MicroRNA-Assisted Hormone Cell Signaling in Colorectal Cancer Resistance

 ,
,  and
and
Abstract
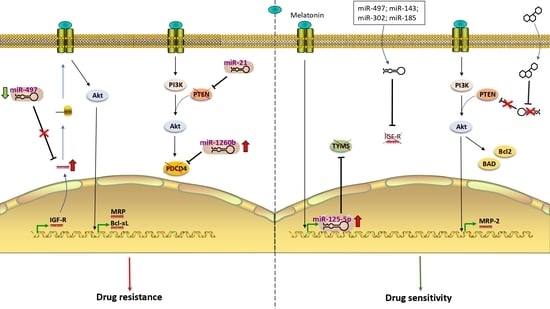
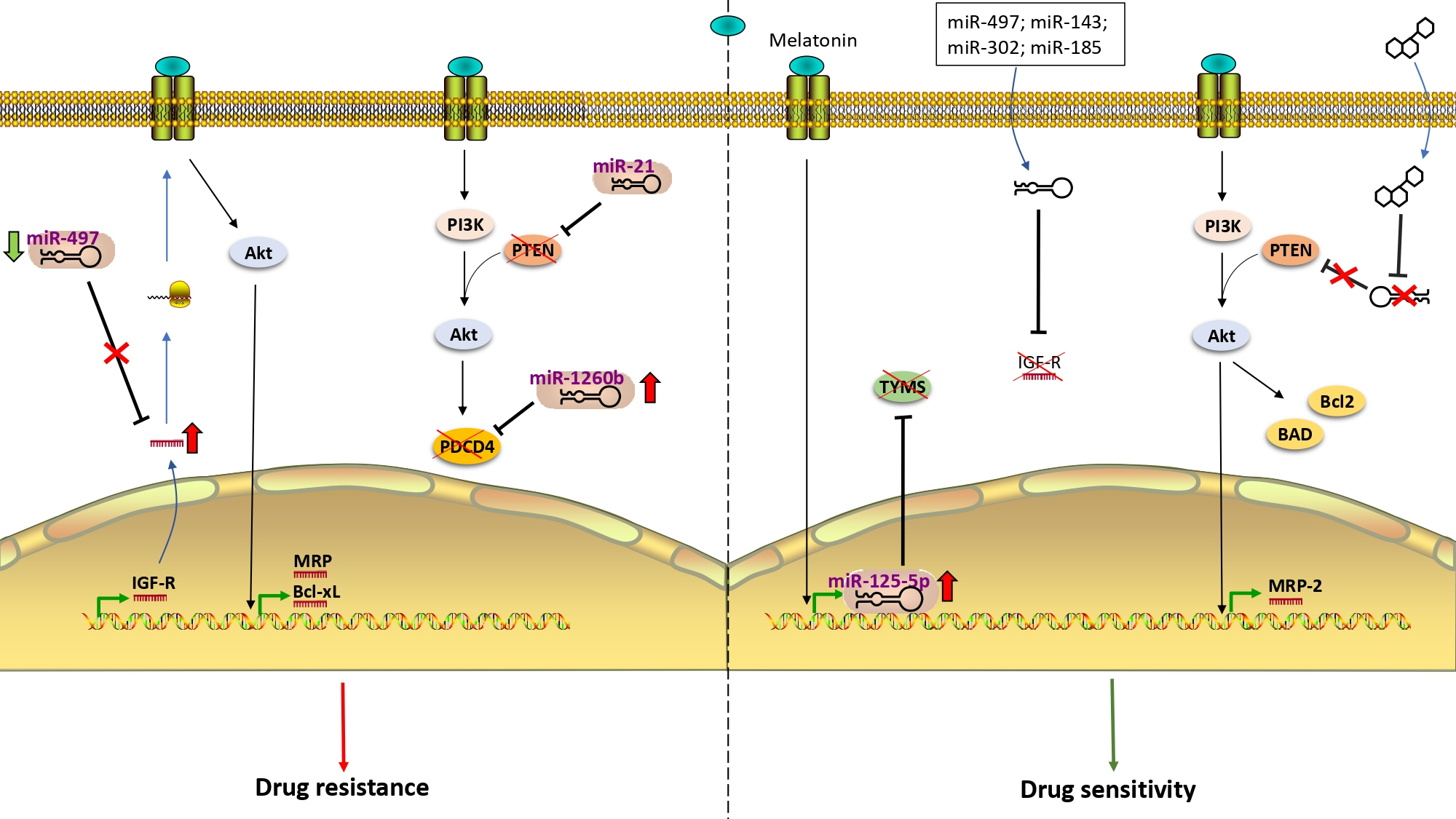
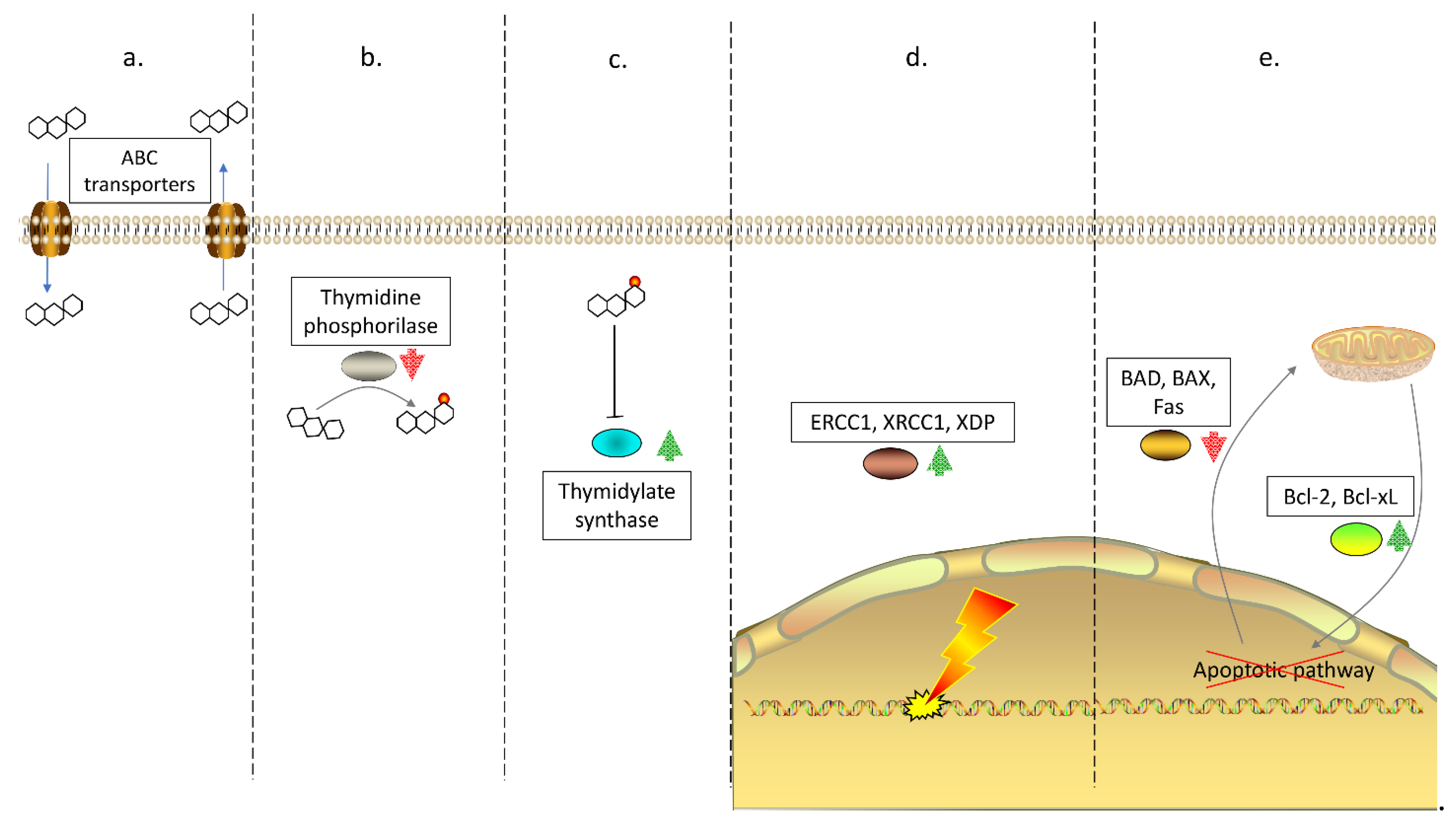
1. Drug Resistance in Colorectal Cancer
2. The Role of Hormone Signaling in CRC Resistance
2.1. Insulin/Insulin-Like Growth Factor Signaling Pathway in CRC
2.2. Thyroid Hormone Signaling Pathway in CRC
2.3. Additional Altered Hormone Signaling in Resistant CRC Cells
3. Role of miRNA in Hormone Signaling in Therapy-Resistant CRC
3.1. miRNAs and Insulin-Like Growth Factor (IGF) Receptor
3.2. miRNAs and IGF-R Downstream Components
3.3. miR-155 and Adrenaline
3.4. miR-7 and Corticotropin-Releasing Hormone Receptor
4. Novel Therapeutic Agents in the Treatment of Drug Resistance in CRC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, A.; Thompson, H.; Adam, J.; Parker, A.; Gammon, L.; Lewis, A.; Bundy, J.G.; Soga, T.; Jalaly, A.; Propper, D.; et al. Remodelling of microRNAs in colorectal cancer by hypoxia alters metabolism profiles and 5-fluorouracil resistance. Hum. Mol. Genet. 2017, 26, 1552–1564. [Google Scholar] [CrossRef]
- Wang, W.; Kandimalla, R.; Huang, H.; Zhu, L.; Li, Y.; Gao, F.; Goel, A.; Wang, X. Molecular subtyping of colorectal cancer: Recent progress, new challenges and emerging opportunities. Semin. Cancer Biol. 2019, 55, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, S.; Huang, X.; Li, L.; Zhang, C.; Pan, Q.; Ren, Z.; Zhou, R.; Ren, Y.; Zi, J.; et al. Drug Resistance in Colorectal Cancer Cell Lines is Partially Associated with Aneuploidy Status in Light of Profiling Gene Expression. J. Proteome Res. 2016, 15, 4047–4059. [Google Scholar] [CrossRef] [PubMed]
- Orsetti, B.; Selves, J.; Bascoul-Mollevi, C.; Lasorsa, L.; Gordien, K.; Bibeau, F.; Massemin, B.; Paraf, F.; Soubeyran, I.; Hostein, I.; et al. Impact of chromosomal instability on colorectal cancer progression and outcome. BMC Cancer 2014, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Humeniuk, R.; Mishra, P.J.; Bertino, J.R.; Banerjee, D. Epigenetic reversal of acquired resistance to 5-fluorouracil treatment. Mol. Cancer Ther. 2009, 8, 1045–1054. [Google Scholar] [CrossRef]
- Cheng, X.; Xu, X.; Chen, D.; Zhao, F.; Wang, W. Therapeutic potential of targeting the Wnt/beta-catenin signaling pathway in colorectal cancer. Biomed. Pharmacother. 2019, 110, 473–481. [Google Scholar] [CrossRef]
- Siraj, A.K.; Parvathareddy, S.K.; Pratheeshkumar, P.; Divya, S.P.; Ahmed, S.O.; Melosantos, R.; Begum, R.; Concepcion, R.M.J.; Al-Sanea, N.; Ashari, L.H.; et al. APC truncating mutations in Middle Eastern Population: Tankyrase inhibitor is an effective strategy to sensitize APC mutant CRC To 5-FU chemotherapy. Biomed. Pharmacother. 2020, 121, 109572. [Google Scholar] [CrossRef]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. 2012, 5, 19–27. [Google Scholar] [PubMed]
- Phipps, A.I.; Buchanan, D.D.; Makar, K.W.; Win, A.K.; Baron, J.A.; Lindor, N.M.; Potter, J.D.; Newcomb, P.A. KRAS-mutation status in relation to colorectal cancer survival: The joint impact of correlated tumour markers. Br. J. Cancer 2013, 108, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, T.M.; Raut, C.P.; Rodriguez-Bigas, M.A. Colorectal carcinogenesis: MSI-H versus MSI-L. Dis. Markers 2004, 20, 199–206. [Google Scholar] [CrossRef]
- Sideris, M.; Papagrigoriadis, S. Molecular biomarkers and classification models in the evaluation of the prognosis of colorectal cancer. Anticancer Res. 2014, 34, 2061–2068. [Google Scholar]
- Aebi, S.; Kurdi-Haidar, B.; Gordon, R.; Cenni, B.; Zheng, H.; Fink, D.; Christen, R.D.; Boland, C.R.; Koi, M.; Fishel, R.; et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996, 56, 3087–3090. [Google Scholar]
- Fink, D.; Nebel, S.; Aebi, S.; Zheng, H.; Cenni, B.; Nehme, A.; Christen, R.D.; Howell, S.B. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996, 56, 4881–4886. [Google Scholar]
- Arnold, C.N.; Goel, A.; Boland, C.R. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int. J. Cancer 2003, 106, 66–73. [Google Scholar] [CrossRef]
- Sahin, I.H.; Akce, M.; Alese, O.; Shaib, W.; Lesinski, G.B.; El-Rayes, B.; Wu, C. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br. J. Cancer 2019, 121, 809–818. [Google Scholar] [CrossRef]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K.; et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. [Google Scholar] [CrossRef]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef] [PubMed]
- Baretti, M.; Azad, N.S. The role of epigenetic therapies in colorectal cancer. Curr. Probl. Cancer 2018, 42, 530–547. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Giovannetti, E.; Cortesi, F.; Mey, V.; Nannizzi, S.; Gallegos Ruiz, M.I.; Ricciardi, S.; Del Tacca, M.; Peters, G.J.; Danesi, R. Epigeneticmechanisms of irinotecansensitivity in colorectalcancercell lines. Mol. Cancer Ther. 2009, 8, 1964–1973. [Google Scholar] [CrossRef]
- Chang, X.; Monitto, C.L.; Demokan, S.; Kim, M.S.; Chang, S.S.; Zhong, X.; Califano, J.A.; Sidransky, D. Identification of hypermethylated genes associated with cisplatin resistance in human cancers. Cancer Res. 2010, 70, 2870–2879. [Google Scholar] [CrossRef]
- Crea, F.; Nobili, S.; Paolicchi, E.; Perrone, G.; Napoli, C.; Landini, I.; Danesi, R.; Mini, E. Epigenetics and chemoresistance in colorectal cancer: An opportunity for treatment tailoring and novel therapeutic strategies. Drug Resist. Updat. 2011, 14, 280–296. [Google Scholar] [CrossRef]
- Plumb, J.A.; Strathdee, G.; Sludden, J.; Kaye, S.B.; Brown, R. Reversal of drug resistance in human tumor xenografts by 2’-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000, 60, 6039–6044. [Google Scholar]
- Lai, M.; Du, G.; Shi, R.; Yao, J.; Yang, G.; Wei, Y.; Zhang, D.; Xu, Z.; Zhang, R.; Li, Y.; et al. MiR-34a inhibits migration and invasion by regulating the SIRT1/p53 pathway in human SW480 cells. Mol. Med. Rep. 2015, 11, 3301–3307. [Google Scholar] [CrossRef][Green Version]
- Zhang, Q.; Wang, J.; Li, N.; Liu, Z.; Chen, Z.; Li, Z.; Lai, Y.; Shen, L.; Gao, J. miR-34a increases the sensitivity of colorectal cancer cells to 5-fluorouracil in vitro and in vivo. Am. J. Cancer Res. 2018, 8, 280–290. [Google Scholar]
- Akao, Y.; Noguchi, S.; Iio, A.; Kojima, K.; Takagi, T.; Naoe, T. Dysregulation of microRNA-34a expression causes drug-resistance to 5-FU in human colon cancer DLD-1 cells. Cancer Lett. 2011, 300, 197–204. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, X.H. MiR-29b reverses oxaliplatin-resistance in colorectal cancer by targeting SIRT1. Oncotarget 2018, 9, 12304–12315. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Wang, Y.; Xi, Y.; Kudo, K.; Bruheim, S.; Botchkina, G.I.; Gavin, E.; Wan, Y.; Formentini, A.; Kornmann, M.; et al. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene 2009, 28, 4065–4074. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Li, Z.; Gao, C.Y.; Cho, C.H. Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J. Gastroenterol. 2016, 22, 6876–6889. [Google Scholar] [CrossRef] [PubMed]
- Herraez, E.; Gonzalez-Sanchez, E.; Vaquero, J.; Romero, M.R.; Serrano, M.A.; Marin, J.J.; Briz, O. Cisplatin-induced chemoresistance in colon cancer cells involves FXR-dependent and FXR-independent up-regulation of ABC proteins. Mol. Pharm. 2012, 9, 2565–2576. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Miura, K.; Kinouchi, M.; Ishida, K.; Fujibuchi, W.; Naitoh, T.; Ogawa, H.; Ando, T.; Yazaki, N.; Watanabe, K.; Haneda, S.; et al. 5-fu metabolism in cancer and orally-administrable 5-fu drugs. Cancers 2010, 2, 1717–1730. [Google Scholar] [CrossRef]
- Ibrahim, S.; Li, G.; Hu, F.; Hou, Z.; Chen, Q.; Li, G.; Luo, X.; Hu, J.; Feng, Y. PIK3R3 promotes chemotherapeutic sensitivity of colorectal cancer through PIK3R3/NF-kB/TP pathway. Cancer Biol. Ther. 2018, 19, 222–229. [Google Scholar] [CrossRef]
- Wang, T.L.; Diaz, L.A., Jr.; Romans, K.; Bardelli, A.; Saha, S.; Galizia, G.; Choti, M.; Donehower, R.; Parmigiani, G.; ShihIe, M.; et al. Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3089–3094. [Google Scholar] [CrossRef]
- Watson, R.G.; Muhale, F.; Thorne, L.B.; Yu, J.; O’Neil, B.H.; Hoskins, J.M.; Meyers, M.O.; Deal, A.M.; Ibrahim, J.G.; Hudson, M.L.; et al. Amplification of thymidylatesynthetase in metastatic colorectal cancer patients pretreated with 5-fluorouracil-based chemotherapy. Eur. J. Cancer 2010, 46, 3358–3364. [Google Scholar] [CrossRef]
- Yu, J.; Miller, R.; Zhang, W.; Sharma, M.; Holtschlag, V.; Watson, M.A.; McLeod, H.L. Copy-number analysis of topoisomerase and thymidylate synthase genes in frozen and FFPE DNAs of colorectal cancers. Pharmacogenomics 2008, 9, 1459–1466. [Google Scholar] [CrossRef]
- Etienne, M.C.; Chazal, M.; Laurent-Puig, P.; Magne, N.; Rosty, C.; Formento, J.L.; Francoual, M.; Formento, P.; Renee, N.; Chamorey, E.; et al. Prognostic value of tumoralthymidylate synthase and p53 in metastatic colorectal cancer patients receiving fluorouracil-based chemotherapy: Phenotypic and genotypic analyses. J. Clin. Oncol. 2002, 20, 2832–2843. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Libra, M.; Navolanic, P.M.; Talamini, R.; Cecchin, E.; Sartor, F.; Tumolo, S.; Masier, S.; Travali, S.; Boiocchi, M.; Toffoli, G. Thymidylatesynthetase mRNA levels are increased in liver metastases of colorectal cancer patients resistant to fluoropyrimidine-based chemotherapy. BMC Cancer 2004, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Hiller-Sturmhofel, S.; Bartke, A. The endocrine system: An overview. Alcohol. Health Res. World 1998, 22, 153–164. [Google Scholar] [PubMed]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenicRas/MAPK and survival PI3K/Akt pathways: A fine balance. Biochem. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef]
- Bray, S.M.; Lee, J.; Kim, S.T.; Hur, J.Y.; Ebert, P.J.; Calley, J.N.; Wulur, I.H.; Gopalappa, T.; Wong, S.S.; Qian, H.R.; et al. Genomic characterization of intrinsic and acquired resistance to cetuximab in colorectal cancer patients. Sci. Rep. 2019, 9, 15365. [Google Scholar] [CrossRef]
- Slattery, M.L.; Wolff, R.K.; Lundgreen, A. A pathway approach to evaluating the association between the CHIEF pathway and risk of colorectal cancer. Carcinogenesis 2015, 36, 49–59. [Google Scholar] [CrossRef]
- Salvatore, L.; Calegari, M.A.; Loupakis, F.; Fassan, M.; Di Stefano, B.; Bensi, M.; Bria, E.; Tortora, G. PTEN in Colorectal Cancer: Shedding Light on Its Role as Predictor and Target. Cancers 2019, 11, 1765. [Google Scholar] [CrossRef]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef]
- Negri, F.V.; Bozzetti, C.; Lagrasta, C.A.; Crafa, P.; Bonasoni, M.P.; Camisa, R.; Pedrazzi, G.; Ardizzoni, A. PTEN status in advanced colorectal cancer treated with cetuximab. Br. J. Cancer 2010, 102, 162–164. [Google Scholar] [CrossRef]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015, 2, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Chocry, M.; Leloup, L.; Kovacic, H. Reversion of resistance to oxaliplatin by inhibition of p38 MAPK in colorectal cancer cell lines: Involvement of the calpain/Nox1 pathway. Oncotarget 2017, 8, 103710–103730. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Mullany, L.E.; Sakoda, L.; Samowitz, W.S.; Wolff, R.K.; Stevens, J.R.; Herrick, J.S. The NF-kappaBsignalling pathway in colorectal cancer: Associations between dysregulated gene and miRNA expression. J. Cancer Res. Clin. Oncol. 2018, 144, 269–283. [Google Scholar] [CrossRef] [PubMed]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef]
- He, L.; Zhu, H.; Zhou, S.; Wu, T.; Wu, H.; Yang, H.; Mao, H.; SekharKathera, C.; Janardhan, A.; Edick, A.M.; et al. Wnt pathway is involved in 5-FU drug resistance of colorectal cancer cells. Exp. Mol. Med. 2018, 50, 101. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, P.G.; Tirro, E.; Pennisi, M.S.; Massimino, M.; Stella, S.; Romano, C.; Manzella, L. The Insulin/IGF System in Colorectal Cancer Development and Resistance to Therapy. Front. Oncol. 2015, 5, 230. [Google Scholar] [CrossRef]
- Abbruzzese, C.; Diodoro, M.G.; Sperduti, I.; Mileo, A.M.; Pattaro, G.; De Salvo, L.; Cosimelli, M.; Perrotti, N.; Paggi, M.G. Detection of phosphorylated insulin receptor in colorectal adenoma and adenocarcinoma: Implications for prognosis and clinical outcome. J. Cell. Physiol. 2015, 230, 562–567. [Google Scholar] [CrossRef]
- Esposito, D.L.; Aru, F.; Lattanzio, R.; Morgano, A.; Abbondanza, M.; Malekzadeh, R.; Bishehsari, F.; Valanzano, R.; Russo, A.; Piantelli, M.; et al. The insulin receptor substrate 1 (IRS1) in intestinal epithelial differentiation and in colorectal cancer. PLoS ONE 2012, 7, e36190. [Google Scholar] [CrossRef]
- Day, E.; Poulogiannis, G.; McCaughan, F.; Mulholland, S.; Arends, M.J.; Ibrahim, A.E.; Dear, P.H. IRS2 is a candidate driver oncogene on 13q34 in colorectal cancer. Int. J. Exp. Pathol. 2013, 94, 203–211. [Google Scholar] [CrossRef]
- Weber, M.M.; Fottner, C.; Liu, S.B.; Jung, M.C.; Engelhardt, D.; Baretton, G.B. Overexpression of the insulin-like growth factor I receptor in human colon carcinomas. Cancer 2002, 95, 2086–2095. [Google Scholar] [CrossRef]
- Lahm, H.; Amstad, P.; Wyniger, J.; Yilmaz, A.; Fischer, J.R.; Schreyer, M.; Givel, J.C. Blockade of the insulin-like growth-factor-I receptor inhibits growth of human colorectal cancer cells: Evidence of a functional IGF-II-mediated autocrine loop. Int. J. Cancer 1994, 58, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Sekharam, M.; Zhao, H.; Sun, M.; Fang, Q.; Zhang, Q.; Yuan, Z.; Dan, H.C.; Boulware, D.; Cheng, J.Q.; Coppola, D. Insulin-like growth factor 1 receptor enhances invasion and induces resistance to apoptosis of colon cancer cells through the Akt/Bcl-x(L) pathway. Cancer Res. 2003, 63, 7708–7716. [Google Scholar] [PubMed]
- Shen, K.; Cui, D.; Sun, L.; Lu, Y.; Han, M.; Liu, J. Inhibition of IGF-IR increases chemosensitivity in human colorectal cancer cells through MRP-2 promoter suppression. J. Cell. Biochem. 2012, 113, 2086–2097. [Google Scholar] [CrossRef] [PubMed]
- L’Heureux, A.; Wieland, D.R.; Weng, C.H.; Chen, Y.H.; Lin, C.H.; Lin, T.H.; Weng, C.H. Association Between Thyroid Disorders and Colorectal Cancer Risk in Adult Patients in Taiwan. JAMA Netw. Open 2019, 2, e193755. [Google Scholar] [CrossRef]
- Dentice, M.; Antonini, D.; Salvatore, D. Type 3 deiodinase and solid tumors: An intriguing pair. Expert Opin. Ther. Targets 2013, 17, 1369–1379. [Google Scholar] [CrossRef]
- Krashin, E.; Piekielko-Witkowska, A.; Ellis, M.; Ashur-Fabian, O. Thyroid Hormones and Cancer: A Comprehensive Review of Preclinical and Clinical Studies. Front. Endocrinol. 2019, 10, 59. [Google Scholar] [CrossRef]
- Dentice, M.; Marsili, A.; Zavacki, A.; Larsen, P.R.; Salvatore, D. The deiodinases and the control of intracellular thyroid hormone signaling during cellular differentiation. Biochim. Biophys. Acta 2013, 1830, 3937–3945. [Google Scholar] [CrossRef]
- Dentice, M.; Luongo, C.; Ambrosio, R.; Sibilio, A.; Casillo, A.; Iaccarino, A.; Troncone, G.; Fenzi, G.; Larsen, P.R.; Salvatore, D. beta-Cateninregulatesdeiodinaselevels and thyroidhormonesignaling in colon cancer cells. Gastroenterology 2012, 143, 1037–1047. [Google Scholar] [CrossRef]
- Catalano, V.; Dentice, M.; Ambrosio, R.; Luongo, C.; Carollo, R.; Benfante, A.; Todaro, M.; Stassi, G.; Salvatore, D. Activated Thyroid Hormone Promotes Differentiation and Chemotherapeutic Sensitization of Colorectal Cancer Stem Cells by Regulating Wnt and BMP4 Signaling. Cancer Res. 2016, 76, 1237–1244. [Google Scholar] [CrossRef]
- Yen, P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [CrossRef]
- Zhu, L.; Tian, G.; Yang, Q.; De, G.; Zhang, Z.; Wang, Y.; Nie, H.; Zhang, Y.; Yang, X.; Li, J. Thyroid hormone receptor beta1 suppresses proliferation and migration by inhibiting PI3K/Akt signaling in human colorectal cancer cells. Oncol. Rep. 2016, 36, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Uchuya-Castillo, J.; Aznar, N.; Frau, C.; Martinez, P.; Le Neve, C.; Marisa, L.; Penalva, L.O.F.; Laurent-Puig, P.; Puisieux, A.; Scoazec, J.Y.; et al. Increased expression of the thyroid hormone nuclear receptor TRalpha1 characterizes intestinal tumors with high Wnt activity. Oncotarget 2018, 9, 30979–30996. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yuan, S.; Tao, F.; Zhang, X.; Zhang, Y.; Sun, X.; Wu, D. Role of Wnt/beta-Catenin Signaling in the Chemoresistance Modulation of Colorectal Cancer. BioMed Res. Int. 2020, 2020, 9390878. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, K.; Maeda, K.; Saigo, C.; Kito, Y.; Yoshida, K.; Takeuchi, T. Adiponectin and Intelectin-1: Important Adipokine Players in Obesity-Related Colorectal Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 866. [Google Scholar] [CrossRef] [PubMed]
- Al-Shibli, S.M.; Harun, N.; Ashour, A.E.; MohdKasmuri, M.H.B.; Mizan, S. Expression of leptin and leptin receptors in colorectal cancer-an immunohistochemical study. PeerJ 2019, 7, e7624. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Tian, Y.; Gong, H.; Guo, S.; Luo, C. Oncogenic role and therapeutic target of leptin signaling in colorectal cancer. Expert Opin. Ther. Targets 2014, 18, 961–971. [Google Scholar] [CrossRef]
- Bartucci, M.; Svensson, S.; Ricci-Vitiani, L.; Dattilo, R.; Biffoni, M.; Signore, M.; Ferla, R.; De Maria, R.; Surmacz, E. Obesity hormone leptin induces growth and interferes with the cytotoxic effects of 5-fluorouracil in colorectal tumor stem cells. Endocr.-Relat. Cancer 2010, 17, 823–833. [Google Scholar] [CrossRef]
- Wang, J.J.; Chong, Q.Y.; Sun, X.B.; You, M.L.; Pandey, V.; Chen, Y.J.; Zhuang, Q.S.; Liu, D.X.; Ma, L.; Wu, Z.S.; et al. AutocrinehGH stimulates oncogenicity, epithelial-mesenchymal transition and cancer stem cell-like behavior in human colorectal carcinoma. Oncotarget 2017, 8, 103900–103918. [Google Scholar] [CrossRef]
- Bogazzi, F.; Ultimieri, F.; Raggi, F.; Russo, D.; Vanacore, R.; Guida, C.; Brogioni, S.; Cosci, C.; Gasperi, M.; Bartalena, L.; et al. Growth hormone inhibits apoptosis in human colonic cancer cell lines: Antagonistic effects of peroxisome proliferator activated receptor-gamma ligands. Endocrinology 2004, 145, 3353–3362. [Google Scholar] [CrossRef]
- Pothoulakis, C.; Torre-Rojas, M.; Duran-Padilla, M.A.; Gevorkian, J.; Zoras, O.; Chrysos, E.; Chalkiadakis, G.; Baritaki, S. CRHR2/Ucn2 signaling is a novel regulator of miR-7/YY1/Fas circuitry contributing to reversal of colorectal cancer cell resistance to Fas-mediated apoptosis. Int. J. Cancer 2018, 142, 334–346. [Google Scholar] [CrossRef]
- Williams, C.; DiLeo, A.; Niv, Y.; Gustafsson, J.A. Estrogen receptor beta as target for colorectal cancer prevention. Cancer Lett. 2016, 372, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Jacenik, D.; Beswick, E.J.; Krajewska, W.M.; Prossnitz, E.R. G protein-coupled estrogen receptor in colon function, immune regulation and carcinogenesis. World J. Gastroenterol. 2019, 25, 4092–4104. [Google Scholar] [CrossRef] [PubMed]
- Pierdominici, M.; Maselli, A.; Varano, B.; Barbati, C.; Cesaro, P.; Spada, C.; Zullo, A.; Lorenzetti, R.; Rosati, M.; Rainaldi, G.; et al. Linking estrogen receptor beta expression with inflammatory bowel disease activity. Oncotarget 2015, 6, 40443–40451. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, F.; Ryan, E.J.; Doherty, G.; Winter, D.C.; Sheahan, K. Estrogen receptors and their implications in colorectal carcinogenesis. Front. Oncol. 2015, 5, 19. [Google Scholar] [CrossRef]
- Elbanna, H.G.; Ebrahim, M.A.; Abbas, A.M.; Zalata, K.; Hashim, M.A. Potential value of estrogen receptor beta expression in colorectal carcinoma: Interaction with apoptotic index. J. Gastrointest. Cancer 2012, 43, 56–62. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Kraus, R.J.; Farrell, M.L.; Jordan, V.C.; Mertz, J.E. Estrogen-related receptor alpha1 transcriptional activities are regulated in part via the ErbB2/HER2 signaling pathway. Mol. Cancer Res. 2007, 5, 71–85. [Google Scholar] [CrossRef]
- Chang, C.Y.; Kazmin, D.; Jasper, J.S.; Kunder, R.; Zuercher, W.J.; McDonnell, D.P. The metabolic regulator ERRalpha, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell 2011, 20, 500–510. [Google Scholar] [CrossRef][Green Version]
- Kallen, J.; Schlaeppi, J.M.; Bitsch, F.; Filipuzzi, I.; Schilb, A.; Riou, V.; Graham, A.; Strauss, A.; Geiser, M.; Fournier, B. Evidence for ligand-independent transcriptional activation of the human estrogen-related receptor alpha (ERRalpha): Crystal structure of ERRalpha ligand binding domain in complex with peroxisome proliferator-activated receptor coactivator-1alpha. J. Biol. Chem. 2004, 279, 49330–49337. [Google Scholar] [CrossRef]
- Vanacker, J.M.; Pettersson, K.; Gustafsson, J.A.; Laudet, V. Transcriptional targets shared by estrogen receptor- related receptors (ERRs) and estrogen receptor (ER) alpha, but not by ERbeta. EMBO J. 1999, 18, 4270–4279. [Google Scholar] [CrossRef]
- Zhou, S.; Xia, H.; Xu, H.; Tang, Q.; Nie, Y.; Gong, Q.Y.; Bi, F. ERRα suppression enhances the cytotoxicity of the MEK inhibitor trametinib against colon cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 1–14. [Google Scholar] [CrossRef]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Wang, Y.L. Editorial: MicroRNAs as New Players in Endocrinology. Front. Endocrinol. 2018, 9, 459. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Ye, G.; Nadeem, L.; Fu, G.; Yang, B.B.; Honarparvar, E.; Dunk, C.; Lye, S.; Peng, C. MicroRNA-378a-5p promotes trophoblast cell survival, migration and invasion by targeting Nodal. J. Cell Sci. 2012, 125, 3124–3132. [Google Scholar] [CrossRef]
- Gao, W.L.; Liu, M.; Yang, Y.; Yang, H.; Liao, Q.; Bai, Y.; Li, Y.X.; Li, D.; Peng, C.; Wang, Y.L. The imprinted H19 gene regulates human placental trophoblast cell proliferation via encoding miR-675 that targets Nodal Modulator 1 (NOMO1). RNA Biol. 2012, 9, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Duarte, R.J.; Cazares-Ordonez, V.; Avila-Chavez, E. The microRNA biogenesis machinery: Regulation by steroid hormones and alterations in cancer. Rev. Investig. Clin. 2014, 66, 460–464. [Google Scholar]
- Guo, S.T.; Jiang, C.C.; Wang, G.P.; Li, Y.P.; Wang, C.Y.; Guo, X.Y.; Yang, R.H.; Feng, Y.; Wang, F.H.; Tseng, H.Y.; et al. MicroRNA-497 targets insulin-like growth factor 1 receptor and has a tumour suppressive role in human colorectal cancer. Oncogene 2013, 32, 1910–1920. [Google Scholar] [CrossRef]
- Qian, X.; Yu, J.; Yin, Y.; He, J.; Wang, L.; Li, Q.; Zhang, L.Q.; Li, C.Y.; Shi, Z.M.; Xu, Q.; et al. MicroRNA-143 inhibits tumor growth and angiogenesis and sensitizes chemosensitivity to oxaliplatin in colorectal cancers. Cell Cycle 2013, 12, 1385–1394. [Google Scholar] [CrossRef]
- Liu, N.; Li, J.; Zhao, Z.; Han, J.; Jiang, T.; Chen, Y.; Hou, N.; Huang, C. MicroRNA-302a enhances 5-fluorouracil-induced cell death in human colon cancer cells. Oncol. Rep. 2017, 37, 631–639. [Google Scholar] [CrossRef]
- Afshar, S.; Najafi, R.; SedighiPashaki, A.; Sharifi, M.; Nikzad, S.; Gholami, M.H.; Khoshghadam, A.; Amini, R.; Karimi, J.; Saidijam, M. MiR-185 enhances radiosensitivity of colorectal cancer cells by targeting IGF1R and IGF2. Biomed. Pharmacother. 2018, 106, 763–769. [Google Scholar] [CrossRef]
- Liwak, U.; Thakor, N.; Jordan, L.E.; Roy, R.; Lewis, S.M.; Pardo, O.E.; Seckl, M.; Holcik, M. Tumor suppressor PDCD4 represses internal ribosome entry site-mediated translation of antiapoptotic proteins and is regulated by S6 kinase 2. Mol. Cell. Biol. 2012, 32, 1818–1829. [Google Scholar] [CrossRef]
- Jansen, A.P.; Camalier, C.E.; Stark, C.; Colburn, N.H. Characterization of programmed cell death 4 in multiple human cancers reveals a novel enhancer of drug sensitivity. Mol. Cancer Ther. 2004, 3, 103–110. [Google Scholar] [PubMed]
- Zhao, J.; Cao, J.; Zhou, L.; Du, Y.; Zhang, X.; Yang, B.; Gao, Y.; Wang, Y.; Ma, N.; Yang, W. MiR-1260b inhibitor enhances the chemosensitivity of colorectal cancer cells to fluorouracil by targeting PDCD4/IGF1. Oncol. Lett. 2018, 16, 5131–5139. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, L.H.; Jacobsen, A.B.; Frankel, L.B.; Wen, J.; Krogh, A.; Lund, A.H. MicroRNA-145 targets YES and STAT1 in colon cancer cells. PLoS ONE 2010, 5, e8836. [Google Scholar] [CrossRef]
- Shi, B.; Sepp-Lorenzino, L.; Prisco, M.; Linsley, P.; de Angelis, T.; Baserga, R. Micro RNA 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells. J. Biol. Chem. 2007, 282, 32582–32590. [Google Scholar] [CrossRef]
- Schetter, A.J.; Okayama, H.; Harris, C.C. The role of microRNAs in colorectal cancer. Cancer J. 2012, 18, 244–252. [Google Scholar] [CrossRef]
- Wu, L.; Shi, B.; Huang, K.; Fan, G. MicroRNA-128 suppresses cell growth and metastasis in colorectal carcinoma by targeting IRS1. Oncol. Rep. 2015, 34, 2797–2805. [Google Scholar] [CrossRef]
- Pu, J.; Bai, D.; Yang, X.; Lu, X.; Xu, L.; Lu, J. Adrenaline promotes cell proliferation and increases chemoresistance in colon cancer HT29 cells through induction of miR-155. Biochem. Biophys. Res. Commun. 2012, 428, 210–215. [Google Scholar] [CrossRef]
- Yao, H.; Duan, Z.; Wang, M.; Awonuga, A.O.; Rappolee, D.; Xie, Y. Adrenaline induces chemoresistance in HT-29 colon adenocarcinoma cells. Cancer Genet. Cytogenet. 2009, 190, 81–87. [Google Scholar] [CrossRef]
- Kerr, L.R.; Hundal, R.; Silva, W.A.; Emerman, J.T.; Weinberg, J. Effects of social housing condition on chemotherapeutic efficacy in a Shionogi carcinoma (SC115) mouse tumor model: Influences of temporal factors, tumor size, and tumor growth rate. Psychosom. Med. 2001, 63, 973–984. [Google Scholar] [CrossRef]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Melichar, B.; Nash, M.A.; Lenzi, R.; Platsoucas, C.D.; Freedman, R.S. Expression of costimulatory molecules CD80 and CD86 and their receptors CD28, CTLA-4 on malignant ascites CD3+ tumour-infiltrating lymphocytes (TIL) from patients with ovarian and other types of peritoneal carcinomatosis. Clin. Exp. Immunol. 2000, 119, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Strater, J.; Hinz, U.; Hasel, C.; Bhanot, U.; Mechtersheimer, G.; Lehnert, T.; Moller, P. Impaired CD95 expression predisposes for recurrence in curatively resected colon carcinoma: Clinical evidence for immunoselection and CD95L mediated control of minimal residual disease. Gut 2005, 54, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, K. Role of apoptosis resistance in immune evasion and metastasis of colorectal cancer. World J. Gastrointest. Oncol. 2010, 2, 399–406. [Google Scholar] [CrossRef]
- Cheng, W.F.; Lee, C.N.; Chang, M.C.; Su, Y.N.; Chen, C.A.; Hsieh, C.Y. Antigen-specific CD8+ T lymphocytes generated from a DNA vaccine control tumors through the Fas-FasL pathway. Mol. Ther. 2005, 12, 960–968. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Huerta-Yepez, S.; Law, I.K.; Baay-Guzman, G.J.; Tirado-Rodriguez, B.; Hoffman, J.M.; Iliopoulos, D.; Hommes, D.W.; Verspaget, H.W.; Chang, L.; et al. Diminished expression of CRHR2 in human colon cancer promotes tumor growth and EMT via persistent IL-6/Stat3 signaling. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 610–630. [Google Scholar] [CrossRef]
- Zhang, N.; Li, X.; Wu, C.W.; Dong, Y.; Cai, M.; Mok, M.T.; Wang, H.; Chen, J.; Ng, S.S.; Chen, M.; et al. microRNA-7 is a novel inhibitor of YY1 contributing to colorectal tumorigenesis. Oncogene 2013, 32, 5078–5088. [Google Scholar] [CrossRef]
- Garnier, A.; Vykoukal, J.; Hubertus, J.; Alt, E.; von Schweinitz, D.; Kappler, R.; Berger, M.; Ilmer, M. Targeting the neurokinin-1 receptor inhibits growth of human colon cancer cells. Int. J. Oncol. 2015, 47, 151–160. [Google Scholar] [CrossRef]
- Lee, Y.S.; Chin, Y.T.; Yang, Y.S.H.; Wei, P.L.; Wu, H.C.; Shih, A.; Lu, Y.T.; Pedersen, J.Z.; Incerpi, S.; Liu, L.F.; et al. The combination of tetraiodothyroacetic acid and cetuximab inhibits cell proliferation in colorectal cancers with different K-ras status. Steroids 2016, 111, 63–70. [Google Scholar] [CrossRef]
- Garcia-Aranda, M.; Redondo, M. Targeting Receptor Kinases in Colorectal Cancer. Cancers 2019, 11, 433. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.L.; Liu, S.T.; Wang, Y.W.; Lin, W.S.; Huang, S.M. Amiodarone promotes cancer cell death through elevated truncated SRSF3 and downregulation of miR-224. Oncotarget 2018, 9, 13390–13406. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Huang, L.; Chen, J.; Lin, M.; Li, W.; Lu, B.; Rutnam, Z.J.; Iwamoto, A.; Wang, Z.; Yang, X.; et al. Gambogic acid inhibits growth, induces apoptosis, and overcomes drug resistance in human colorectal cancer cells. Int. J. Oncol. 2015, 47, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Agnihotri, N. Piperlongumine, a piper alkaloid targets Ras/PI3K/Akt/mTOR signaling axis to inhibit tumor cell growth and proliferation in DMH/DSS induced experimental colon cancer. Biomed. Pharmacother. 2019, 109, 1462–1477. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. microRNA Therapeutics in Cancer—An Emerging Concept. EBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
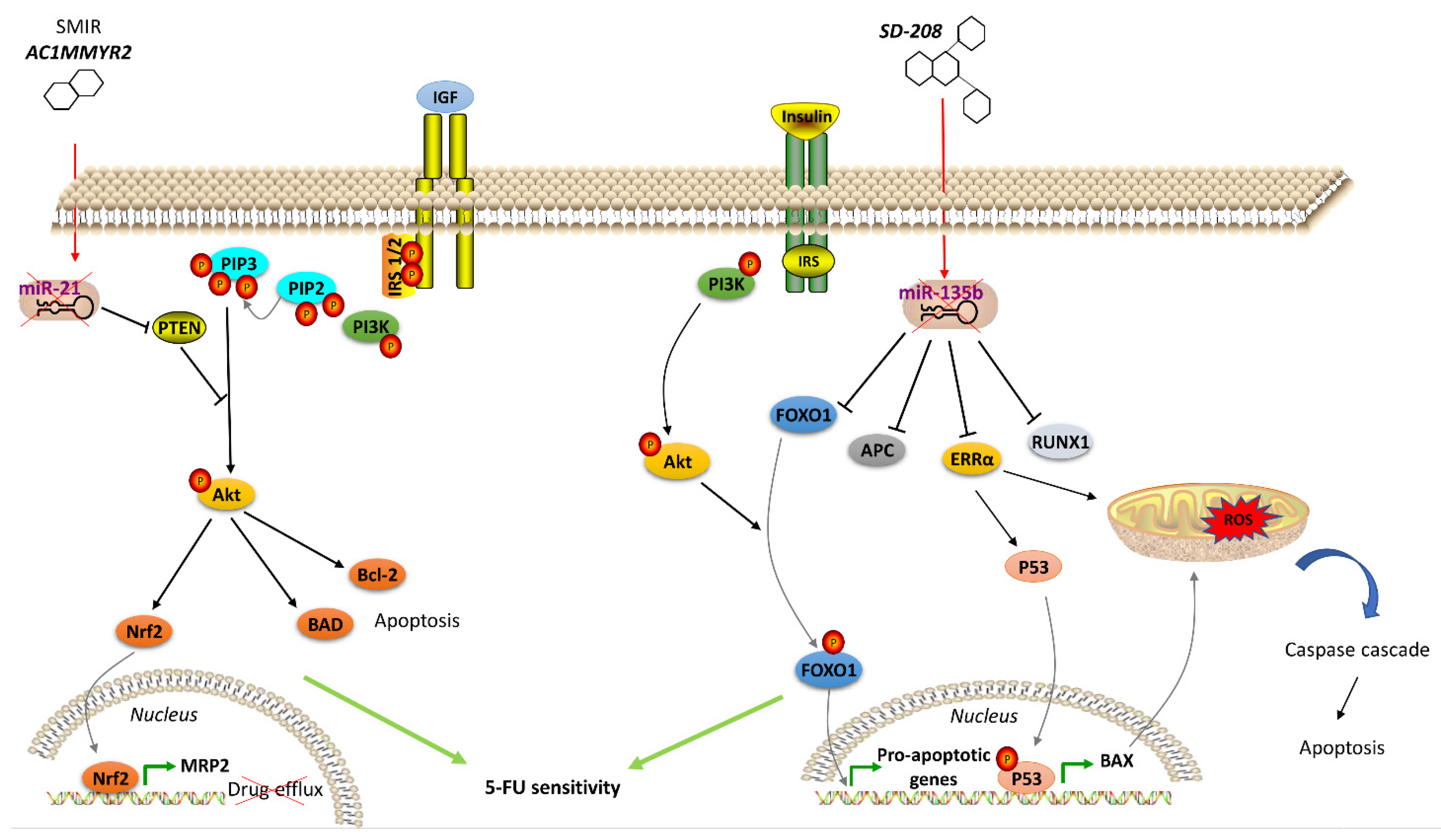
- Shi, Z.; Zhang, J.; Qian, X.; Han, L.; Zhang, K.; Chen, L.; Liu, J.; Ren, Y.; Yang, M.; Zhang, A.; et al. AC1MMYR2, an inhibitor of dicer-mediated biogenesis of Oncomir miR-21, reverses epithelial-mesenchymal transition and suppresses tumor growth and progression. Cancer Res. 2013, 73, 5519–5531. [Google Scholar] [CrossRef] [PubMed]
- Akbari, A.; Ghahremani, M.H.; Mobini, G.R.; Abastabar, M.; Akhtari, J.; Bolhassani, M.; Heidari, M. Down-regulation of miR-135b in colon adenocarcinoma induced by a TGF-beta receptor I kinase inhibitor (SD-208). Iran. J. Basic Med. Sci. 2015, 18, 856–861. [Google Scholar]
- Qin, Y.; Li, L.; Wang, F.; Zhou, X.; Liu, Y.; Yin, Y.; Qi, X. Knockdown of Mir-135b Sensitizes Colorectal Cancer Cells to Oxaliplatin-Induced Apoptosis Through Increase of FOXO1. Cell. Physiol. Biochem. 2018, 48, 1628–1637. [Google Scholar] [CrossRef]
- Valeri, N.; Braconi, C.; Gasparini, P.; Murgia, C.; Lampis, A.; Paulus-Hock, V.; Hart, J.R.; Ueno, L.; Grivennikov, S.I.; Lovat, F.; et al. MicroRNA-135b promotes cancer progression by acting as a downstream effector of oncogenic pathways in colon cancer. Cancer Cell 2014, 25, 469–483. [Google Scholar] [CrossRef]
- Ostermann, A.L.; Wunderlich, C.M.; Schneiders, L.; Vogt, M.C.; Woeste, M.A.; Belgardt, B.F.; Niessen, C.M.; Martiny, B.; Schauss, A.C.; Frommolt, P.; et al. Intestinal insulin/IGF1 signalling through FoxO1 regulates epithelial integrity and susceptibility to colon cancer. Nat. Metab. 2019, 1, 371–389. [Google Scholar] [CrossRef]
- Xin, Z.; Jiang, S.; Jiang, P.; Yan, X.; Fan, C.; Di, S.; Wu, G.; Yang, Y.; Reiter, R.J.; Ji, G. Melatonin as a treatment for gastrointestinal cancer: A review. J. Pineal Res. 2015, 58, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Sakatani, A.; Sonohara, F.; Goel, A. Melatonin-mediated downregulation of thymidylate synthase as a novel mechanism for overcoming 5-fluorouracil associated chemoresistance in colorectal cancer cells. Carcinogenesis 2019, 40, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, R.; Van Roosbroeck, K.; Calin, G.A. Cell-to-cell communication: microRNAs as hormones. Mol. Oncol. 2017, 11, 1673–1686. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.J.; Huang, X.H.; Zheng, C.C.; Yin, X.F.; Li, B.; He, Q.Y. Liensinine perchlorate inhibits colorectal cancer tumorigenesis by inducing mitochondrial dysfunction and apoptosis. Food Funct. 2018, 9, 5536–5546. [Google Scholar] [CrossRef]
- Zheng, Z.; Xu, L.; Zhang, S.; Li, W.; Tou, F.; He, Q.; Rao, J.; Shen, Q. Peiminine inhibits colorectal cancer cell proliferation by inducing apoptosis and autophagy and modulating key metabolic pathways. Oncotarget 2017, 8, 47619–47631. [Google Scholar] [CrossRef]
- Zheng, X.; Shi, C.; Shujun, F.; Yukun, L.; Juan, Z.; Hui, L.; Ying, Z.; Xi, Z. Function and mechanism of microRNA-20a in colorectal cancer. Exp. Ther. Med. 2020, 19, 1605–1616. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| miRNA | Expression | Targets | Resistance | Reference |
|---|---|---|---|---|
| miR-21 | ↑ | PDCD4 TIAM1 PTEN TGFBR2 CDC25A | 5-FU | [120] |
| miR-143 | ↓ | NF-kB Bcl-2; IGF-1R | 5-FU | [94,121] |
| miR-365; miR-224 | ↓ | - | 5-FU | [122] |
| miR-34a | ↓ | SIRT1 | 5-FU | [123] |
| miR-494 | ↓ | DPYD | 5-FU | [124] |
| miR-302 | ↓ | IGF-1R | 5-FU | [133] |
| miR-10b | ↑ | BIM | 5-FU | [134] |
| miR-375-3p | ↓ | TYMS | 5-FU | [135] |
| miR-20a | ↑ | BNIP2 | 5-FU OxaliplatinTeniposide | [136] |
| miR-320 | ↓ | FOXM1 | 5-FU Oxaliplatin | [127] |
| miR-34a | ↑ | - | Oxaliplatin | [129] |
| let-7g | ↓ | Cyclin D Myc E2F | S-1 | [125] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massaro, C.; Safadeh, E.; Sgueglia, G.; Stunnenberg, H.G.; Altucci, L.; Dell’Aversana, C. MicroRNA-Assisted Hormone Cell Signaling in Colorectal Cancer Resistance. Cells 2021, 10, 39. https://doi.org/10.3390/cells10010039
Massaro C, Safadeh E, Sgueglia G, Stunnenberg HG, Altucci L, Dell’Aversana C. MicroRNA-Assisted Hormone Cell Signaling in Colorectal Cancer Resistance. Cells. 2021; 10(1):39. https://doi.org/10.3390/cells10010039
Chicago/Turabian StyleMassaro, Crescenzo, Elham Safadeh, Giulia Sgueglia, Hendrik G. Stunnenberg, Lucia Altucci, and Carmela Dell’Aversana. 2021. "MicroRNA-Assisted Hormone Cell Signaling in Colorectal Cancer Resistance" Cells 10, no. 1: 39. https://doi.org/10.3390/cells10010039
APA StyleMassaro, C., Safadeh, E., Sgueglia, G., Stunnenberg, H. G., Altucci, L., & Dell’Aversana, C. (2021). MicroRNA-Assisted Hormone Cell Signaling in Colorectal Cancer Resistance. Cells, 10(1), 39. https://doi.org/10.3390/cells10010039