TIME Is a Great Healer—Targeting Myeloid Cells in the Tumor Immune Microenvironment to Improve Triple-Negative Breast Cancer Outcomes


Abstract
1. Introduction
2. Macrophages in Normal Mammary Glands
3. Triple-Negative Breast Cancer
4. The TIME and T Cells
5. Tumor-Associated Macrophages in Breast Cancer
6. Preclinical Models of TNBC
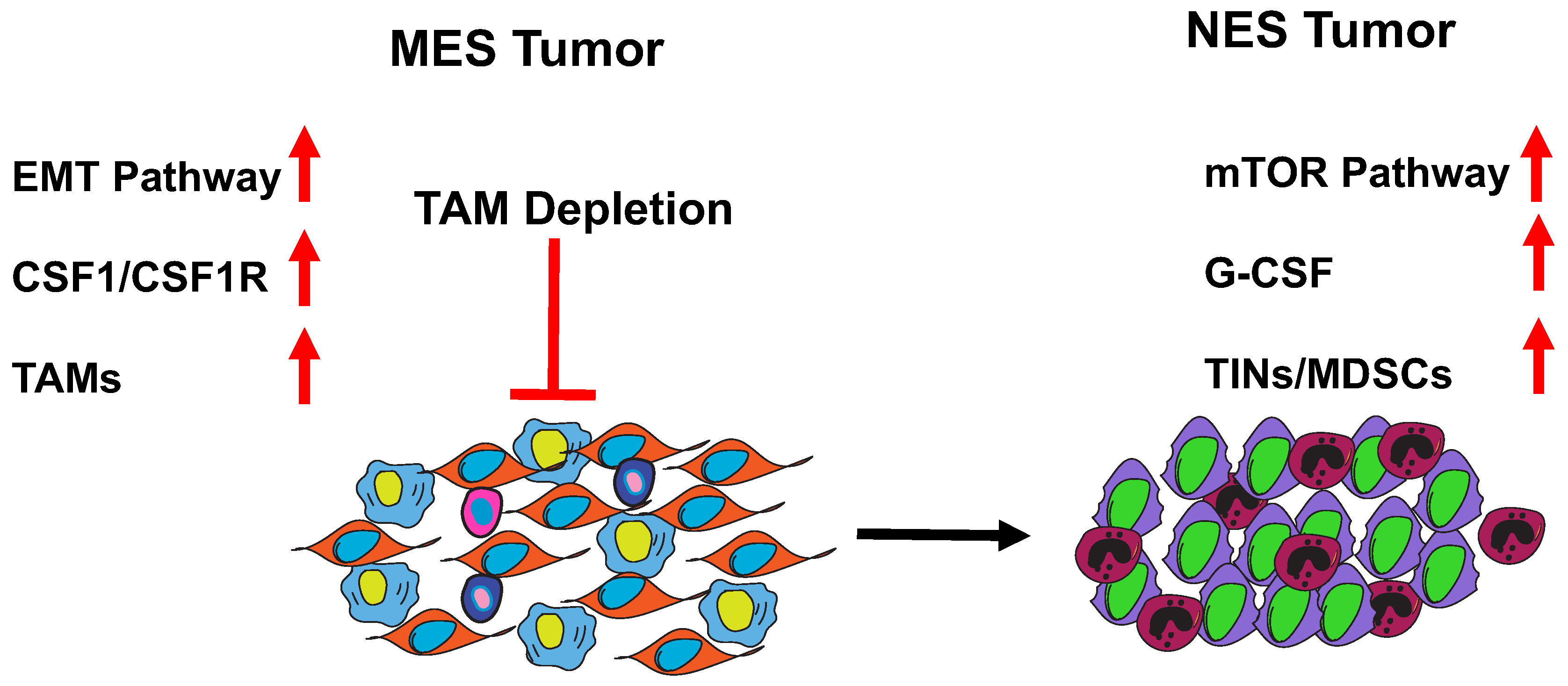
7. MES to NES in TNBC
8. Role of MDSCs in Breast Cancer and Metastasis
9. Clinical Relevance and Biomarkers for TAMs and MDSCs
10. Therapeutic Interventions for TAMs and MDSCs
11. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaufmann, S.H. Immunology’s Foundation: The 100-Year Anniversary of the Nobel Prize to Paul Ehrlich and Elie Metchnikoff. Nat. Immunol. 2008, 9, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; van de Laar, L. A Hitchhiker’s Guide to Myeloid Cell Subsets: Practical Implementation of a Novel Mononuclear Phagocyte Classification System. Front. Immunol. 2015, 6, 406. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Perdiguero, E.G.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef]
- Watson, C.J.; Khaled, W.T. Mammary development in the embryo and adult: A journey of morphogenesis and commitment. Development 2008, 135, 995–1003. [Google Scholar] [CrossRef]
- Jappinen, N.; Felix, I.; Lokka, E.; Tyystjarvi, S.; Pynttari, A.; Lahtela, T.; Gerke, H.; Elima, K.; Rantakari, P.; Salmi, M. Fetal-derived macrophages dominate in adult mammary glands. Nat. Commun. 2019, 10, 281. [Google Scholar] [CrossRef]
- Dawson, C.A.; Pal, B.; Vaillant, F.; Gandolfo, L.C.; Liu, Z.; Bleriot, C.; Ginhoux, F.; Smyth, G.K.; Lindeman, G.J.; Mueller, S.N.; et al. Tissue-resident ductal macrophages survey the mammary epithelium and facilitate tissue remodelling. Nat. Cell Biol. 2020, 22, 546–558. [Google Scholar] [CrossRef]
- Engebraaten, O.; Vollan, H.K.M.; Borresen-Dale, A.L. Triple-negative breast cancer and the need for new therapeutic targets. Am. J. Pathol. 2013, 183, 1064–1074. [Google Scholar] [CrossRef]
- Dietze, E.C.; Sistrunk, C.; Miranda-Carboni, G.; O’Regan, R.; Seewaldt, V.L. Triple-negative breast cancer in African-American women: Disparities versus biology. Nat. Rev. Cancer 2015, 15, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients with Advanced Non‒Small-Cell Lung Cancer Treated with Pembrolizumab: Results from the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Powles, T.; Donskov, F.; Plimack, E.R.; Barthelemy, P.; Hammers, H.J.; et al. Survival outcomes and independent response assessment with nivolumab plus ipilimumab versus sunitinib in patients with advanced renal cell carcinoma: 42-month follow-up of a randomized phase 3 clinical trial. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Franzoi, M.A.; de Azambuja, E. Atezolizumab in metastatic triple-negative breast cancer: IMpassion130 and 131 trials—How to explain different results? ESMO Open 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Chui, S.Y.; Emens, L.A. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. Reply. N Engl. J. Med. 2019, 380, 987–988. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Nanda, R.; Liu, M.C.; Yau, C.; Shatsky, R.; Pusztai, L.; Wallace, A.; Chien, A.J.; Forero-Torres, A.; Ellis, E.; Han, H.; et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women with Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol. 2020, 6, 676–684. [Google Scholar] [CrossRef]
- Loi, S.; Michiels, S.; Salgado, R.; Sirtaine, N.; Jose, V.; Fumagalli, D.; Kellokumpu-Lehtinen, P.L.; Bono, P.; Kataja, V.; Desmedt, C.; et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Ann. Oncol. 2014, 25, 1544–1550. [Google Scholar] [CrossRef]
- Gruosso, T.; Gigoux, M.; Manem, V.S.K.; Bertos, N.; Zuo, D.; Perlitch, I.; Saleh, S.M.I.; Zhao, H.; Souleimanova, M.; Johnson, R.M.; et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J. Clin. Investig. 2019, 129, 1785–1800. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Liu, M.; Kuo, F.; Capistrano, K.J.; Kang, D.; Nixon, B.G.; Shi, W.; Chou, C.; Do, M.H.; Stamatiades, E.G.; Gao, S.; et al. TGF-beta suppresses type 2 immunity to cancer. Nature 2020, 587, 115–120. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guerin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Arwert, E.N.; Harney, A.S.; Entenberg, D.; Wang, Y.; Sahai, E.; Pollard, J.W.; Condeelis, J.S. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep. 2018, 23, 1239–1248. [Google Scholar] [CrossRef]
- Patsialou, A.; Bravo-Cordero, J.J.; Wang, Y.; Entenberg, D.; Liu, H.; Clarke, M.; Condeelis, J.S. Intravital multiphoton imaging reveals multicellular streaming as a crucial component of in vivo cell migration in human breast tumors. Intravital 2013, 2, e25294. [Google Scholar] [CrossRef]
- Hao, Q.; Vadgama, J.V.; Wang, P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun. Signal. 2020, 18, 82. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Monkkonen, J.; Kellokumpu-Lehtinen, P.L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 101. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schroder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yao, Y.; Gong, C.; Yu, F.; Su, S.; Chen, J.; Liu, B.; Deng, H.; Wang, F.; Lin, L.; et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 2011, 19, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Olson, O.C.; Kim, H.; Quail, D.F.; Foley, E.A.; Joyce, J.A. Tumor-Associated Macrophages Suppress the Cytotoxic Activity of Antimitotic Agents. Cell Rep. 2017, 19, 101–113. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L.; et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Mouse 4T1 breast tumor model. Curr. Protoc. Immunol. 2001. [Google Scholar] [CrossRef]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell Biol. 1992, 12, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Pollard, J.W. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007, 67, 5064–5066. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D.; Silwal-Pandit, L.; Dannenfelser, R.; Langerød, A.; Vollan, H.K.M.; Vaske, C.; Siegel, J.U.; Troyanskaya, O.; Chin, S.-F.; Caldas, C.; et al. Lymphocyte Invasion in IC10/Basal-Like Breast Tumors Is Associated with Wild-Type TP53. Mol. Cancer Res. 2015, 13, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; Coffelt, S.B.; Duits, D.E.M.; van Miltenburg, M.H.; Slagter, M.; de Rink, I.; Henneman, L.; Kas, S.M.; Prekovic, S.; Hau, C.S.; et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019, 572, 538–542. [Google Scholar] [CrossRef]
- Medina, D.; Kittrell, F.S.; Shepard, A.; Stephens, L.C.; Jiang, C.; Lu, J.; Allred, D.C.; McCarthy, M.; Ullrich, R.L. Biological and genetic properties of the p53 null preneoplastic mammary epithelium. FASEB J. 2002, 16, 881–883. [Google Scholar] [CrossRef]
- Herschkowitz, J.I.; Zhao, W.; Zhang, M.; Usary, J.; Murrow, G.; Edwards, D.; Knezevic, J.; Greene, S.B.; Darr, D.; Troester, M.A.; et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2778–2783. [Google Scholar] [CrossRef]
- Tekpli, X.; Lien, T.; Rossevold, A.H.; Nebdal, D.; Borgen, E.; Ohnstad, H.O.; Kyte, J.A.; Vallon-Christersson, J.; Fongaard, M.; Due, E.U.; et al. An independent poor-prognosis subtype of breast cancer defined by a distinct tumor immune microenvironment. Nat. Commun. 2019, 10, 5499. [Google Scholar] [CrossRef]
- Seth, S.; Huo, L.; Rauch, G.M.; Adrada, B.E.; Piwnica-Worms, H.; Thompson, A.M.; Mittendorf, E.A.; Litton, J.K.; Symmans, W.F.; Draetta, G.F.; et al. Delineating longitudinal patterns of response to neoadjuvant systemic therapy (NAST) in triple-negative breast cancer (TNBC): Profiling results from a randomized, TNBC enrolling trial to confirm molecular profiling improves survival (ARTEMIS.; NCT02276443). J. Clin. Oncol. 2019, 37, 586. [Google Scholar] [CrossRef]
- Kim, I.S.; Gao, Y.; Welte, T.; Wang, H.; Liu, J.; Janghorban, M.; Sheng, K.; Niu, Y.; Goldstein, A.; Zhao, N.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 2019, 21, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kambayashi, Y.; Aiba, S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 2012, 1, 1433–1434. [Google Scholar] [CrossRef]
- Welte, T.; Kim, I.S.; Tian, L.; Gao, X.; Wang, H.; Li, J.; Holdman, X.B.; Herschkowitz, J.I.; Pond, A.; Xie, G.; et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol. 2016, 18, 632–644. [Google Scholar] [CrossRef]
- Markowitz, J.; Wesolowski, R.; Papenfuss, T.; Brooks, T.R.; Carson, W.E., 3rd. Myeloid-derived suppressor cells in breast cancer. Breast Cancer Res. Treat. 2013, 140, 13–21. [Google Scholar] [CrossRef]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Kruger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4+CD25+Foxp3+ T cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef]
- Redd, P.S.; Ibrahim, M.L.; Klement, J.D.; Sharman, S.K.; Paschall, A.V.; Yang, D.; Nayak-Kapoor, A.; Liu, K. SETD1B Activates iNOS Expression in Myeloid-Derived Suppressor Cells. Cancer Res. 2017, 77, 2834–2843. [Google Scholar] [CrossRef]
- Ochoa, A.C.; Zea, A.H.; Hernandez, C.; Rodriguez, P.C. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin. Cancer Res. 2007, 13, 721s–726s. [Google Scholar] [CrossRef]
- Ma, X.; Wang, M.; Yin, T.; Zhao, Y.; Wei, X. Myeloid-Derived Suppressor Cells Promote Metastasis in Breast Cancer after the Stress of Operative Removal of the Primary Cancer. Front. Oncol. 2019, 9, 855. [Google Scholar] [CrossRef]
- Yuan, Z.Y.; Luo, R.Z.; Peng, R.J.; Wang, S.S.; Xue, C. High infiltration of tumor-associated macrophages in triple-negative breast cancer is associated with a higher risk of distant metastasis. Onco Targets Ther. 2014, 7, 1475–1480. [Google Scholar] [CrossRef] [PubMed]
- Bergenfelz, C.; Larsson, A.M.; von Stedingk, K.; Gruvberger-Saal, S.; Aaltonen, K.; Jansson, S.; Jernstrom, H.; Janols, H.; Wullt, M.; Bredberg, A.; et al. Systemic Monocytic-MDSCs Are Generated from Monocytes and Correlate with Disease Progression in Breast Cancer Patients. PLos ONE 2015, 10, e0127028. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, J.V.; Flotte, T.J.; Yi, E.S.; Kroneman, T.; Suman, V.; Paludo, J.; McWilliams, R.R.; Kottschade, L.A.; Yan, Y.; Block, M.S.; et al. Quantitative assessment of tumor-infiltrating neutrophils to predict immunotherapy responses in metastatic melanoma. J. Clin. Oncol. 2019, 37, e21039. [Google Scholar] [CrossRef]
- Yan, H.H.; Jiang, J.; Pang, Y.; Achyut, B.R.; Lizardo, M.; Liang, X.; Hunter, K.; Khanna, C.; Hollander, C.; Yang, L. CCL9 Induced by TGFβ Signaling in Myeloid Cells Enhances Tumor Cell Survival in the Premetastatic Organ. Cancer Res. 2015, 75, 5283–5298. [Google Scholar] [CrossRef]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.C.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 2020, 52, 357–373.e9. [Google Scholar] [CrossRef]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPalpha Axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef]
- Dove, A. CD18 trials disappoint again. Nat. Biotechnol. 2000, 18, 817–818. [Google Scholar] [CrossRef]
- Panni, R.Z.; Herndon, J.M.; Zuo, C.; Hegde, S.; Hogg, G.D.; Knolhoff, B.L.; Breden, M.A.; Li, X.; Krisnawan, V.E.; Khan, S.Q.; et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Du Four, S.; Maenhout, S.K.; De Pierre, K.; Renmans, D.; Niclou, S.P.; Thielemans, K.; Neyns, B.; Aerts, J.L. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology 2015, 4, e998107. [Google Scholar] [CrossRef]
- Xin, H.; Zhang, C.; Herrmann, A.; Du, Y.; Figlin, R.; Yu, H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009, 69, 2506–2513. [Google Scholar] [CrossRef]
- Uddin, M.M.; Zou, Y.; Sharma, T.; Gatla, H.R.; Vancurova, I. Proteasome inhibition induces IKK-dependent interleukin-8 expression in triple negative breast cancer cells: Opportunity for combination therapy. PLos ONE 2018, 13, e0201858. [Google Scholar] [CrossRef]
- Daurkin, I.; Eruslanov, E.; Vieweg, J.; Kusmartsev, S. Generation of antigen-presenting cells from tumor-infiltrated CD11b myeloid cells with DNA demethylating agent 5-aza-2′-deoxycytidine. Cancer Immunol. Immunother. 2010, 59, 697–706. [Google Scholar] [CrossRef]
- Lamy, M.; Ferreira, A.; Dias, J.S.; Braga, S.; Silva, G.; Barbas, A. Notch-out for breast cancer therapies. New Biotechnol. 2017, 39, 215–221. [Google Scholar] [CrossRef]
- Janghorban, M.; Xin, L.; Rosen, J.M.; Zhang, X.H.-F. Notch Signaling as a Regulator of the Tumor Immune Response: To Target or Not to Target? Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef]
- Welte, T.; Zhang, X.H.; Rosen, J.M. Repurposing Antiestrogens for Tumor Immunotherapy. Cancer Discov. 2017, 7, 17–19. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Mikyskova, R.; Indrova, M.; Pollakova, V.; Bieblova, J.; Simova, J.; Reinis, M. Cyclophosphamide-induced myeloid-derived suppressor cell population is immunosuppressive but not identical to myeloid-derived suppressor cells induced by growing TC-1 tumors. J. Immunother. 2012, 35, 374–384. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Target | Drug | In Combination with | Phase | Clinical Trial Identifier |
|---|---|---|---|---|
| CSF1R/CSF1 | Ipatasertib | Atezo or pac or nab-Paclitaxel | 1b | NCT03800836 * |
| Pexidartinib | nAB-Paclitaxel Eribulin | 1b 1a/1b | NCT01525602 NCT01596751 | |
| LY3022855 | Durvalumab OR tremelimumab | 1b | NCT02718911 | |
| Emactuzumab | Selicrelumab | 1b | NCT02760797 | |
| Lacnotuzumab | Spartalizumab | 1b/2 | NCT02807844 | |
| CCL2/CCR2 | Carlumab | 1b | NCT01204996 | |
| CD47/SIRPα | IBI322 | Pembrolizumab | 1a/1b | NCT04328831 * |
| ALX148 | Pembrolizumab, trastuzumab or chemo | 1 | NCT03013218 * | |
| Hu5F9-G4 | 1 | NCT02216409 | ||
| ARGINASE | INCB001158 | 1 | NCT02903914 * | |
| MDSC DIFFERENTIATION | Decitabine | 1 | NCT00030615 | |
| MDSC SURVIVAL | Entinostat | Ipilimumab and Nivolumab | 1 | NCT02453620 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Zhang, X.H.F.; Rosen, J.M. TIME Is a Great Healer—Targeting Myeloid Cells in the Tumor Immune Microenvironment to Improve Triple-Negative Breast Cancer Outcomes. Cells 2021, 10, 11. https://doi.org/10.3390/cells10010011
Singh S, Zhang XHF, Rosen JM. TIME Is a Great Healer—Targeting Myeloid Cells in the Tumor Immune Microenvironment to Improve Triple-Negative Breast Cancer Outcomes. Cells. 2021; 10(1):11. https://doi.org/10.3390/cells10010011
Chicago/Turabian StyleSingh, Swarnima, Xiang H. F. Zhang, and Jeffrey M. Rosen. 2021. "TIME Is a Great Healer—Targeting Myeloid Cells in the Tumor Immune Microenvironment to Improve Triple-Negative Breast Cancer Outcomes" Cells 10, no. 1: 11. https://doi.org/10.3390/cells10010011
APA StyleSingh, S., Zhang, X. H. F., & Rosen, J. M. (2021). TIME Is a Great Healer—Targeting Myeloid Cells in the Tumor Immune Microenvironment to Improve Triple-Negative Breast Cancer Outcomes. Cells, 10(1), 11. https://doi.org/10.3390/cells10010011