor suppressors and oncogenes from pan-cancer genome-sequencing data. Bioinformatics 2015, 31, 3561–3568. [Google Scholar] [CrossRef]
- Wrzeszczynski, K.O.; Varadan, V.; Byrnes, J.; Lum, E.; Kamalakaran, S.; Levine, D.A.; Dimitrova, N.; Zhang, M.Q.; Lucito, R. Identification of Tumor Suppressors and Oncogenes from Genomic and Epigenetic Features in Ovarian Cancer. PLoS ONE 2011, 6, e28503. [Google Scholar] [CrossRef]
- Pavel, A.B.; Vasile, C.I. Identifying cancer type specific oncogenes and tumor suppressors using limited size data. J. Bioinform. Comput. Biol. 2016, 14, 1650031. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Finding the Cancer-Critical Genes. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK26816/ (accessed on 3 November 2019).
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef]
- Liu, K.; Ling, S.; Lin, W.-C. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell. Biol. 2011, 31, 4464–4481. [Google Scholar] [CrossRef] [PubMed]
- Chicas, A.; Molina, P.; Bargonetti, J. Mutant p53 forms a complex with Sp1 on HIV-LTR DNA. Biochem. Biophys. Res. Commun. 2000, 279, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Bargonetti, J.; Chicas, A.; White, D.; Prives, C. p53 represses Sp1 DNA binding and HIV-LTR directed transcription. Cell. Mol. Biol. Noisy-Gd. Fr. 1997, 43, 935–949. [Google Scholar]
- Torgeman, A.; Mor-Vaknin, N.; Zelin, E.; Ben-Aroya, Z.; Löchelt, M.; Flügel, R.M.; Aboud, M. Sp1-p53 heterocomplex mediates activation of HTLV-I long terminal repeat by 12-O-tetradecanoylphorbol-13-acetate that is antagonized by protein kinase C. Virology 2001, 281, 10–20. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 Cooperates with ETS and Selectively Up-regulates Human MDR1 Not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef]
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; Del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem. 2002, 277, 18817–18826. [Google Scholar] [CrossRef]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef]
- Li, Y.; Prives, C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene 2007, 26, 2220–2225. [Google Scholar] [CrossRef]
- Chan, W.M.; Siu, W.Y.; Lau, A.; Poon, R.Y.C. How Many Mutant p53 Molecules Are Needed To Inactivate a Tetramer? Mol. Cell. Biol. 2004, 24, 3536–3551. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.-C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef]
- Haupt, S.; di Agostino, S.; Mizrahi, I.; Alsheich-Bartok, O.; Voorhoeve, M.; Damalas, A.; Blandino, G.; Haupt, Y. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res. 2009, 69, 4818–4826. [Google Scholar] [CrossRef]
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Restle, A.; Färber, M.; Baumann, C.; Böhringer, M.; Scheidtmann, K.H.; Müller-Tidow, C.; Wiesmüller, L. Dissecting the role of p53 phosphorylation in homologous recombination provides new clues for gain-of-function mutants. Nucleic Acids Res. 2008, 36, 5362–5375. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Pfister, N.T.; Fomin, V.; Regunath, K.; Zhou, J.Y.; Zhou, W.; Silwal-Pandit, L.; Freed-Pastor, W.A.; Laptenko, O.; Neo, S.P.; Bargonetti, J.; et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 2015, 29, 1298–1315. [Google Scholar] [CrossRef]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Fontemaggi, G.; Fazi, F.; Di Agostino, S.; Padula, F.; Biagioni, F.; Muti, P.; Strano, S.; Blandino, G. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2012, 19, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef] [PubMed]
- Neilsen, P.M.; Noll, J.E.; Mattiske, S.; Bracken, C.P.; Gregory, P.A.; Schulz, R.B.; Lim, S.P.; Kumar, R.; Suetani, R.J.; Goodall, G.J.; et al. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene 2013, 32, 2992–3000. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, B.; Miao, L.; Mei, Y.; Wu, M. Mutant p53-R273H gains new function in sustained activation of EGFR signaling via suppressing miR-27a expression. Cell Death Dis. 2013, 4, e574. [Google Scholar] [CrossRef]
- Su, X.; Chakravarti, D.; Cho, M.S.; Liu, L.; Gi, Y.J.; Lin, Y.-L.; Leung, M.L.; El-Naggar, A.; Creighton, C.J.; Suraokar, M.B.; et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010, 467, 986–990. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Genes | |
|---|---|---|
| Protein-coding | Transcription factors | FOXL2, RUNX1, DNMT1, DNMT3A, ETS2, ETV6, EZH2, FOXO1, FOXO3, GLI1, HDAC1, FOXO4, MXI1, NOTCH1, NOTCH2, NOTCH3, PAX5, RARB, SKIL, TCF3, WT1, ZBTB16, NR4A3, NCOA4, KLF4, LITAF, YAP1, SALL4, HOPX, LHX4, FUS |
| Kinases | BCR, CDKN1B, MAP3K8, FLT3 | |
| Protein binding | RHOA, ECT2, IDH1, NPM1, PHB, PML, PTPN11, SPOP, RASSF1, ARHGEF12, SIRT1, SUZ12, WHSC1L1, WDR11, RB1, CBL, DMBT1 | |
| Noncoding RNA (ncRNA) | MIR106A, MIR107, MIR125B1, MIR146A, MIR150, MIR155, MIR17, MIR18A, MIR194-1, MIR194-2, MIR196A2, MIR20A, MIR203A, MIR210, MIR214, MIR222, MIR223, MIR24-1, MIR27A, MIR18B | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Datta, N.; Chakraborty, S.; Basu, M.; Ghosh, M.K. Tumor Suppressors Having Oncogenic Functions: The Double Agents. Cells 2021, 10, 46. https://doi.org/10.3390/cells10010046
Datta N, Chakraborty S, Basu M, Ghosh MK. Tumor Suppressors Having Oncogenic Functions: The Double Agents. Cells. 2021; 10(1):46. https://doi.org/10.3390/cells10010046
Chicago/Turabian StyleDatta, Neerajana, Shrabastee Chakraborty, Malini Basu, and Mrinal K. Ghosh. 2021. "Tumor Suppressors Having Oncogenic Functions: The Double Agents" Cells 10, no. 1: 46. https://doi.org/10.3390/cells10010046
APA StyleDatta, N., Chakraborty, S., Basu, M., & Ghosh, M. K. (2021). Tumor Suppressors Having Oncogenic Functions: The Double Agents. Cells, 10(1), 46. https://doi.org/10.3390/cells10010046