Microglial Turnover in Ageing-Related Neurodegeneration: Therapeutic Avenue to Intervene in Disease Progression





Abstract
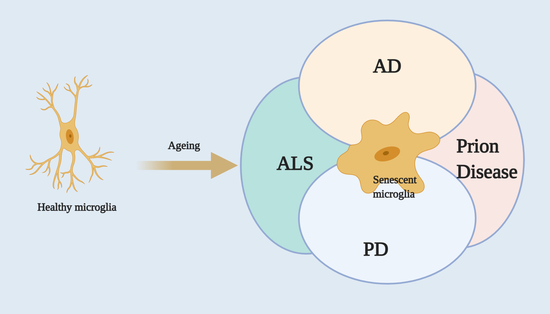
{kind=link}
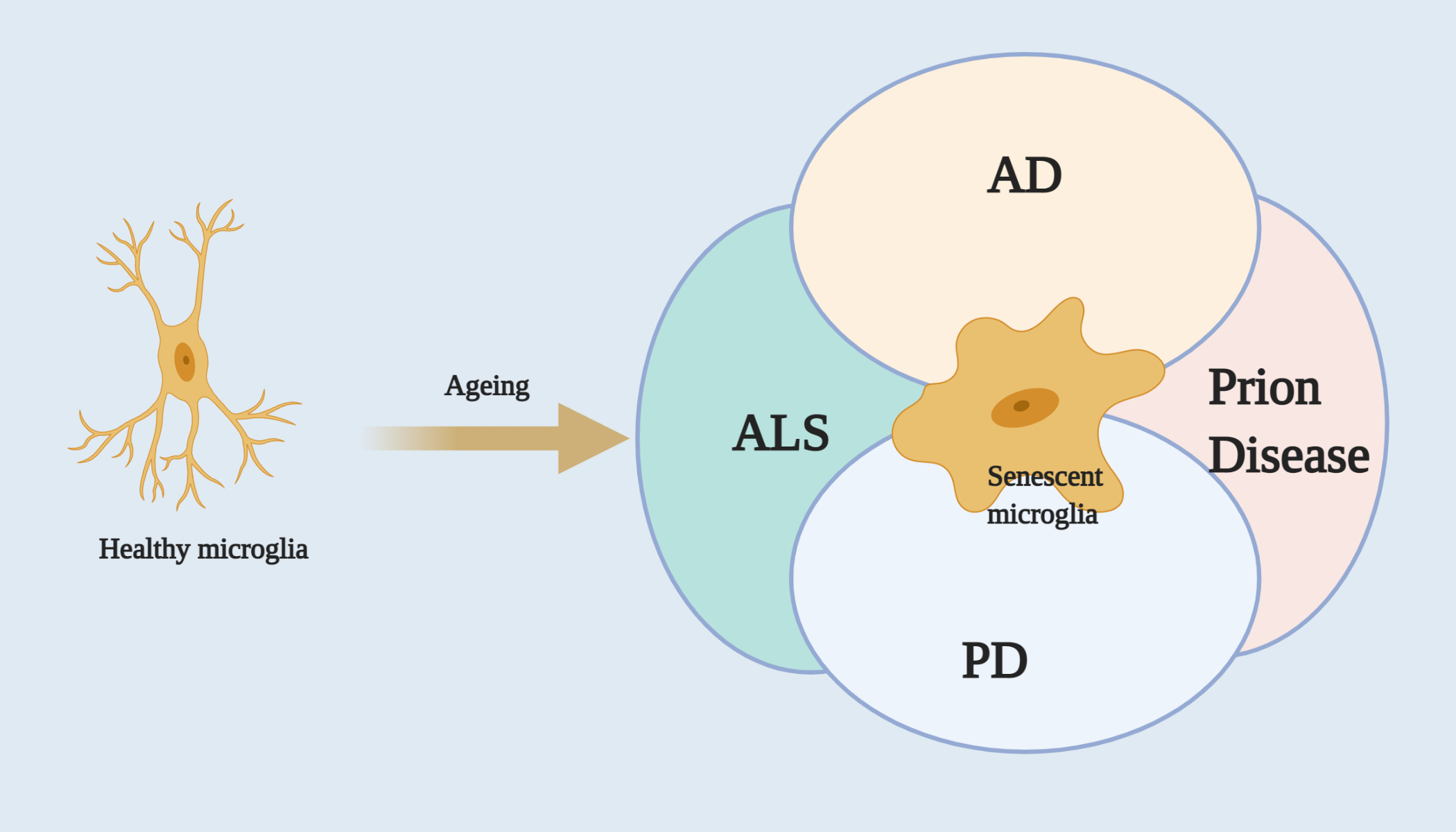
{kind=link}
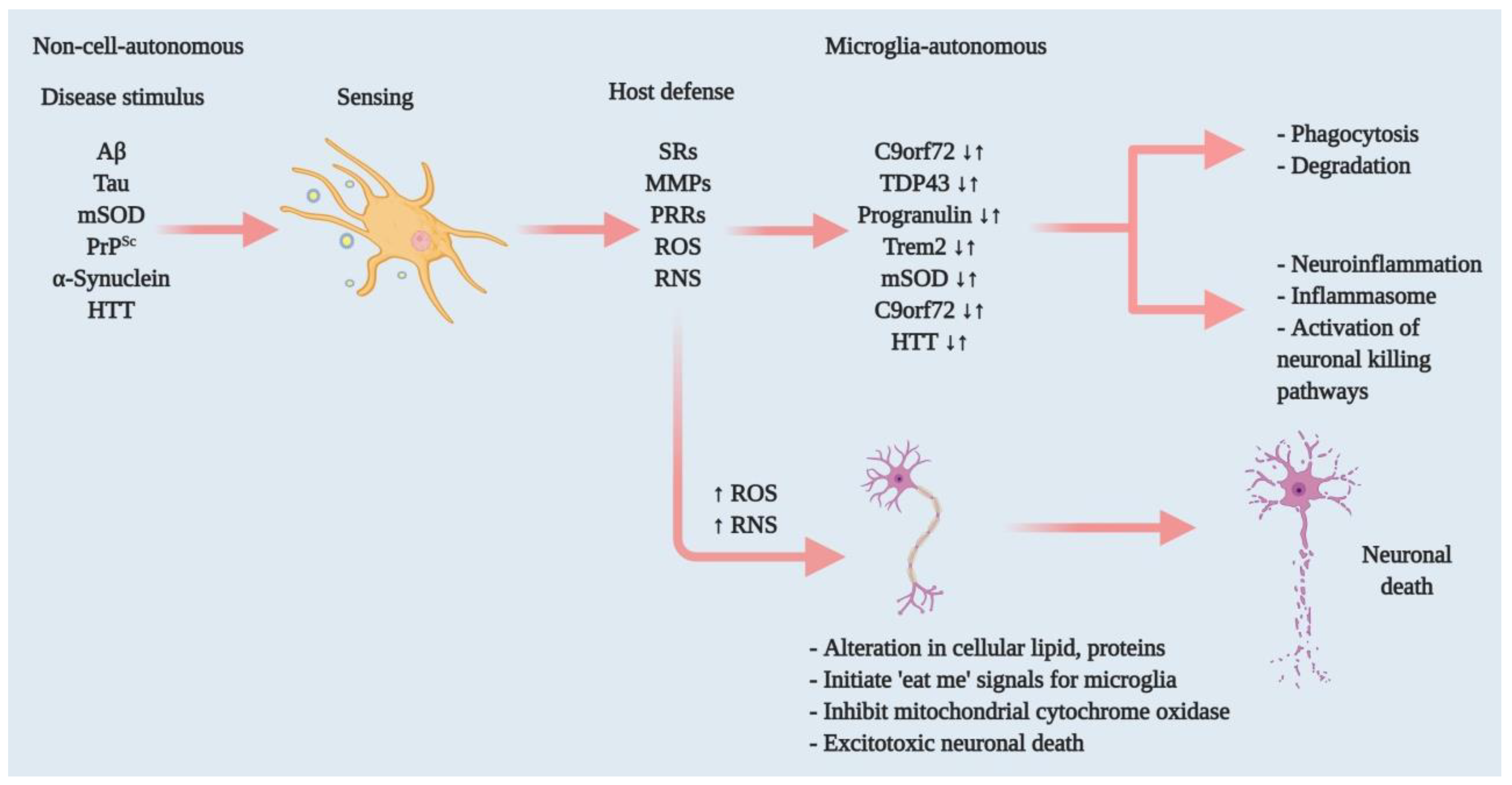
{kind=link}
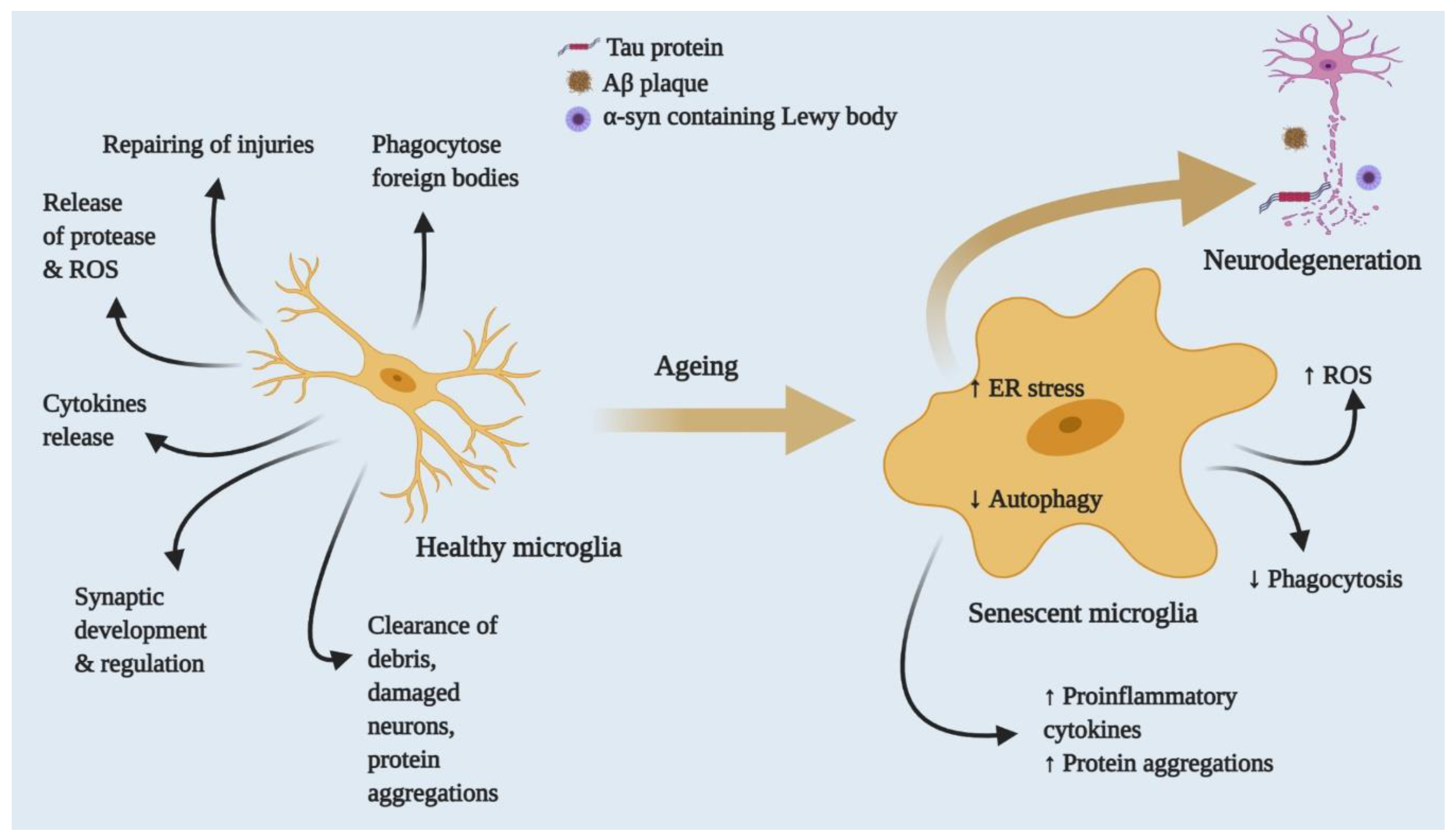{kind=link}
1. Introduction
2. Molecular and Functional Background of Microglia
3. Microglia and Ageing
4. Microglia and Neurodegenerative Diseases: Functional Relation
4.1. Alzheimer’s Disease
4.2. Parkinson’s Disease
4.3. Amyotrophic Lateral Sclerosis
4.4. Multiple Sclerosis
4.5. Huntington’s Disease
4.6. Frontotemporal Dementia
4.7. Prion Disease
5. Microglia as a Therapeutic Possibility in Neurodegenerative Diseases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Crotti, A.; Ransohoff, R.M. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 2016, 44, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015, 43, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762. [Google Scholar] [CrossRef]
- Hefendehl, J.K.; Neher, J.J.; Sühs, R.B.; Kohsaka, S.; Skodras, A.; Jucker, M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 2014, 13, 60–69. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System during Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Gómez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 2481–2493. [Google Scholar] [CrossRef]
- Füger, P.; Hefendehl, J.K.; Veeraraghavalu, K.; Wendeln, A.C.; Schlosser, C.; Obermüller, U.; Wegenast-Braun, B.M.; Neher, J.J.; Martus, P.; Kohsaka, S.; et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 2017, 20, 1371–1376. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Gordon, S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 1992, 48, 405–415. [Google Scholar] [CrossRef]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernandez-Klett, F.; Lin, G.; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; Staszewski, O.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, R.S.; Hayes, N.L. New neurons: Extraordinary evidence or extraordinary conclusion? Science 2000, 288, 771. [Google Scholar] [CrossRef]
- Rakic, P. Neurogenesis in adult primates. Prog. Brain Res. 2002, 138, 3–14. [Google Scholar] [CrossRef]
- Knuesel, I.; Chicha, L.; Britschgi, M.; Schobel, S.A.; Bodmer, M.; Hellings, J.A.; Toovey, S.; Prinssen, E.P. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 2014, 10, 643–660. [Google Scholar] [CrossRef]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- El Khoury, J. Neurodegeneration and the neuroimmune system. Nat. Med. 2010, 16, 1369–1370. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; El Khoury, J. Microglia in Health and Disease. Cold Spring Harb. Perspect. Biol. 2015, 8, a020560. [Google Scholar] [CrossRef]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Fourgeaud, L.; Través, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagórska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Bittner, T.; Jung, C.K.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 13, 411–413. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef]
- Healy, L.M.; Perron, G.; Won, S.Y.; Michell-Robinson, M.A.; Rezk, A.; Ludwin, S.K.; Moore, C.S.; Hall, J.A.; Bar-Or, A.; Antel, J.P. MerTK Is a Functional Regulator of Myelin Phagocytosis by Human Myeloid Cells. J. Immunol. 2016, 196, 3375–3384. [Google Scholar] [CrossRef]
- Pasciuto, E.; Burton, O.T.; Roca, C.P.; Lagou, V.; Rajan, W.D.; Theys, T.; Mancuso, R.; Tito, R.Y.; Kouser, L.; Callaerts-Vegh, Z.; et al. Microglia Require CD4 T Cells to Complete the Fetal-to-Adult Transition. Cell 2020, 182, 625–640. [Google Scholar] [CrossRef]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438. [Google Scholar] [CrossRef]
- Ge, Y.; Grossman, R.I.; Babb, J.S.; Rabin, M.L.; Mannon, L.J.; Kolson, D.L. Age-related total gray matter and white matter changes in normal adult brain. Part I: Volumetric MR imaging analysis. Ajnr. Am. J. Neuroradiol. 2002, 23, 1327–1333. [Google Scholar]
- Sowell, E.R.; Peterson, B.S.; Thompson, P.M.; Welcome, S.E.; Henkenius, A.L.; Toga, A.W. Mapping cortical change across the human life span. Nat. Neurosci. 2003, 6, 309–315. [Google Scholar] [CrossRef]
- Salat, D.H.; Buckner, R.L.; Snyder, A.Z.; Greve, D.N.; Desikan, R.S.; Busa, E.; Morris, J.C.; Dale, A.M.; Fischl, B. Thinning of the cerebral cortex in aging. Cerebral Cortex 2004, 14, 721–730. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef]
- Dröge, W.; Schipper, H.M. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell 2007, 6, 361–370. [Google Scholar] [CrossRef]
- Vijg, J.; Campisi, J. Puzzles, promises and a cure for ageing. Nature 2008, 454, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Bailey, W.M.; Braun, K.J.; Gensel, J.C. Age decreases macrophage IL-10 expression: Implications for functional recovery and tissue repair in spinal cord injury. Exp. Neurol. 2015, 273, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J. Gene expression profiling in fetal, aged, and Alzheimer hippocampus: A continuum of stress-related signaling. Neurochem. Res. 2004, 29, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R.W. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1329–1331. [Google Scholar] [CrossRef]
- Ott, C.; König, J.; Höhn, A.; Jung, T.; Grune, T. Macroautophagy is impaired in old murine brain tissue as well as in senescent human fibroblasts. Redox Biol. 2016, 10, 266–273. [Google Scholar] [CrossRef]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef]
- Wong, W.T. Microglial aging in the healthy CNS: Phenotypes, drivers, and rejuvenation. Front. Cell. Neurosci. 2013, 7, 22. [Google Scholar] [CrossRef]
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sánchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Koellhoffer, E.C.; McCullough, L.D.; Ritzel, R.M. Old Maids: Aging and Its Impact on Microglia Function. Int. J. Mol. Sci. 2017, 18, 769. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Wu, Z. Microglia-aging: Roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behav. Brain Res. 2009, 201, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, S.T.; Graham, D.I. Aging-associated changes in human brain. J. Neuropathol. Exp. Neurol. 1997, 56, 1269–1275. [Google Scholar] [CrossRef]
- Gray, D.A.; Woulfe, J. Lipofuscin and aging: A matter of toxic waste. Sci. Aging Knowl. Environ. Sage Ke 2005, 2005, re1. [Google Scholar] [CrossRef]
- Ahmed, Z.; Sheng, H.; Xu, Y.F.; Lin, W.L.; Innes, A.E.; Gass, J.; Yu, X.; Wuertzer, C.A.; Hou, H.; Chiba, S.; et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 2010, 177, 311–324. [Google Scholar] [CrossRef]
- Wehrspaun, C.C.; Haerty, W.; Ponting, C.P. Microglia recapitulate a hematopoietic master regulator network in the aging human frontal cortex. Neurobiol. Aging 2015, 36, 2443.e9–2443.e20. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. Embo Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Frautschy, S.A.; Yang, F.; Irrizarry, M.; Hyman, B.; Saido, T.C.; Hsiao, K.; Cole, G.M. Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. 1998, 152, 307–317. [Google Scholar]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. New Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.S.; Jo, S.A. Mechanisms of Amyloid-β Peptide Clearance: Potential Therapeutic Targets for Alzheimer’s Disease. Biomol. Ther. 2012, 20, 245–255. [Google Scholar] [CrossRef]
- Frenkel, D.; Wilkinson, K.; Zhao, L.; Hickman, S.E.; Means, T.K.; Puckett, L.; Farfara, D.; Kingery, N.D.; Weiner, H.L.; El Khoury, J. Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat. Commun. 2013, 4, 2030. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, J.; Hickman, S.E.; Thomas, C.A.; Cao, L.; Silverstein, S.C.; Loike, J.D. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 1996, 382, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Mishra, V.K.; Shih, H.H.; Parveen, F.; Lenzen, D.; Ito, E.; Chan, T.F.; Ke, L.Y. Identifying the Therapeutic Significance of Mesenchymal Stem Cells. Cells 2020, 9, 1145. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Hickman, S.E.; El Khoury, J. The neuroimmune system in Alzheimer’s disease: The glass is half full. J. Alzheimer’s Dis. Jad 2013, 33 (Suppl. 1), S295–S302. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Neuropathology of Parkinson disease. Parkinsonism Relat. Disord. 2018, 46 (Suppl. 1), S30–S33. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.G.; Béraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal. 2015, 8, ra45. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, R.; Cookson, M.R. Pathways to Parkinsonism Redux: Convergent pathobiological mechanisms in genetics of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, R32–R44. [Google Scholar] [CrossRef] [PubMed]
- Daher, J.P.; Volpicelli-Daley, L.A.; Blackburn, J.P.; Moehle, M.S.; West, A.B. Abrogation of α-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc. Natl. Acad. Sci. USA 2014, 111, 9289–9294. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 6–17. [Google Scholar] [CrossRef]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 2004, 55, 221–235. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar] [CrossRef]
- Apolloni, S.; Amadio, S.; Montilli, C.; Volonté, C.; D’Ambrosi, N. Ablation of P2 × 7 receptor exacerbates gliosis and motoneuron death in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 4102–4116. [Google Scholar] [CrossRef]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef]
- Harraz, M.M.; Marden, J.J.; Zhou, W.; Zhang, Y.; Williams, A.; Sharov, V.S.; Nelson, K.; Luo, M.; Paulson, H.; Schöneich, C.; et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J. Clin. Investig. 2008, 118, 659–670. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Henkel, J.S.; Zhang, W.; Urushitani, M.; Julien, J.P.; Appel, S.H. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010, 58, 231–243. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; Zarrow, J.; Grant, S.; et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Jawaid, A.; Henstridge, C.M.; Valeri, A.; Merlini, M.; Robinson, J.L.; Lee, E.B.; Rose, J.; Appel, S.; Lee, V.M.; et al. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron 2017, 95, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, R.; Lu, H.; Butovsky, O.; Ohno, N.; Rietsch, A.M.; Cialic, R.; Wu, P.M.; Doykan, C.E.; Lin, J.; Cotleur, A.C.; et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 2014, 211, 1533–1549. [Google Scholar] [CrossRef]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef]
- Ghosh, R.; Tabrizi, S.J. Huntington disease. Handb. Clin. Neurol. 2018, 147, 255–278. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Keller, C.; Cortes Ramirez, E.P. Huntington’s disease—neuropathology. Handb. Clin. Neurol. 2011, 100, 83–100. [Google Scholar] [CrossRef]
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; DiFiglia, M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172. [Google Scholar] [CrossRef]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain A J. Neurol. 2007, 130, 1759–1766. [Google Scholar] [CrossRef]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Crotti, A.; Benner, C.; Kerman, B.E.; Gosselin, D.; Lagier-Tourenne, C.; Zuccato, C.; Cattaneo, E.; Gage, F.H.; Cleveland, D.W.; Glass, C.K. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat. Neurosci. 2014, 17, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M.; Bigio, E.H.; Cairns, N.J.; Alafuzoff, I.; Kril, J.; Kovacs, G.G.; Ghetti, B.; Halliday, G.; Holm, I.E.; et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2010, 119, 1–4. [Google Scholar] [CrossRef]
- Chang, M.C.; Srinivasan, K.; Friedman, B.A.; Suto, E.; Modrusan, Z.; Lee, W.P.; Kaminker, J.S.; Hansen, D.V.; Sheng, M. Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J. Exp. Med. 2017, 214, 2611–2628. [Google Scholar] [CrossRef]
- Arrant, A.E.; Onyilo, V.C.; Unger, D.E.; Roberson, E.D. Progranulin Gene Therapy Improves Lysosomal Dysfunction and Microglial Pathology Associated with Frontotemporal Dementia and Neuronal Ceroid Lipofuscinosis. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 2341–2358. [Google Scholar] [CrossRef]
- Walker, L.C.; Jucker, M. Neurodegenerative diseases: Expanding the prion concept. Annu. Rev. Neurosci. 2015, 38, 87–103. [Google Scholar] [CrossRef]
- Yamasaki, T.; Suzuki, A.; Hasebe, R.; Horiuchi, M. Flow Cytometric Detection of PrP(Sc) in Neurons and Glial Cells from Prion-Infected Mouse Brains. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Falsig, J.; Julius, C.; Margalith, I.; Schwarz, P.; Heppner, F.L.; Aguzzi, A. A versatile prion replication assay in organotypic brain slices. Nat. Neurosci. 2008, 11, 109–117. [Google Scholar] [CrossRef]
- Sorce, S.; Nuvolone, M.; Keller, A.; Falsig, J.; Varol, A.; Schwarz, P.; Bieri, M.; Budka, H.; Aguzzi, A. The role of the NADPH oxidase NOX2 in prion pathogenesis. PLoS Pathog. 2014, 10, e1004531. [Google Scholar] [CrossRef]
- Aguzzi, A.; Zhu, C. Microglia in prion diseases. J. Clin. Investig. 2017, 127, 3230–3239. [Google Scholar] [CrossRef] [PubMed]
- Hafner-Bratkovič, I.; Benčina, M.; Fitzgerald, K.A.; Golenbock, D.; Jerala, R. NLRP3 inflammasome activation in macrophage cell lines by prion protein fibrils as the source of IL-1β and neuronal toxicity. Cell. Mol. Life Sci. Cmls 2012, 69, 4215–4228. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.L.; Shi, Q.; Zhang, J.; Zhang, B.Y.; Gong, H.S.; Guo, Y.; Wang, S.B.; Xu, Y.; Wang, K.; Chen, C.; et al. Abnormal activation of microglia accompanied with disrupted CX3CR1/CX3CL1 pathway in the brains of the hamsters infected with scrapie agent 263K. J. Mol. Neurosci. Mn 2013, 51, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.M.; Field, R.H.; Perry, V.H.; Murray, C.L.; Cunningham, C. Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia 2010, 58, 2017–2030. [Google Scholar] [CrossRef]
- Sakai, K.; Hasebe, R.; Takahashi, Y.; Song, C.H.; Suzuki, A.; Yamasaki, T.; Horiuchi, M. Absence of CD14 delays progression of prion diseases accompanied by increased microglial activation. J. Virol. 2013, 87, 13433–13445. [Google Scholar] [CrossRef]
- Kouadir, M.; Yang, L.; Tan, R.; Shi, F.; Lu, Y.; Zhang, S.; Yin, X.; Zhou, X.; Zhao, D. CD36 participates in PrP(106-126)-induced activation of microglia. PLoS ONE 2012, 7, e30756. [Google Scholar] [CrossRef]
- Shinjo, R.; Imagama, S.; Ito, Z.; Ando, K.; Nishida, Y.; Ishiguro, N.; Kadomatsu, K. Keratan sulfate expression is associated with activation of a subpopulation of microglia/macrophages in Wallerian degeneration. Neurosci. Lett. 2014, 579, 80–85. [Google Scholar] [CrossRef]
- Doty, K.R.; Guillot-Sestier, M.V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293. [Google Scholar] [CrossRef]
- Ryan, K.J.; White, C.C.; Patel, K.; Xu, J.; Olah, M.; Replogle, J.M.; Frangieh, M.; Cimpean, M.; Winn, P.; McHenry, A.; et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Prokop, S.; Miller, K.R.; Drost, N.; Handrick, S.; Mathur, V.; Luo, J.; Wegner, A.; Wyss-Coray, T.; Heppner, F.L. Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer’s disease-like mice. J. Exp. Med. 2015, 212, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Varvel, N.H.; Grathwohl, S.A.; Degenhardt, K.; Resch, C.; Bosch, A.; Jucker, M.; Neher, J.J. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-β deposition in mouse models of Alzheimer’s disease. J. Exp. Med. 2015, 212, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Goñi, F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron 2015, 85, 1162–1176. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Werner, G.; Bohrmann, B.; Liesz, A.; Mazaheri, F.; Capell, A.; Feederle, R.; Knuesel, I.; Kleinberger, G.; Haass, C. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. Embo Mol. Med. 2016, 8, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Green, K.N. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. BrainBehav. Immun. 2017, 61, 1–11. [Google Scholar] [CrossRef]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef]
- Olmos-Alonso, A.; Schetters, S.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain A J. Neurol. 2016, 139, 891–907. [Google Scholar] [CrossRef]
- Luo, J.; Elwood, F.; Britschgi, M.; Villeda, S.; Zhang, H.; Ding, Z.; Zhu, L.; Alabsi, H.; Getachew, R.; Narasimhan, R.; et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J. Exp. Med. 2013, 210, 157–172. [Google Scholar] [CrossRef]
- Savage, J.C.; Jay, T.; Goduni, E.; Quigley, C.; Mariani, M.M.; Malm, T.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 6532–6543. [Google Scholar] [CrossRef]
- Luz, I.; Liraz, O.; Michaelson, D.M. An Anti-apoE4 Specific Monoclonal Antibody Counteracts the Pathological Effects of apoE4 In Vivo. Curr. Alzheimer Res. 2016, 13, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.S. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, L.; Zhang, S.Y.; Casanova, J.L.; Sancho-Shimizu, V. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med. 2016, 22, 511–527. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azam, S.; Haque, M.E.; Kim, I.-S.; Choi, D.-K. Microglial Turnover in Ageing-Related Neurodegeneration: Therapeutic Avenue to Intervene in Disease Progression. Cells 2021, 10, 150. https://doi.org/10.3390/cells10010150
Azam S, Haque ME, Kim I-S, Choi D-K. Microglial Turnover in Ageing-Related Neurodegeneration: Therapeutic Avenue to Intervene in Disease Progression. Cells. 2021; 10(1):150. https://doi.org/10.3390/cells10010150
Chicago/Turabian StyleAzam, Shofiul, Md. Ezazul Haque, In-Su Kim, and Dong-Kug Choi. 2021. "Microglial Turnover in Ageing-Related Neurodegeneration: Therapeutic Avenue to Intervene in Disease Progression" Cells 10, no. 1: 150. https://doi.org/10.3390/cells10010150
APA StyleAzam, S., Haque, M. E., Kim, I.-S., & Choi, D.-K. (2021). Microglial Turnover in Ageing-Related Neurodegeneration: Therapeutic Avenue to Intervene in Disease Progression. Cells, 10(1), 150. https://doi.org/10.3390/cells10010150