PE_PGRS33, an Important Virulence Factor of Mycobacterium tuberculosis and Potential Target of Host Humoral Immune Response

 ,
, 
Abstract
1. Introduction
2. Vaccines in TB: Current Status
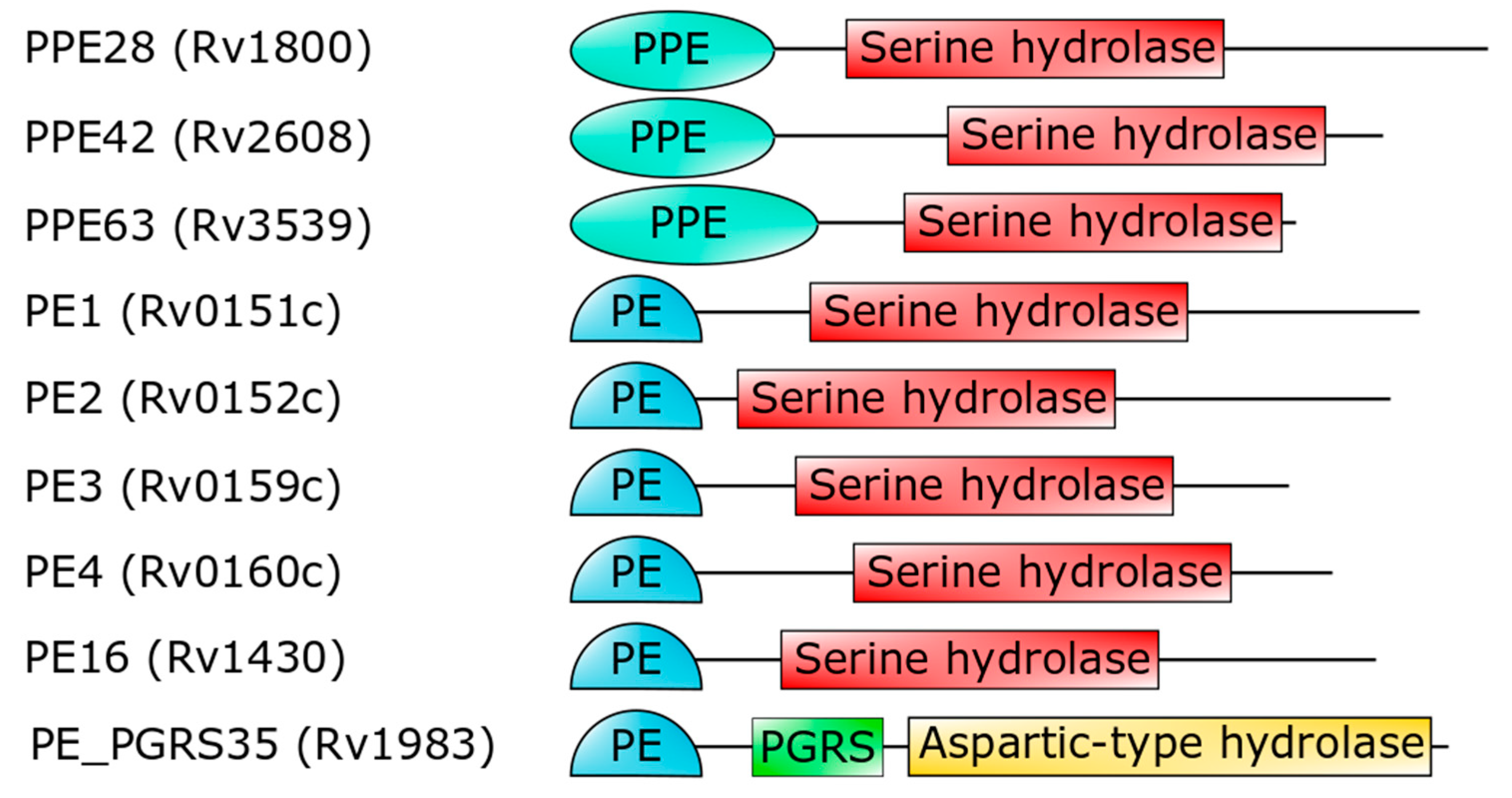
3. The PE_PGRS Family of Surface Proteins
4. Structural Features of PE_PGRS33, the PE_PGRS Prototype
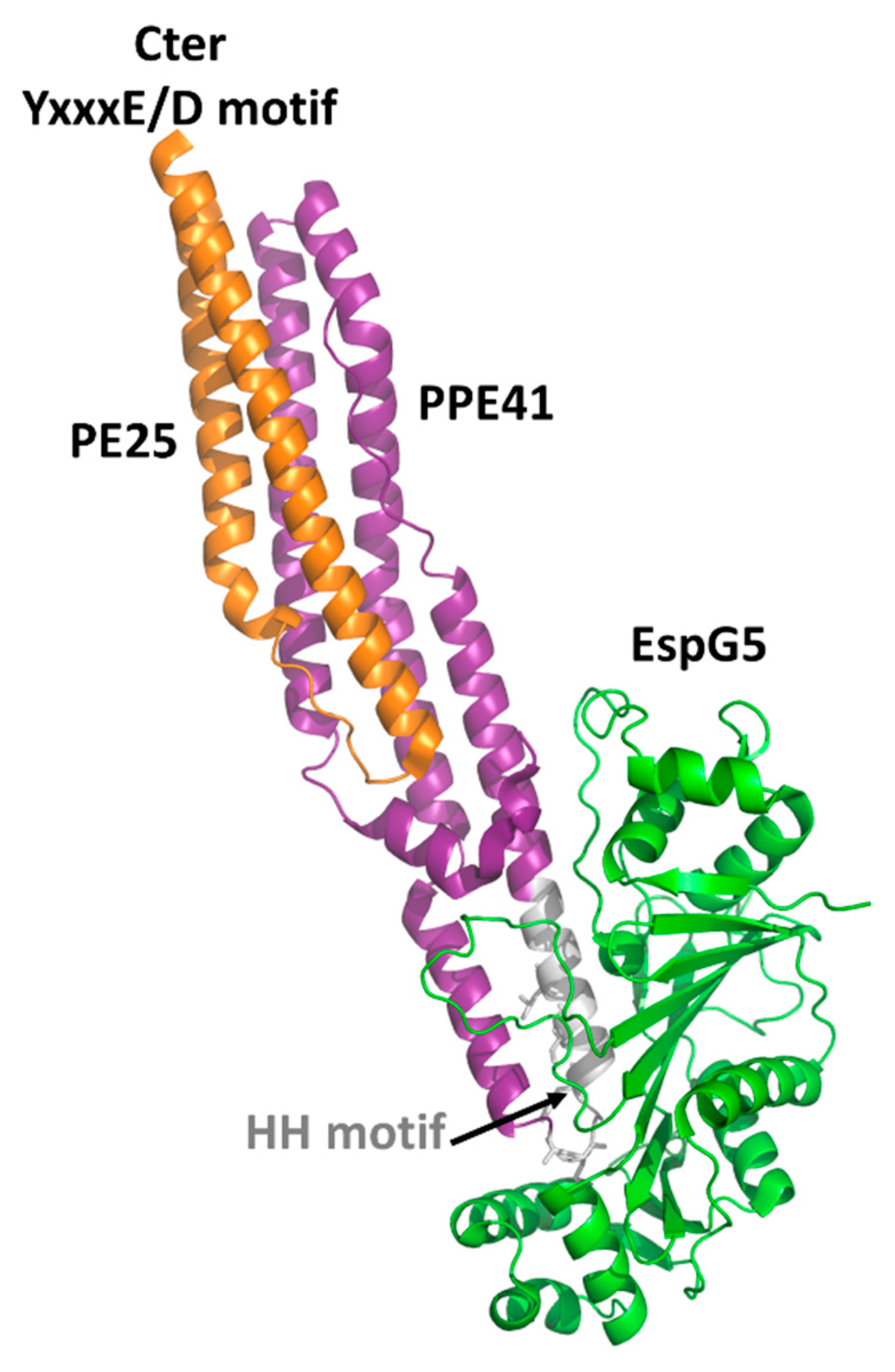
4.1. The PE Domain
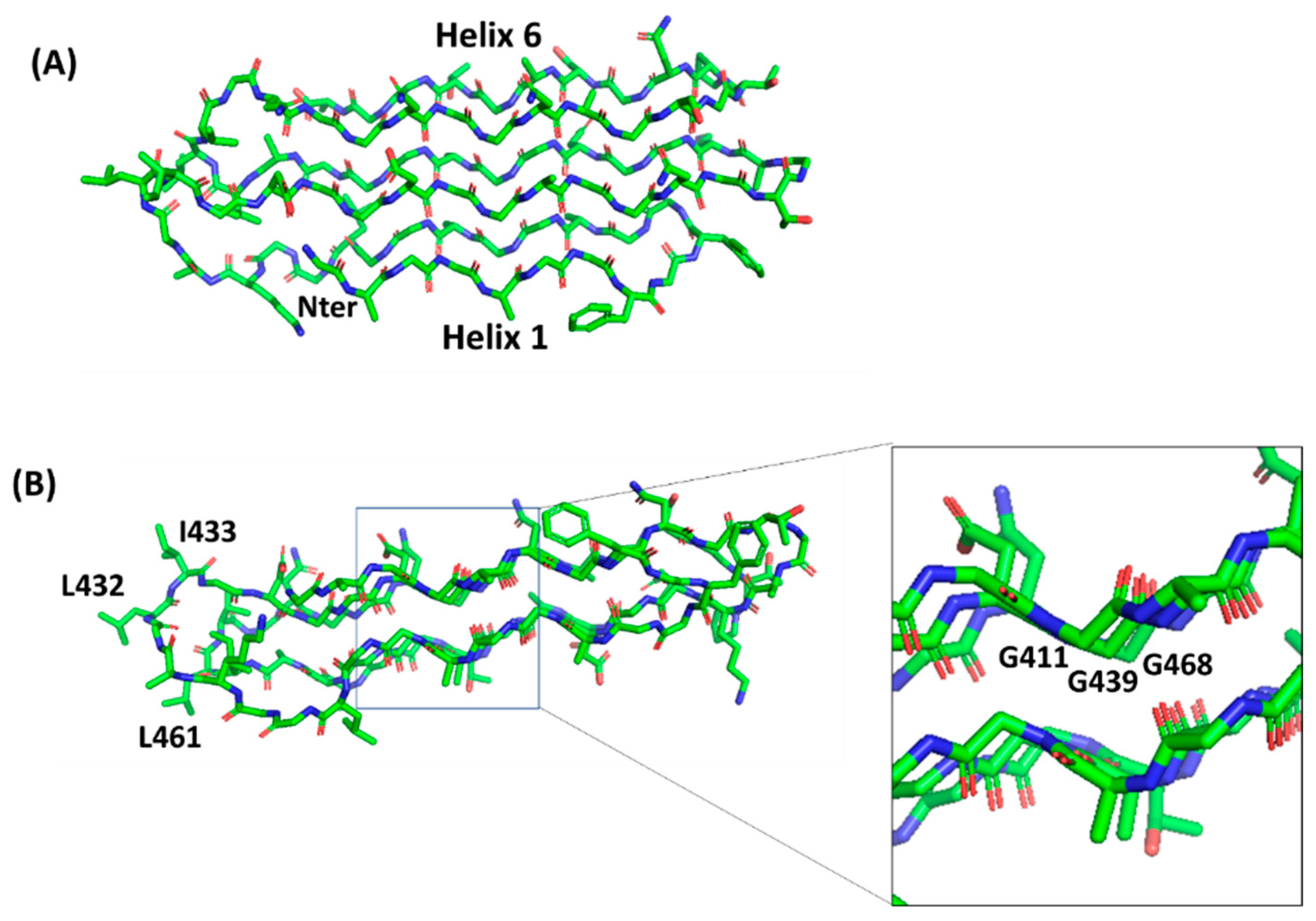
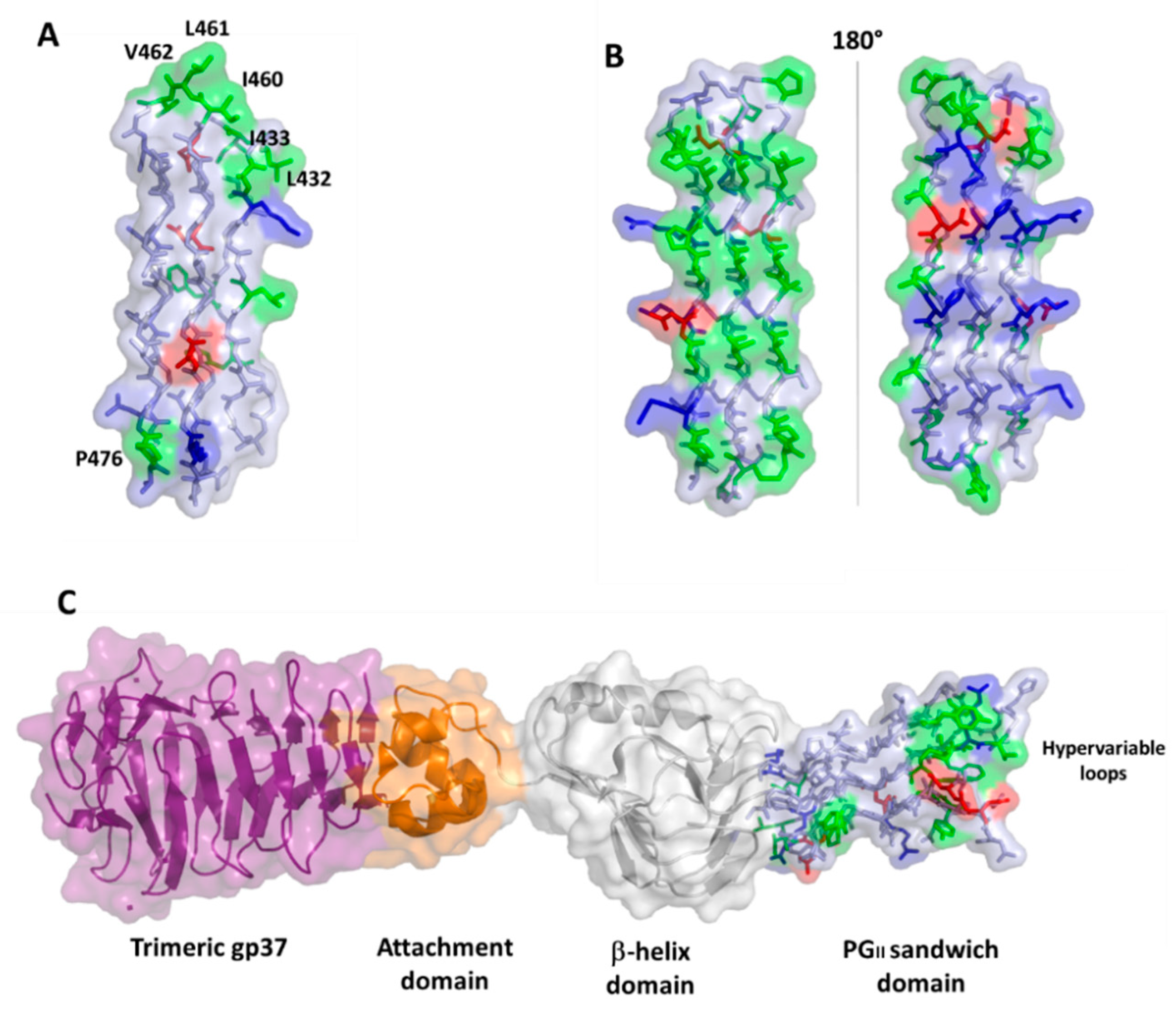
4.2. PGRS Domain Contains Multiple PGII Modules
5. PE_PGRS33 as a Promising Target of the Humoral Response
6. PGRS PGII Sandwich Structure Tolerates Polymorphism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allele * | Polymorphism * | Amino Acid Variation | Position (aa) | PGII | Clade/ Lineage | Functional Notes | Clinical Notes |
|---|---|---|---|---|---|---|---|
| allele 1 (allele 11) [72] | Wild type | - | - | - | 4 LAM/ Haarlem | Associated with cavitary TB | |
| allele 26 (allele 3) [72] | S4 D14 I4 | −28 Gly X +1 Gly Ala | 239 338–498 413 | 3, 4, 5 | 1 EAI | Impaired entry in macrophages compared to wt allele. Hypervirulent in mice | Associated with noncavitary TB |
| allele 45 1 (allele 5) [72] | S22 a S4 I4 | Gly → Ser - +1 Gly Ala | 233 239 413 | 2, 4 | Animal lineage | - | |
| allele 461 (allele 6) [72] | D2 S4 I4 | −4 Gly X 717 +1 Gly Ala | 140–163 239 413 | 1, 4 | 1 EAI | - | |
| allele 471 (allele 18) [72] | S4 D19 b I4 | - −3 Gly X +1 Gly Ala | 239 257–270 413 | 2, 4 | 3 Delhi/CAS | - | |
| allele 481 (ΔG184-G213) [74] | D20 c | −30 aa | 184–213 | 1, 2 | - | Weaker immunostimulatory activity (reduced TNF-α) | - |
| allele 491 (ΔL237-G327) [74] | D21 d | −91 aa | 237–327 | 2, 3 | - | - | |
| allele 501 (ΔL372-A403) [74] | D22 e | −32 aa | 372–403 | 4 | - | - | |
| allele 511 (ΔG196-D243) [74] | D22 f | −48 aa | 196–243 | 1, 2 | - | - | |
| allele 521 (SA440) [74] | ID2 g | −36 aa +11 aa | 236–271 272 | 2 | - | - | |
| allele 22 (SA455) [74] | I1 S4 I4 | +2 Gly Ala Gly - +1 Gly Ala | 199 239 413 | 1, 4 | - | Associated with cavitary TB | |
| allele 40 | D8 | −47 aa | 213–260 | 2 | - | - | Associated with non-cavitary TB |
| allele 531 (33260) [71] | D23 h | −238 aa | 260–498 | 2, 3, 4, 5 | - | Sufficient to complement the Mtb Δ33 mutant in the in vitro macrophage invasion assay | - |
| allele 541 (Group 2) [106] | S4 I6 i | - + Ala Gly | 408 | 4 | - | - | Associated with extrapulmonary TB |
7. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TB | Tuberculosis |
| WHO | World Health Organization |
| Mtb | Mycobacterium tuberculosis |
| BCG | Bacille Calmette and Guérin |
| DC | Dendritic cell |
| IFN | Interferon |
| TLR | Toll-like receptor |
| PDB | Protein Data Bank |
| Ms | Mycobacterium smegmatis |
| CDR | Complementarity-determining region |
| ER | Endoplasmic reticulum |
| rmsd | root mean square deviations |
| IL | Interleukin |
| TNF-α | Tumor necrosis factor–α |
References
- Harding, E. WHO global progress report on tuberculosis elimination. Lancet Respir. Med. 2020, 8, 19. [Google Scholar] [CrossRef]
- World Health Organization. The End TB Strategy. Available online: https://www.who.int/tb/strategy/en/ (accessed on 14 January 2021).
- Barry, C.E., 3rd; Boshoff, H.I.; Dartois, V.; Dick, T.; Ehrt, S.; Flynn, J.; Schnappinger, D.; Wilkinson, R.J.; Young, D. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009, 7, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Delogu, G.; Sali, M.; Fadda, G. The biology of mycobacterium tuberculosis infection. Mediterr. J. Hematol. Infect. Dis. 2013, 5, e2013070. [Google Scholar] [CrossRef] [PubMed]
- Nikitushkin, V.D.; Demina, G.R.; Shleeva, M.O.; Guryanova, S.V.; Ruggiero, A.; Berisio, R.; Kaprelyants, A.S. A product of RpfB and RipA joint enzymatic action promotes the resuscitation of dormant mycobacteria. Febs. J. 2015, 282, 2500–2511. [Google Scholar] [CrossRef]
- Ruggiero, A.; Squeglia, F.; Romano, M.; Vitagliano, L.; De Simone, A.; Berisio, R. The structure of Resuscitation promoting factor B from M. tuberculosis reveals unexpected ubiquitin-like domains. Biochim. Biophys. Acta 2016, 1860, 445–451. [Google Scholar] [CrossRef]
- Squeglia, F.; Romano, M.; Ruggiero, A.; Vitagliano, L.; De Simone, A.; Berisio, R. Carbohydrate recognition by RpfB from Mycobacterium tuberculosis unveiled by crystallographic and molecular dynamics analyses. Biophys. J. 2013, 104, 2530–2539. [Google Scholar] [CrossRef]
- Squeglia, F.; Ruggiero, A.; Berisio, R. Chemistry of Peptidoglycan in Mycobacterium tuberculosis Life Cycle: An off-the-wall Balance of Synthesis and Degradation. Chemistry 2018, 24, 2533–2546. [Google Scholar] [CrossRef]
- Squeglia, F.; Ruggiero, A.; Berisio, R. Exit from mycobacterial dormancy: A structural perspective. Curr. Med. Chem. 2015, 22, 1698–1709. [Google Scholar] [CrossRef]
- Gengenbacher, M.; Kaufmann, S.H. Mycobacterium tuberculosis: Success through dormancy. Fems. Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef]
- Orme, I.M.; Basaraba, R.J. The formation of the granuloma in tuberculosis infection. Semin. Immunol. 2014, 26, 601–609. [Google Scholar] [CrossRef]
- Dheda, K.; Gumbo, T.; Maartens, G.; Dooley, K.E.; Murray, M.; Furin, J.; Nardell, E.A.; Warren, R.M.; Lancet Respiratory Medicine Drug-Resistant Tuberculosis Commission Group. The Lancet Respiratory Medicine Commission: 2019 update: Epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant and incurable tuberculosis. Lancet Respir. Med. 2019, 7, 820–826. [Google Scholar] [CrossRef]
- Tran, V.; Liu, J.; Behr, M.A. BCG Vaccines. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Mangtani, P.; Abubakar, I.; Ariti, C.; Beynon, R.; Pimpin, L.; Fine, P.E.M.; Rodrigues, L.C.; Smith, P.G.; Lipman, M.; Whiting, P.F.; et al. Protection by BCG Vaccine Against Tuberculosis: A Systematic Review of Randomized Controlled Trials. Clin. Infect. Dis. 2014, 58, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Dodd, P.J.; White, R.G. The potential impact of BCG vaccine supply shortages on global paediatric tuberculosis mortality. BMC Med. 2016, 14, 138. [Google Scholar] [CrossRef] [PubMed]
- Nguipdop-Djomo, P.; Heldal, E.; Rodrigues, L.C.; Abubakar, I.; Mangtani, P. BCG vaccination: A long-lasting protection against tuberculosis?—Authors’ reply. Lancet Infect. Dis. 2016, 16, 408–409. [Google Scholar] [CrossRef]
- Schrager, L.K.; Harris, R.C.; Vekemans, J. Research and development of new tuberculosis vaccines: A review. F1000Research 2018, 7, 1732. [Google Scholar] [CrossRef]
- Delogu, G.; Manganelli, R.; Brennan, M.J. Critical research concepts in tuberculosis vaccine development. Clin. Microbiol. Infect. 2014, 20, 59–65. [Google Scholar] [CrossRef][Green Version]
- Schrager, L.K.; Vekemens, J.; Drager, N.; Lewinsohn, D.M.; Olesen, O.F. The status of tuberculosis vaccine development. Lancet Infect. Dis. 2020, 20, e28–e37. [Google Scholar] [CrossRef]
- Mendez-Samperio, P. Global Efforts in the Development of Vaccines for Tuberculosis: Requirements for Improved Vaccines Against Mycobacterium tuberculosis. Scand. J. Immunol. 2016, 84, 204–210. [Google Scholar] [CrossRef]
- Gong, W.; Liang, Y.; Wu, X. The current status, challenges, and future developments of new tuberculosis vaccines. Hum. Vaccin. Immunother. 2018, 14, 1697–1716. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E.; Weiner, J.; von Reyn, C.F. Novel approaches to tuberculosis vaccine development. Int. J. Infect. Dis. 2017, 56, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Brazier, B.; McShane, H. Towards new TB vaccines. Semin. Immunopathol. 2020, 42, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Soundarya, J.S.V.; Ranganathan, U.D.; Tripathy, S.P. Current trends in tuberculosis vaccine. Med. J. Armed Forces India 2019, 75, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Samperio, P. Development of tuberculosis vaccines in clinical trials: Current status. Scand. J. Immunol. 2018, 88, e12710. [Google Scholar] [CrossRef] [PubMed]
- Delogu, G.; Provvedi, R.; Sali, M.; Manganelli, R. Mycobacterium tuberculosis virulence: Insights and impact on vaccine development. Future Microbiol. 2015, 10, 1177–1194. [Google Scholar] [CrossRef]
- Tameris, M.D.; Hatherill, M.; Landry, B.S.; Scriba, T.J.; Snowden, M.A.; Lockhart, S.; Shea, J.E.; McClain, J.B.; Hussey, G.D.; Hanekom, W.A.; et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: A randomised, placebo-controlled phase 2b trial. Lancet 2013, 381, 1021–1028. [Google Scholar] [CrossRef]
- Boer, M.C.; van Meijgaarden, K.E.; Goletti, D.; Vanini, V.; Prins, C.; Ottenhoff, T.H.; Joosten, S.A. KLRG1 and PD-1 expression are increased on T-cells following tuberculosis-treatment and identify cells with different proliferative capacities in BCG-vaccinated adults. Tuberc. (Edinb) 2016, 97, 163–171. [Google Scholar] [CrossRef]
- Fletcher, H.A.; Schrager, L. TB vaccine development and the End TB Strategy: Importance and current status. Trans. R Soc. Trop. Med. Hyg. 2016, 110, 212–218. [Google Scholar] [CrossRef]
- Lindenstrom, T.; Knudsen, N.P.; Agger, E.M.; Andersen, P. Control of chronic mycobacterium tuberculosis infection by CD4 KLRG1- IL-2-secreting central memory cells. J. Immunol. 2013, 190, 6311–6319. [Google Scholar] [CrossRef]
- Andersen, P.; Scriba, T.J. Moving tuberculosis vaccines from theory to practice. Nat. Rev. Immunol. 2019, 19, 550–562. [Google Scholar] [CrossRef]
- Comas, I.; Chakravartti, J.; Small, P.M.; Galagan, J.; Niemann, S.; Kremer, K.; Ernst, J.D.; Gagneux, S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 2010, 42, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Coscolla, M.; Copin, R.; Sutherland, J.; Gehre, F.; de Jong, B.; Owolabi, O.; Mbayo, G.; Giardina, F.; Ernst, J.D.; Gagneux, S.M. tuberculosis T Cell Epitope Analysis Reveals Paucity of Antigenic Variation and Identifies Rare Variable TB Antigens. Cell Host Microbe 2015, 18, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Moguche, A.O.; Musvosvi, M.; Penn-Nicholson, A.; Plumlee, C.R.; Mearns, H.; Geldenhuys, H.; Smit, E.; Abrahams, D.; Rozot, V.; Dintwe, O.; et al. Antigen Availability Shapes T Cell Differentiation and Function during Tuberculosis. Cell Host Microbe 2017, 21, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuizen, N.E.; Kulkarni, P.S.; Shaligram, U.; Cotton, M.F.; Rentsch, C.A.; Eisele, B.; Grode, L.; Kaufmann, S.H.E. The Recombinant Bacille Calmette-Guerin Vaccine VPM1002: Ready for Clinical Efficacy Testing. Front. Immunol. 2017, 8, 1147. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo-Asensio, J.; Marinova, D.; Martin, C.; Aguilo, N. MTBVAC: Attenuating the Human Pathogen of Tuberculosis (TB) Toward a Promising Vaccine against the TB Epidemic. Front. Immunol. 2017, 8, 1803. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Chen, Q.F.; Cui, X.H.; Yu, Y.; Li, Y.P. Mycobacterium vaccae vaccine to prevent tuberculosis in high risk people: A meta-analysis. J. Infect. 2010, 60, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Chen, Q.F.; Li, Y.P.; Wu, S.M. Mycobacterium vaccae as adjuvant therapy to anti-tuberculosis chemotherapy in never-treated tuberculosis patients: A meta-analysis. PLoS ONE 2011, 6, e23826. [Google Scholar] [CrossRef]
- Saqib, M.; Khatri, R.; Singh, B.; Gupta, A.; Kumar, A.; Bhaskar, S. Mycobacterium indicus pranii as a booster vaccine enhances BCG induced immunity and confers higher protection in animal models of tuberculosis. Tuberc. (Edinb) 2016, 101, 164–173. [Google Scholar] [CrossRef] [PubMed]
- von Reyn, C.F.; Lahey, T.; Arbeit, R.D.; Landry, B.; Kailani, L.; Adams, L.V.; Haynes, B.C.; Mackenzie, T.; Wieland-Alter, W.; Connor, R.I.; et al. Safety and immunogenicity of an inactivated whole cell tuberculosis vaccine booster in adults primed with BCG: A randomized, controlled trial of DAR-901. PLoS ONE 2017, 12, e0175215. [Google Scholar] [CrossRef]
- Vilaplana, C.; Montane, E.; Pinto, S.; Barriocanal, A.M.; Domenech, G.; Torres, F.; Cardona, P.J.; Costa, J. Double-blind, randomized, placebo-controlled Phase I Clinical Trial of the therapeutical antituberculous vaccine RUTI. Vaccine 2010, 28, 1106–1116. [Google Scholar] [CrossRef]
- Day, C.L.; Tameris, M.; Mansoor, N.; van Rooyen, M.; de Kock, M.; Geldenhuys, H.; Erasmus, M.; Makhethe, L.; Hughes, E.J.; Gelderbloem, S.; et al. Induction and regulation of T-cell immunity by the novel tuberculosis vaccine M72/AS01 in South African adults. Am. J. Respir. Crit. Care Med. 2013, 188, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Luabeya, A.K.; Kagina, B.M.; Tameris, M.D.; Geldenhuys, H.; Hoff, S.T.; Shi, Z.; Kromann, I.; Hatherill, M.; Mahomed, H.; Hanekom, W.A.; et al. First-in-human trial of the post-exposure tuberculosis vaccine H56:IC31 in Mycobacterium tuberculosis infected and non-infected healthy adults. Vaccine 2015, 33, 4130–4140. [Google Scholar] [CrossRef] [PubMed]
- Penn-Nicholson, A.; Tameris, M.; Smit, E.; Day, T.A.; Musvosvi, M.; Jayashankar, L.; Vergara, J.; Mabwe, S.; Bilek, N.; Geldenhuys, H.; et al. Safety and immunogenicity of the novel tuberculosis vaccine ID93 + GLA-SE in BCG-vaccinated healthy adults in South Africa: A randomised, double-blind, placebo-controlled phase 1 trial. Lancet Respir. Med. 2018, 6, 287–298. [Google Scholar] [CrossRef]
- Tkachuk, A.P.; Gushchin, V.A.; Potapov, V.D.; Demidenko, A.V.; Lunin, V.G.; Gintsburg, A.L. Multi-subunit BCG booster vaccine GamTBvac: Assessment of immunogenicity and protective efficacy in murine and guinea pig TB models. PLoS ONE 2017, 12, e0176784. [Google Scholar] [CrossRef]
- Nemes, E.; Hesseling, A.C.; Tameris, M.; Mauff, K.; Downing, K.; Mulenga, H.; Rose, P.; van der Zalm, M.; Mbaba, S.; Van As, D.; et al. Safety and Immunogenicity of Newborn MVA85A Vaccination and Selective, Delayed Bacille Calmette-Guerin for Infants of Human Immunodeficiency Virus-Infected Mothers: A Phase 2 Randomized, Controlled Trial. Clin. Infect. Dis. 2018, 66, 554–563. [Google Scholar] [CrossRef]
- Smaill, F.; Xing, Z. Human type 5 adenovirus-based tuberculosis vaccine: Is the respiratory route of delivery the future? Expert Rev. Vaccines 2014, 13, 927–930. [Google Scholar] [CrossRef]
- Chen, T.; Blanc, C.; Eder, A.Z.; Prados-Rosales, R.; Souza, A.C.; Kim, R.S.; Glatman-Freedman, A.; Joe, M.; Bai, Y.; Lowary, T.L.; et al. Association of Human Antibodies to Arabinomannan With Enhanced Mycobacterial Opsonophagocytosis and Intracellular Growth Reduction. J. Infect. Dis. 2016, 214, 300–310. [Google Scholar] [CrossRef]
- Chen, T.; Blanc, C.; Liu, Y.; Ishida, E.; Singer, S.; Xu, J.; Joe, M.; Jenny-Avital, E.R.; Chan, J.; Lowary, T.L.; et al. Capsular glycan recognition provides antibody-mediated immunity against tuberculosis. J. Clin. Investig. 2020, 130, 1808–1822. [Google Scholar] [CrossRef]
- Prados-Rosales, R.; Carreno, L.; Cheng, T.; Blanc, C.; Weinrick, B.; Malek, A.; Lowary, T.L.; Baena, A.; Joe, M.; Bai, Y.; et al. Enhanced control of Mycobacterium tuberculosis extrapulmonary dissemination in mice by an arabinomannan-protein conjugate vaccine. PLoS Pathog. 2017, 13, e1006250. [Google Scholar] [CrossRef]
- Prados-Rosales, R.; Carreno, L.J.; Weinrick, B.; Batista-Gonzalez, A.; Glatman-Freedman, A.; Xu, J.; Chan, J.; Jacobs, W.R., Jr.; Porcelli, S.A.; Casadevall, A. The Type of Growth Medium Affects the Presence of a Mycobacterial Capsule and Is Associated with Differences in Protective Efficacy of BCG Vaccination Against Mycobacterium tuberculosis. J. Infect. Dis. 2016, 214, 426–437. [Google Scholar] [CrossRef]
- Lu, L.L.; Chung, A.W.; Rosebrock, T.R.; Ghebremichael, M.; Yu, W.H.; Grace, P.S.; Schoen, M.K.; Tafesse, F.; Martin, C.; Leung, V.; et al. A Functional Role for Antibodies in Tuberculosis. Cell 2016, 167, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.L.; Smith, M.T.; Yu, K.K.Q.; Luedemann, C.; Suscovich, T.J.; Grace, P.S.; Cain, A.; Yu, W.H.; McKitrick, T.R.; Lauffenburger, D.; et al. IFN-gamma-independent immune markers of Mycobacterium tuberculosis exposure. Nat. Med. 2019, 25, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A. Antibodies to Mycobacterium tuberculosis. N. Engl. J. Med. 2017, 376, 283–285. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Mohareer, K.; Tundup, S.; Hasnain, S.E. Transcriptional regulation of Mycobacterium tuberculosis PE/PPE genes: A molecular switch to virulence? J. Mol. Microbiol. Biotechnol. 2011, 21, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Meena, L.S. An overview to understand the role of PE_PGRS family proteins in Mycobacterium tuberculosis H37 Rv and their potential as new drug targets. Biotechnol. Appl. Biochem. 2015, 62, 145–153. [Google Scholar] [CrossRef]
- Vallecillo, A.J.; Espitia, C. Expression of Mycobacterium tuberculosis pe_pgrs33 is repressed during stationary phase and stress conditions, and its transcription is mediated by sigma factor A. Microb. Pathog. 2009, 46, 119–127. [Google Scholar] [CrossRef]
- De Maio, F.; Berisio, R.; Manganelli, R.; Delogu, G. PE_PGRS proteins of Mycobacterium tuberculosis: A specialized molecular task force at the forefront of host-pathogen interaction. Virulence 2020, 11, 898–915. [Google Scholar] [CrossRef]
- Brennan, M.J.; Delogu, G.; Chen, Y.; Bardarov, S.; Kriakov, J.; Alavi, M.; Jacobs, W.R., Jr. Evidence that mycobacterial PE_PGRS proteins are cell surface constituents that influence interactions with other cells. Infect. Immun. 2001, 69, 7326–7333. [Google Scholar] [CrossRef]
- Copin, R.; Coscolla, M.; Seiffert, S.N.; Bothamley, G.; Sutherland, J.; Mbayo, G.; Gagneux, S.; Ernst, J.D. Sequence diversity in the pe_pgrs genes of Mycobacterium tuberculosis is independent of human T cell recognition. MBio 2014, 5. [Google Scholar] [CrossRef]
- Banu, S.; Honore, N.; Saint-Joanis, B.; Philpott, D.; Prevost, M.C.; Cole, S.T. Are the PE-PGRS proteins of Mycobacterium tuberculosis variable surface antigens? Mol. Microbiol. 2002, 44, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, A.; Delogu, G.; Colone, M.; Sali, M.; Stringaro, A.; Arancia, G.; Fadda, G.; Palu, G.; Manganelli, R. PE is a functional domain responsible for protein translocation and localization on mycobacterial cell wall. Mol. Microbiol. 2007, 66, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Chatrath, S.; Gupta, V.K.; Garg, L.C. The PGRS domain is responsible for translocation of PE_PGRS30 to cell poles while the PE and the C-terminal domains localize it to the cell wall. Febs. Lett. 2014, 588, 990–994. [Google Scholar] [CrossRef]
- Shah, S.; Briken, V. Modular Organization of the ESX-5 Secretion System in Mycobacterium tuberculosis. Front. Cell Infect. Microbiol. 2016, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, A.M.; Bestebroer, J.; Savage, N.D.; de Punder, K.; van Zon, M.; Wilson, L.; Korbee, C.J.; van der Sar, A.M.; Ottenhoff, T.H.; van der Wel, N.N.; et al. Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and inflammasome activation. J. Immunol. 2011, 187, 4744–4753. [Google Scholar] [CrossRef]
- Gey van Pittius, N.C.; Sampson, S.L.; Lee, H.; Kim, Y.; van Helden, P.D.; Warren, R.M. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol. Biol. 2006, 6, 95. [Google Scholar] [CrossRef]
- Kanji, A.; Hasan, Z.; Ali, A.; McNerney, R.; Mallard, K.; Coll, F.; Hill-Cawthorne, G.; Nair, M.; Clark, T.G.; Zaver, A.; et al. Characterization of genomic variations in SNPs of PE_PGRS genes reveals deletions and insertions in extensively drug resistant (XDR) M. tuberculosis strains from Pakistan. Int. J. Mycobacteriol. 2015, 4, 73–79. [Google Scholar] [CrossRef]
- Delogu, G.; Sanguinetti, M.; Pusceddu, C.; Bua, A.; Brennan, M.J.; Zanetti, S.; Fadda, G. PE_PGRS proteins are differentially expressed by Mycobacterium tuberculosis in host tissues. Microbes Infect. 2006, 8, 2061–2067. [Google Scholar] [CrossRef]
- Kruh, N.A.; Troudt, J.; Izzo, A.; Prenni, J.; Dobos, K.M. Portrait of a pathogen: The Mycobacterium tuberculosis proteome in vivo. PLoS ONE 2010, 5, e13938. [Google Scholar] [CrossRef]
- Palucci, I.; Camassa, S.; Cascioferro, A.; Sali, M.; Anoosheh, S.; Zumbo, A.; Minerva, M.; Iantomasi, R.; De Maio, F.; Di Sante, G.; et al. PE_PGRS33 Contributes to Mycobacterium tuberculosis Entry in Macrophages through Interaction with TLR2. PLoS ONE 2016, 11, e0150800. [Google Scholar] [CrossRef]
- Camassa, S.; Palucci, I.; Iantomasi, R.; Cubeddu, T.; Minerva, M.; De Maio, F.; Jouny, S.; Petruccioli, E.; Goletti, D.; Ria, F.; et al. Impact of pe_pgrs33 Gene Polymorphisms on Mycobacterium tuberculosis Infection and Pathogenesis. Front. Cell Infect. Microbiol. 2017, 7, 137. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.; Sharma, T.; Singh, Y.; Kohli, S.; Manjunath, P.; Singh, A.; Semmler, T.; Wieler, L.H.; Tedin, K.; Ehtesham, N.Z.; et al. The PGRS Domain of Mycobacterium tuberculosis PE_PGRS Protein Rv0297 Is Involved in Endoplasmic Reticulum Stress-Mediated Apoptosis through Toll-Like Receptor 4. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Pathak, S.K.; Banerjee, A.; Pathak, S.; Bhattacharyya, A.; Yang, Z.; Talarico, S.; Kundu, M.; Basu, J. Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-alpha. J. Biol. Chem. 2007, 282, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Bansal, K.; Elluru, S.R.; Narayana, Y.; Chaturvedi, R.; Patil, S.A.; Kaveri, S.V.; Bayry, J.; Balaji, K.N. PE_PGRS antigens of Mycobacterium tuberculosis induce maturation and activation of human dendritic cells. J. Immunol. 2010, 184, 3495–3504. [Google Scholar] [CrossRef] [PubMed]
- Sayes, F.; Pawlik, A.; Frigui, W.; Groschel, M.I.; Crommelynck, S.; Fayolle, C.; Cia, F.; Bancroft, G.J.; Bottai, D.; Leclerc, C.; et al. CD4+ T Cells Recognizing PE/PPE Antigens Directly or via Cross Reactivity Are Protective against Pulmonary Mycobacterium tuberculosis Infection. PLoS Pathog. 2016, 12, e1005770. [Google Scholar] [CrossRef]
- Espitia, C.; Laclette, J.P.; Mondragon-Palomino, M.; Amador, A.; Campuzano, J.; Martens, A.; Singh, M.; Cicero, R.; Zhang, Y.; Moreno, C. The PE-PGRS glycine-rich proteins of Mycobacterium tuberculosis: A new family of fibronectin-binding proteins? Microbiol. (Read.) 1999, 145, 3487–3495. [Google Scholar] [CrossRef]
- Koh, K.W.; Soh, S.E.; Seah, G.T. Strong antibody responses to Mycobacterium tuberculosis PE-PGRS62 protein are associated with latent and active tuberculosis. Infect. Immun. 2009, 77, 3337–3343. [Google Scholar] [CrossRef]
- Singh, K.K.; Zhang, X.; Patibandla, A.S.; Chien, P., Jr.; Laal, S. Antigens of Mycobacterium tuberculosis expressed during preclinical tuberculosis: Serological immunodominance of proteins with repetitive amino acid sequences. Infect. Immun. 2001, 69, 4185–4191. [Google Scholar] [CrossRef]
- Minerva, M.; De Maio, F.; Camassa, S.; Battah, B.; Ivana, P.; Manganelli, R.; Sanguinetti, M.; Sali, M.; Delogu, G. Evaluation of PE_PGRS33 as a potential surface target for humoral responses against Mycobacterium tuberculosis. Pathog. Dis. 2017, 75. [Google Scholar] [CrossRef]
- Gastelum-Avina, P.; Velazquez, C.; Espitia, C.; Lares-Villa, F.; Garibay-Escobar, A. A PE_PGRS33 protein of Mycobacterium tuberculosis: An ideal target for future tuberculosis vaccine design. Expert Rev. Vaccines 2015, 14, 699–711. [Google Scholar] [CrossRef]
- Cascioferro, A.; Daleke, M.H.; Ventura, M.; Dona, V.; Delogu, G.; Palu, G.; Bitter, W.; Manganelli, R. Functional dissection of the PE domain responsible for translocation of PE_PGRS33 across the mycobacterial cell wall. PLoS ONE 2011, 6, e27713. [Google Scholar] [CrossRef]
- Korotkova, N.; Freire, D.; Phan, T.H.; Ummels, R.; Creekmore, C.C.; Evans, T.J.; Wilmanns, M.; Bitter, W.; Parret, A.H.; Houben, E.N.; et al. Structure of the Mycobacterium tuberculosis type VII secretion system chaperone EspG5 in complex with PE25-PPE41 dimer. Mol. Microbiol. 2014, 94, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Ekiert, D.C.; Cox, J.S. Structure of a PE-PPE-EspG complex from Mycobacterium tuberculosis reveals molecular specificity of ESX protein secretion. Proc. Natl. Acad. Sci. USA 2014, 111, 14758–14763. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cheng, H.F.; Zhou, J.; Chan, C.Y.; Lau, K.F.; Tsui, S.K.; Au, S.W. Structural basis of the PE-PPE protein interaction in Mycobacterium tuberculosis. J. Biol. Chem. 2017, 292, 16880–16890. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.; Sawaya, M.R.; Wang, S.; Phillips, M.; Cascio, D.; Eisenberg, D. Toward the structural genomics of complexes: Crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2006, 103, 8060–8065. [Google Scholar] [CrossRef]
- Daleke, M.H.; Ummels, R.; Bawono, P.; Heringa, J.; Vandenbroucke-Grauls, C.M.; Luirink, J.; Bitter, W. General secretion signal for the mycobacterial type VII secretion pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 11342–11347. [Google Scholar] [CrossRef]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54. [Google Scholar] [CrossRef]
- Burggraaf, M.J.; Speer, A.; Meijers, A.S.; Ummels, R.; van der Sar, A.M.; Korotkov, K.V.; Bitter, W.; Kuijl, C. Type VII Secretion Substrates of Pathogenic Mycobacteria Are Processed by a Surface Protease. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Tanneeru, K.; Guruprasad, L. The PE-PPE domain in mycobacterium reveals a serine alpha/beta hydrolase fold and function: An in-silico analysis. PLoS ONE 2011, 6, e16745. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.J.; Delogu, G. The PE multigene family: A ‘molecular mantra’ for mycobacteria. Trends Microbiol. 2002, 10, 246–249. [Google Scholar] [CrossRef]
- Delogu, G.; Pusceddu, C.; Bua, A.; Fadda, G.; Brennan, M.J.; Zanetti, S. Rv1818c-encoded PE_PGRS protein of Mycobacterium tuberculosis is surface exposed and influences bacterial cell structure. Mol. Microbiol. 2004, 52, 725–733. [Google Scholar] [CrossRef]
- Bykov, S.; Asher, S. Raman studies of solution polyglycine conformations. J. Phys. Chem. B 2010, 114, 6636–6641. [Google Scholar] [CrossRef]
- Adzhubei, A.A.; Sternberg, M.J.; Makarov, A.A. Polyproline-II helix in proteins: Structure and function. J. Mol. Biol. 2013, 425, 2100–2132. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Pai, M.; Linkins, L.A. Direct oral anticoagulants for treatment of HIT: Update of Hamilton experience and literature review. Blood 2017, 130, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Pentelute, B.L.; Gates, Z.P.; Tereshko, V.; Dashnau, J.L.; Vanderkooi, J.M.; Kossiakoff, A.A.; Kent, S.B. X-ray structure of snow flea antifreeze protein determined by racemic crystallization of synthetic protein enantiomers. J. Am. Chem. Soc. 2008, 130, 9695–9701. [Google Scholar] [CrossRef] [PubMed]
- Swanson, N.A.; Cingolani, G. A Tail of Phage Adhesins. Structure 2018, 26, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Dunne, M.; Denyes, J.M.; Arndt, H.; Loessner, M.J.; Leiman, P.G.; Klumpp, J. Salmonella Phage S16 Tail Fiber Adhesin Features a Rare Polyglycine Rich Domain for Host Recognition. Structure 2018, 26, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Dheenadhayalan, V.; Delogu, G.; Brennan, M.J. Expression of the PE_PGRS 33 protein in Mycobacterium smegmatis triggers necrosis in macrophages and enhanced mycobacterial survival. Microbes Infect. 2006, 8, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Balaji, K.N.; Goyal, G.; Narayana, Y.; Srinivas, M.; Chaturvedi, R.; Mohammad, S. Apoptosis triggered by Rv1818c, a PE family gene from Mycobacterium tuberculosis is regulated by mitochondrial intermediates in T cells. Microbes Infect. 2007, 9, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Parada, C.; Acosta-Gio, E.; Espitia, C. The PGRS Domain from PE_PGRS33 of Mycobacterium tuberculosis is Target of Humoral Immune Response in Mice and Humans. Front. Immunol. 2014, 5, 236. [Google Scholar] [CrossRef] [PubMed]
- Chaitra, M.G.; Shaila, M.S.; Nayak, R. Evaluation of T-cell responses to peptides with MHC class I-binding motifs derived from PE_PGRS 33 protein of Mycobacterium tuberculosis. J. Med. Microbiol. 2007, 56, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Massire, C.; Li, H.; Cummins, L.L.; Li, F.; Jin, J.; Fan, X.; Wang, S.; Shao, L.; Zhang, S.; et al. Molecular characterization of drug-resistant Mycobacterium tuberculosis isolates circulating in China by multilocus PCR and electrospray ionization mass spectrometry. J. Clin. Microbiol. 2011, 49, 2719–2721. [Google Scholar] [CrossRef]
- Talarico, S.; Cave, M.D.; Foxman, B.; Marrs, C.F.; Zhang, L.; Bates, J.H.; Yang, Z. Association of Mycobacterium tuberculosis PE PGRS33 polymorphism with clinical and epidemiological characteristics. Tuberc. (Edinb) 2007, 87, 338–346. [Google Scholar] [CrossRef]
- Irene, C.; Fantappie, L.; Caproni, E.; Zerbini, F.; Anesi, A.; Tomasi, M.; Zanella, I.; Stupia, S.; Prete, S.; Valensin, S.; et al. Bacterial outer membrane vesicles engineered with lipidated antigens as a platform for Staphylococcus aureus vaccine. Proc. Natl. Acad. Sci. USA 2019, 116, 21780–21788. [Google Scholar] [CrossRef]
- Prados-Rosales, R.; Brown, L.; Casadevall, A.; Montalvo-Quiros, S.; Luque-Garcia, J.L. Isolation and identification of membrane vesicle-associated proteins in Gram-positive bacteria and mycobacteria. MethodsX 2014, 1, 124–129. [Google Scholar] [CrossRef]







| TB Vaccine Candidate | Antigen | Adjuvant | Clinical Phase |
|---|---|---|---|
| VPM 1002 [35] | Live attenuated Mycobacterium tuberculosis | - | 3 |
| MTBVAC [36] | Live attenuated M. tuberculosis | - | 2a |
| Vaccae [37,38] | Heat-killed M. vaccae | - | 3 |
| MIP [39] | Heat-killed M. indicus pranii | - | 3 |
| DAR-901 [40] | Heat-killed M. obuense | - | 2b |
| RUTI [41] | Cell wall fragments of M. tuberculosis | - | 2a |
| M72/AS01 [42] | Protein subunit (Rv1196 and Rv0125) | AS01 | 2b |
| H56/IC31 [43] | Protein subunit (ESAT-6, Ag85B, and Rv2660c) | IC31 | 2b |
| ID93 + GLA-SE [44] | Protein subunit (Rv2608, Rv3619, Rv3620, and Rv1813) | GLA-SE | 2a |
| GamTBvac [45] | Protein subunit (Ag85A and ESAT-6-CFP10) | CpG ODN | 1 |
| TB/Flu-04L[19] | Recombinant influenza vector expressing (Ag85A and ESAT-6) | Flu-04L | 2a |
| ChAdOx1 85A/MVA85 [46] | Recombinant simian adenovirus expressing (Ag85A) | ChAdOx1, MVA | 1 |
| Ad5Ag85A [47] | Human adenovirus serotype 5 expressing (Ag85A) | Ad5 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kramarska, E.; Squeglia, F.; De Maio, F.; Delogu, G.; Berisio, R. PE_PGRS33, an Important Virulence Factor of Mycobacterium tuberculosis and Potential Target of Host Humoral Immune Response. Cells 2021, 10, 161. https://doi.org/10.3390/cells10010161
Kramarska E, Squeglia F, De Maio F, Delogu G, Berisio R. PE_PGRS33, an Important Virulence Factor of Mycobacterium tuberculosis and Potential Target of Host Humoral Immune Response. Cells. 2021; 10(1):161. https://doi.org/10.3390/cells10010161
Chicago/Turabian StyleKramarska, Eliza, Flavia Squeglia, Flavio De Maio, Giovanni Delogu, and Rita Berisio. 2021. "PE_PGRS33, an Important Virulence Factor of Mycobacterium tuberculosis and Potential Target of Host Humoral Immune Response" Cells 10, no. 1: 161. https://doi.org/10.3390/cells10010161
APA StyleKramarska, E., Squeglia, F., De Maio, F., Delogu, G., & Berisio, R. (2021). PE_PGRS33, an Important Virulence Factor of Mycobacterium tuberculosis and Potential Target of Host Humoral Immune Response. Cells, 10(1), 161. https://doi.org/10.3390/cells10010161