Methylome and Epialleles in Rice Epilines Selected for Energy Use Efficiency

 , ,
, ,
Abstract
1. Introduction
2. Material and Methods
2.1. Plant Material and Growth Conditions
2.2. Selection for Respiration Rate and Energy Use Efficiency
2.3. Field Trials
2.4. RNA Sequencing Analysis
2.5. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.6. Whole Genome Bisulfite Sequencing (WGBS)
2.6.1. Detection of Differential Cytosine Methylation
2.6.2. DNA Methylome and Transcriptome Integration
2.7. Targeted Bisulfite Sequencing
2.8. Chromatin Immunoprecipitation Sequencing (ChIP-seq)
2.8.1. Chromatin Immunoprecipitation
2.8.2. ChIP Sequencing
3. Results
3.1. Rice Epilines Selected for Energy Use Efficiency Show an Enhanced Seed Yield
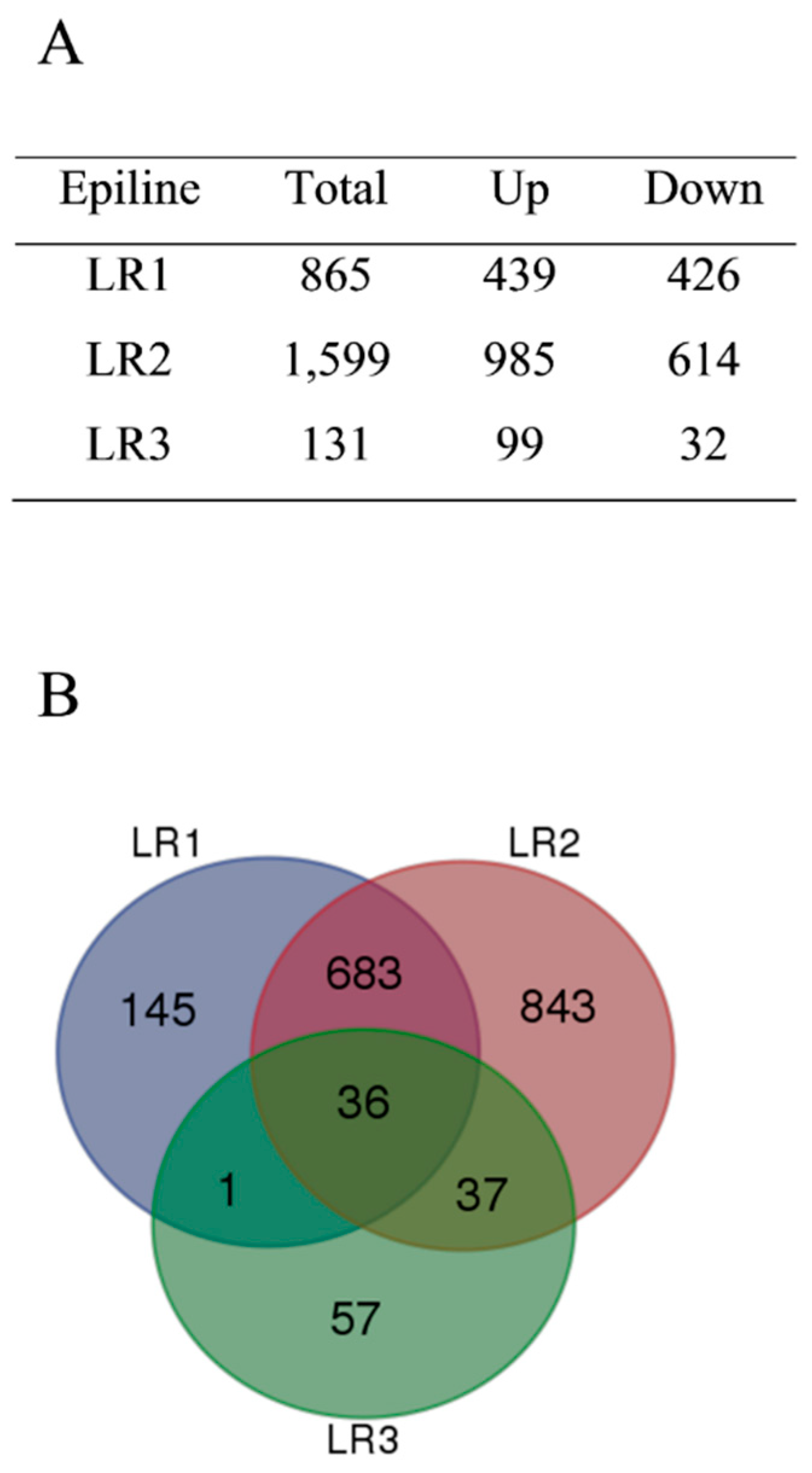
3.2. Altered Transcriptomes in Rice Epilines
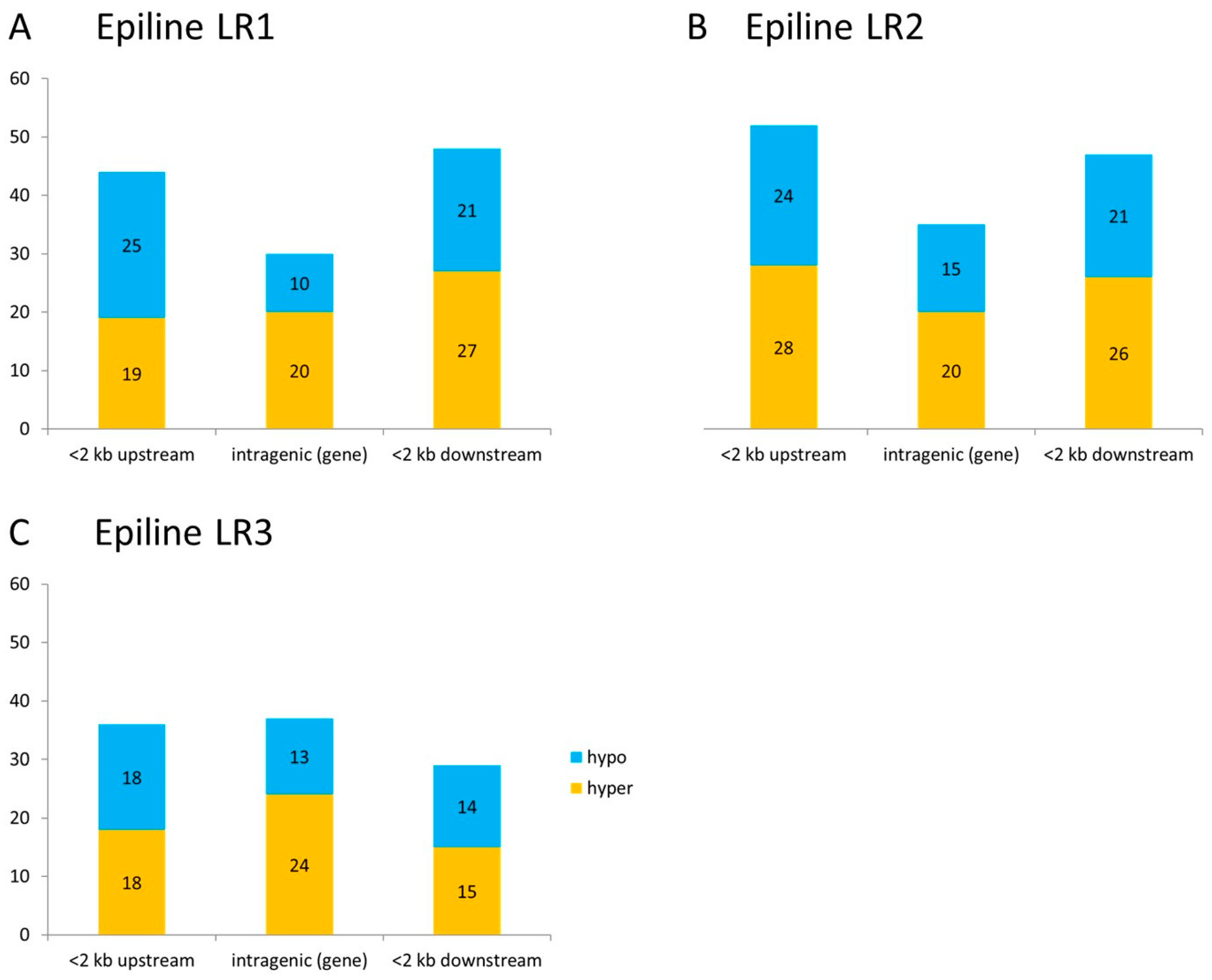
3.3. Differentially Methylated Regions in Rice Epilines
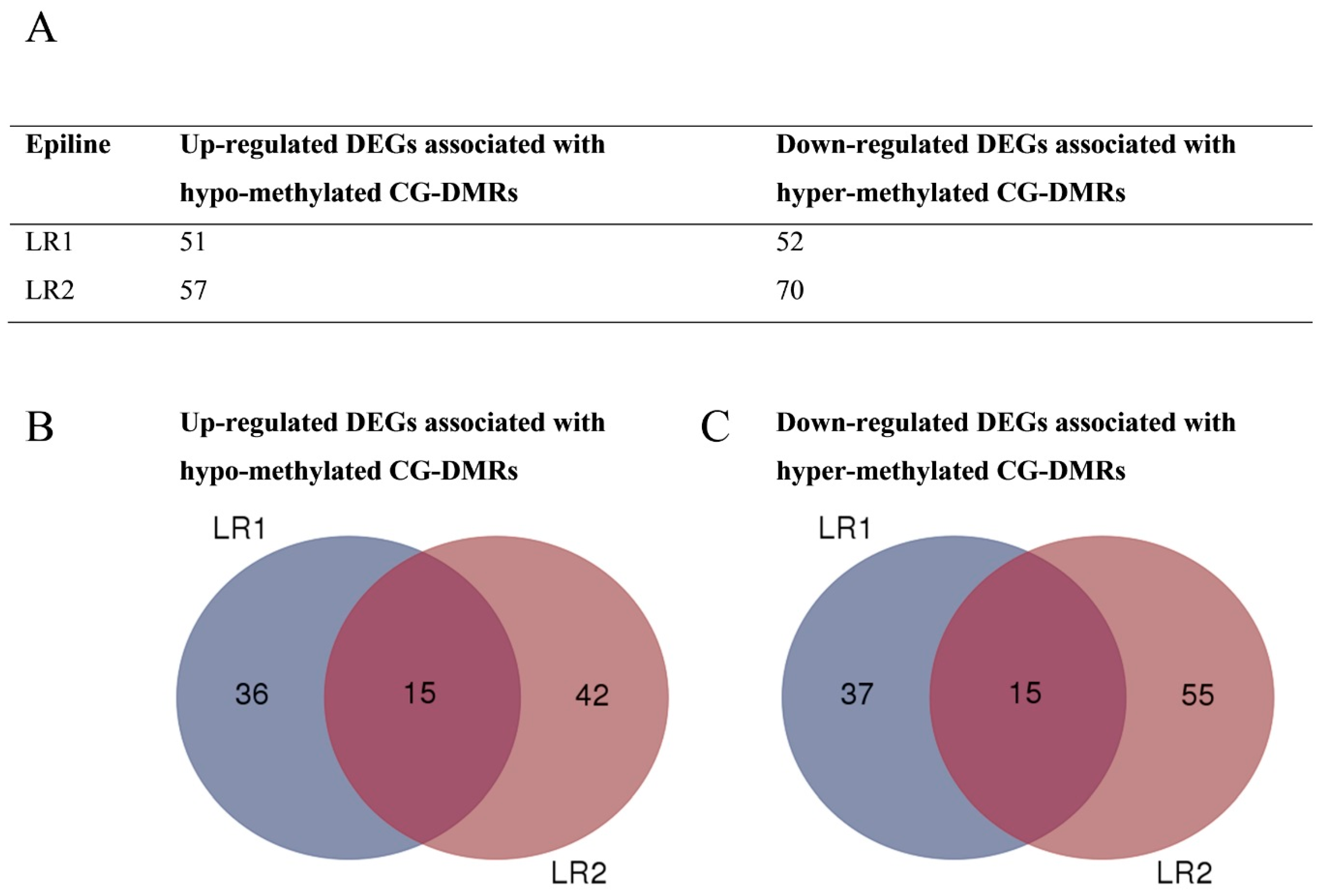
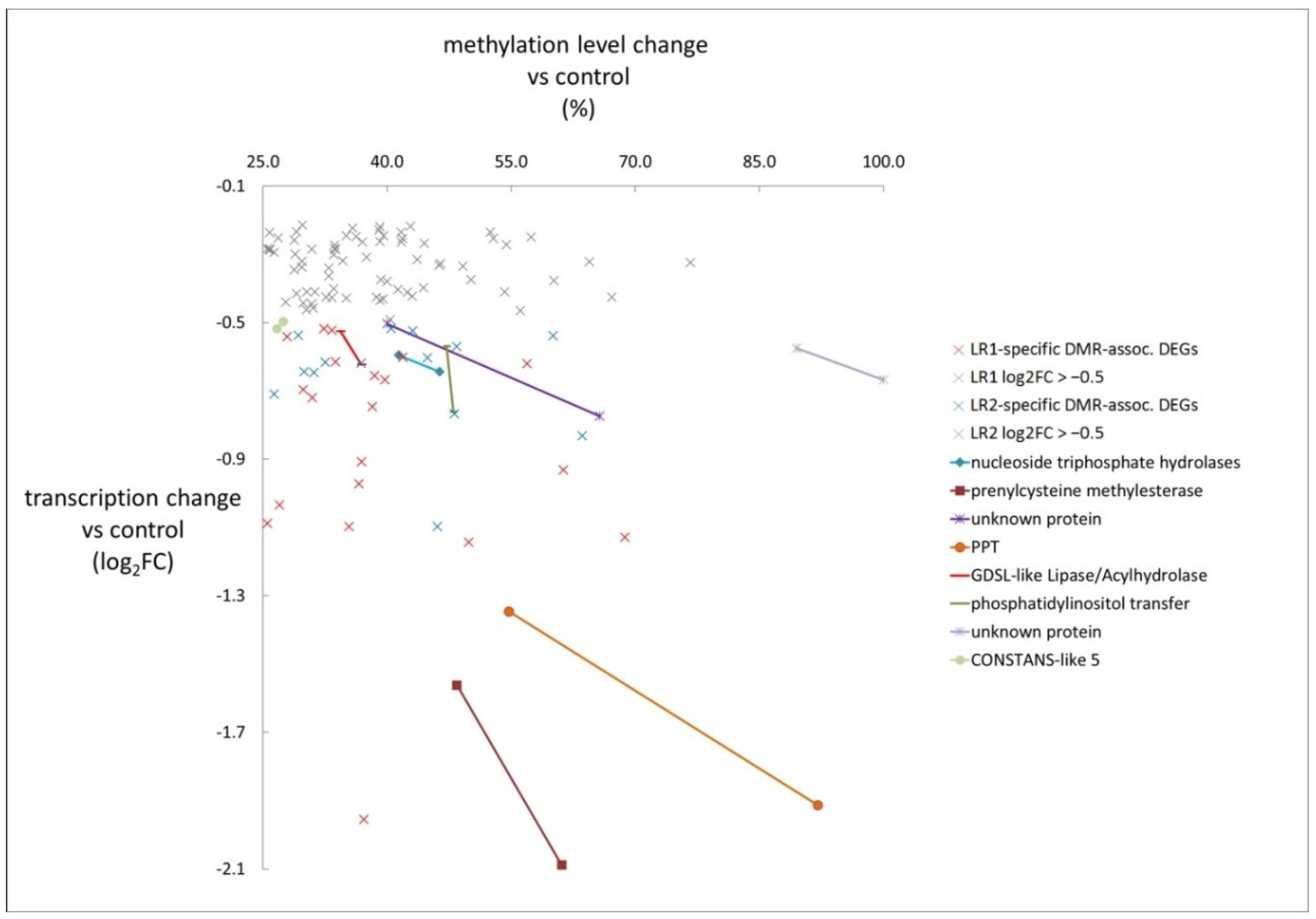
3.4. DMRs Upstream of DEGs Identify Putative Epialleles
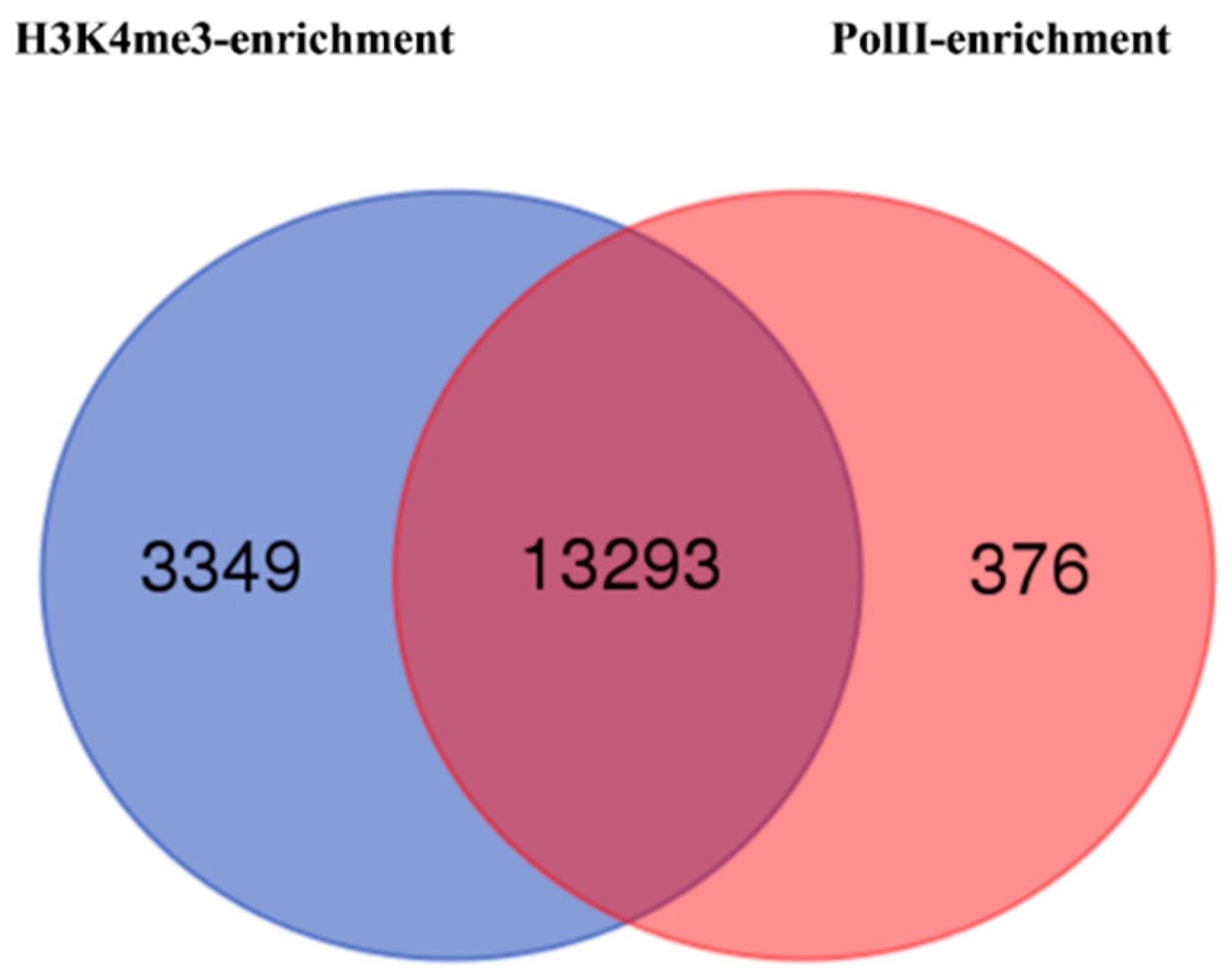
3.5. Histone H3 Lysine-4 Trimethylation and RNA Polymerase II (PolII) Enrichments in Epiline LR2 Identify Activated Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gallusci, P.; Dai, Z.W.; Génard, M.; Gauffretau, A.; Leblanc-Fournier, N.; Richard-Molard, C.; Vile, D.; Brunel-Muguet, S. Epigenetics for plant improvement: Current knowledge and modeling avenues. Trends Plant Sci. 2017, 22, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M.; Schmitz, R.J. Exploiting induced and natural epigenetic variation for crop improvement. Nat. Rev. Genet. 2017, 18, 563–575. [Google Scholar] [CrossRef] [PubMed]
- De Block, M.; Van Lijsebettens, M. Energy efficiency and energy homeostasis as genetic and epigenetic components of plant performance and crop productivity. Curr. Opin. Plant Biol. 2011, 14, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Hauben, M.; Haesendonckx, B.; Standaert, E.; Van Der Kelen, K.; Azmi, A.; Akpo, H.; Van Breusegem, F.; Guisez, Y.; Bots, M.; Lambert, B.; et al. Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc. Natl. Acad. Sci. USA 2009, 106, 20109–20114. [Google Scholar] [CrossRef] [PubMed]
- Verkest, A.; Byzova, M.; Martens, C.; Willems, P.; Verwulgen, T.; Slabbinck, B.; Rombaut, D.; Van de Velde, J.; Vandepoele, K.; Standaert, E.; et al. Selection for improved energy use efficiency and drought tolerance in canola results in distinct transcriptome and epigenome changes. Plant Physiol. 2015, 168, 1338–1350. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Hense, S.; Minoche, A.E.; Dohm, J.C.; Himmelbauer, H.; Schmidt, T.; Zakrzewski, F. Cytosine methylation of an ancient satellite family in the wild beet Beta procumbens. Cytogenet. Genome Res. 2014, 143, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, F.; Schubert, V.; Viehoever, P.; Minoche, A.E.; Dohm, J.C.; Himmelbauer, H.; Weisshaar, B.; Schmidt, T. The CHH motif in sugar beet satellite DNA: A modulator for cytosine methylation. Plant J. 2014, 78, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Blevins, T.; Pontvianne, F.; Cocklin, R.; Podicheti, R.; Chandrasekhara, C.; Yerneni, S.; Braun, C.; Lee, B.; Rusch, D.; Mockaitis, K.; et al. A two-step process for epigenetic inheritance in Arabidopsis. Mol. Cell 2014, 54, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Calarco, J.P.; Borges, F.; Donoghue, M.T.A.; Van Ex, F.; Jullien, P.E.; Lopes, T.; Gardner, R.; Berger, F.; Feijó, J.A.; Becker, J.D.; et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 2012, 151, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Silveira, A.B.; Trontin, C.; Cortijo, S.; Barau, J.; Del Bem, L.E.; Loudet, O.; Colot, V.; Vincentz, M. Extensive natural epigenetic variation at a de novo originated gene. PLoS Genet. 2013, 9, e1003437. [Google Scholar] [CrossRef] [PubMed]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Perrella, G.; Lopez-Vernaza, M.A.; Carr, C.; Sani, E.; Gosselé, V.; Verduyn, C.; Kellermeier, F.; Hannah, M.A.; Amtmann, A. Histone deacetylase complex1 expression level titrates plant growth and abscisic acid sensitivity in Arabidopsis. Plant Cell 2013, 25, 3491–3505. [Google Scholar] [CrossRef] [PubMed]
- Roudier, F.; Ahmed, I.; Bérard, C.; Sarazin, A.; Mary-Huard, T.; Cortijo, S.; Bouyer, D.; Caillieux, E.; Duvernois-Berthet, E.; Al-Shikhley, L.; et al. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 2011, 30, 1928–1938. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; To, T.K.; Ishida, J.; Morosawa, T.; Kawashima, M.; Matsui, A.; Toyoda, T.; Kimura, H.; Shinozaki, K.; Seki, M. Alterations of lysine modifications on the histone H3 N-tail under drought stress conditions in Arabidopsis thaliana. Plant Cell Physiol. 2008, 49, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Fromm, M.; Avramova, Z. Multiple exposures to drought ‘train’ transcriptional responses in Arabidopsis. Nat. Commun. 2012, 3, 740. [Google Scholar] [CrossRef] [PubMed]
- Du, J.M.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Wendte, J.M.; Schmitz, R.J. Specifications of targeting heterochromatin modifications in plants. Mol. Plant 2018, 11, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Van Bel, M.; Woloszynska, M.; Slabbinck, B.; Martens, C.; De Block, M.; Coppens, F.; Van Lijsebettens, M. Plant-RRBS, a bisulfite and next-generation sequencing-based methylome profiling method enriching for coverage of cytosine positions. BMC Plant Biol. 2017, 17, 115. [Google Scholar] [CrossRef] [PubMed]
- De Block, M.; De Brouwer, D. A simple and robust in vitro assay to quantify the vigour of oilseed rape lines and hybrids. Plant Physiol. Biochem. 2002, 40, 845–852. [Google Scholar] [CrossRef]
- Parkinson, H.; Sarkans, U.; Kolesnikov, N.; Abeygunawardena, N.; Burdett, T.; Dylag, M.; Emam, I.; Farne, A.; Hastings, E.; Holloway, E.; et al. ArrayExpress update—An archive of microarray and high-throughput sequencing-based functional genomics experiments. Nucleic Acids Res. 2011, 39, D1002–D1004. [Google Scholar] [CrossRef] [PubMed]
- FastQC. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 13 July 2018).
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.-S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Proost, S.; Van Bel, M.; Sterck, L.; Billiau, K.; Van Parys, T.; Van de Peer, Y.; Vandepoele, K. PLAZA: A comparative genomics resource to study gene and genome evolution in plants. Plant Cell 2009, 21, 3718–3731. [Google Scholar] [CrossRef] [PubMed]
- Van Bel, M.; Proost, S.; Wischnitzki, E.; Movahedi, S.; Scheerlinck, C.; Van de Peer, Y.; Vandepoele, K. Dissecting plant genomes with the PLAZA comparative genomics platform. Plant Physiol. 2012, 158, 590–600. [Google Scholar] [CrossRef] [PubMed]
- PLAZA. Available online: https://bioinformatics.psb.ugent.be/plaza (accessed on 13 July 2018).
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, C.A.; Feng, X.; Schoft, V.K.; Hsieh, T.-F.; Uzawa, R.; Rodrigues, J.A.; Zemach, A.; Chumak, N.; Machlicova, A.; Nishimura, T.; et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 2012, 337, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.D.; Schmitz, R.J.; Ecker, J.R. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 2012, 28, 583–585. [Google Scholar] [CrossRef] [PubMed]
- MethPrimer. Available online: http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi (accessed on 13 July 2018).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Reizel, Y.; Spiro, A.; Sabag, O.; Skversky, Y.; Hecht, M.; Keshet, I.; Berman, B.P.; Cedar, H. Gender-specific postnatal demethylation and establishment of epigenetic memory. Genes Dev. 2015, 29, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Jiang, Y.; Meydan, C.; Shank, K.; Pandey, S.; Barreyro, L.; Antony-Debre, I.; Viale, A.; Socci, N.; Sun, Y.; et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 2015, 27, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, Z.; Qin, R.; Qiu, Y.; Wang, J.-L.; Cui, X.; Gu, L.; Zhang, X.; Guo, X.; Wang, D.; et al. Identification and characterization of an epi-allele of FIE1 reveals a regulatory linkage between two epigenetic marks in rice. Plant Cell 2012, 24, 4407–4421. [Google Scholar] [CrossRef] [PubMed]
- Van Lijsebettens, M.; Grasser, K.D. Transcript elongation factors: Shaping transcriptomes after transcript initiation. Trends Plant Sci. 2014, 19, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Small, I. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 2014, 65, 415–442. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Linneweber, C.; Small, I. Pentatricopeptide repeat proteins: A socket set for organelle gene expression. Trends Plant Sci. 2008, 13, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Heber, U.; Walker, D. Concerning a dual function of coupled cyclic electron transport in leaves. Plant Physiol. 1992, 100, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- Miyake, C. Alternative electron flows (water-water cycle and cyclic electron flow around PSI) in photosynthesis: Molecular mechanisms and physiological functions. Plant Cell physiol. 2010, 51, 1951–1963. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Badger, M.R. Photoprotection in plants: A new light on photosystem II damage. Trends Plant Sci. 2011, 16, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Livingston, A.K.; Cruz, J.A.; Kohzuma, K.; Dhingra, A.; Kramer, D.M. An Arabidopsis mutant with high cyclic electron flow around photosystem I (hcef) involving the NADPH dehydrogenase complex. Plant Cell 2010, 22, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Suorsa, M.; Sirpiö, S.; Aro, E.-M. Towards characterization of the chloroplast NAD(P)H dehydrogenase complex. Mol. Plant 2009, 2, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Shikanai, T. Central role of cyclic electron transport around photosystem I in the regulation of photosynthesis. Curr. Opin. Biotechnol. 2014, 26, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Yamori, W.; Sakata, N.; Suzuki, Y.; Shikanai, T.; Makino, A. Cyclic electron flow around photosystem I via chloroplast NAD(P)H dehydrogenase (NDH) complex performs a significant physiological role during photosynthesis and plant growth at low temperature in rice. Plant J. 2011, 68, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Yamori, W.; Shikanai, T.; Makino, A. Photosystem I cyclic electron flow via chloroplast NADH dehydrogenase-like complex performs a physiological role for photosynthesis at low light. Sci. Rep. 2015, 5, 13908. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Nakamura, T.; Sugita, M.; Shimizu, T.; Shikanai, T. A pentatricopeptide repeat protein is a site recognition factor in chloroplast RNA editing. J. Biol. Chem. 2006, 281, 37661–37667. [Google Scholar] [CrossRef] [PubMed]
- Yagi, Y.; Tachikawa, M.; Noguchi, H.; Satoh, S.; Obokata, J.; Nakamura, T. Pentatricopeptide repeat proteins involved in plant organellar RNA editing. RNA Biol. 2013, 10, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Endo, T.; Peltier, G.; Tasaka, M.; Shikanai, T. A nucleus-encoded factor, CRR2, is essential for the expression of chloroplast ndhB in Arabidopsis. Plant J. 2003, 36, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Myouga, F.; Motohashi, R.; Shinozaki, K.; Shikanai, T. Conserved domain structure of pentatricopeptide repeat proteins involved in chloroplast RNA editing. Proc. Natl. Acad. Sci. USA 2007, 104, 8178–8183. [Google Scholar] [CrossRef] [PubMed]
- Kotera, E.; Tasaka, M.; Shikanai, T. A pentatricopeptide repeat protein is essential for RNA editing in chloroplasts. Nature 2005, 433, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Shi, N.; Li, Q.; Mi, H. An active supercomplex of NADPH dehydrogenase mediated cyclic electron flow around Photosystem I from the panicle chloroplast of Oryza sativa. Acta Biochim. Biophys. Sin. 2014, 46, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Cao, X.; Jacobsen, S.E. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 2003, 299, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W.-L.; Zilberman, D.; Xie, Z.; Johansen, L.K.; Carrington, J.C.; Jacobsen, S.E. RNA silencing genes control de novo DNA methylation. Science 2004, 303, 1336. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Rowley, M.J.; Böhmdorfer, G.; Wierzbicki, A.T. A SWI/SNF chromatin-remodeling complex acts in noncoding RNA-mediated transcriptional silencing. Mol. Cell 2013, 49, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Du, J.; Hale, C.J.; Gallego-Bartolome, J.; Feng, S.; Vashisht, A.A.; Chory, J.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, B.T.; Lee, K.; Rohr, N.A.; Hall, D.W.; Schmitz, R.J. Stable inheritance of DNA methylation allows creation of epigenotype maps and the study of epiallele inheritance patterns in the absence of genetic variation. Genome Biol. 2017, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, O.; Reinders, J.; Čaikovski, M.; Smathajitt, C.; Paszkowski, J. Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell 2007, 130, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Bond, D.M.; Baulcombe, D.C. Small RNAs and heritable epigenetic variation in plants. Trends Cell Biol. 2014, 24, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Antosz, W.; Pfab, A.; Ehrnsberger, H.F.; Holzinger, P.; Köllen, K.; Mortensen, S.A.; Bruckmann, A.; Schubert, T.; Längst, G.; Griesenbeck, J.; et al. The composition of the Arabidopsis RNA polymerase II transcript elongation complex reveals the interplay between elongation and mRNA processing factors. Plant Cell 2017, 29, 854–870. [Google Scholar] [CrossRef] [PubMed]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.T.; Petruk, S.; Sedkov, Y.; Cho, E.; Tillib, S.; Canaani, E.; Mazo, A. Modulation of heat shock gene expression by the TAC1 chromatin-modifying complex. Nat. Cell Biol. 2004, 6, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Gibert, J.-M.; Peronnet, F.; Schlötterer, C. Phenotypic plasticity in Drosophila pigmentation caused by temperature sensitivity of a chromatin regulator network. PLoS Genet. 2007, 3, e30. [Google Scholar] [CrossRef] [PubMed]
- Jobson, M.A.; Jordan, J.M.; Sandrof, M.A.; Hibshman, J.D.; Lennox, A.L.; Baugh, L.R. Transgenerational effects of early life starvation on growth, reproduction, and stress resistance in Caenorhabditis elegans. Genetics 2015, 201, 201–212. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Line | In Vitro Assays | Soil | Field Trial b | ||||
|---|---|---|---|---|---|---|---|
| Average Cellular Respiration (S3) | NAD(P)H Content (S3) | Energy Use Efficiency a (S3) | Photo-Respiration (S5) | Leaf Respiration (S5) | Seed Yield | ||
| (S6) | (S7) | ||||||
| Control | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| LR1 | 88 ** | 101 | 115 ** | 106 | 81 * | 116 * | 105 |
| LR2 | 86 *** | 100 | 116 ** | 106 | 80 * | 110 * | 101 |
| LR3 | 95 ** | 97 | 102 | 112 ** | 96 | 99 | 101 |
| Line | Mean Methylated Cytosines a | ||
|---|---|---|---|
| CG | CHG | CHH | |
| (%) | |||
| Control | 45.8 | 16.6 | 3.2 |
| LR1 | 44.7 | 17.3 | 3.0 |
| LR2 | 45.8 | 17.1 | 3.1 |
| LR3 | 42.2 | 16.2 | 2.9 |
| Epiline | CG | CHG | CHH | |||
|---|---|---|---|---|---|---|
| Hyper-Methylated | Hypo-Methylated | Hyper-Methylated | Hypo-Methylated | Hyper-Methylated | Hypo-Methylated | |
| 25% Methylation Level Difference (DMRs) | ||||||
| LR1 | 6601 | 5460 | 4953 | 5047 | 2165 | 3329 |
| LR2 | 4682 | 4588 | 3649 | 2926 | 2428 | 3238 |
| LR3 | 6020 | 5735 | 4049 | 4361 | 1792 | 3571 |
| 80% Methylation Level Difference (hDMRs) | ||||||
| LR1 | 115 | 98 | 2 | 1 | 0 | 1 |
| LR2 | 114 | 104 | 4 | 3 | 2 | 2 |
| LR3 | 116 | 102 | 2 | 2 | 0 | 2 |
| DMR | DEG | DEG Parameter | DMR Parameter | Gene |
|---|---|---|---|---|
| Gene ID | log2FC | Methylation Level Difference (%) | Description | |
| Hyper-methylated | OSINDICA_01G41530 | −0.60 | 41.4 | nucleoside triphosphate hydrolases |
| −0.64 | 46.4 | |||
| OSINDICA_01G43010 | −1.56 | 48.4 | prenylcysteine methylesterase | |
| −2.09 | 61.1 | |||
| OSINDICA_03G51310 | −0.50 | 40.0 | unknown protein | |
| −0.77 | 65.7 | |||
| OSINDICA_05G06440 | −1.35 | 54.7 | phosphoenolpyruvate/phosphate translocator (PPT) | |
| −1.91 | 92.1 | |||
| OSINDICA_06G29930 | −0.62 | 36.9 | GDSL-like Lipase/acylhydrolase | |
| −0.53 | 34.4 | |||
| OSINDICA_06G36260 | −0.57 | 47.2 | phosphatidylinositol transfer family protein | |
| −0.76 | 48.0 | |||
| OSINDICA_09G30400 | −0.57 | 89.5 | unknown protein | |
| −0.67 | 100.0 | |||
| Hypo-methylated | OSINDICA_02G44050 | 0.70 | −34.2 | brassinosteroid-responsive RING-H2 |
| 0.98 | −37.3 | |||
| OSINDICA_01G35920 | 1.12 | −54.4 | sugar transporter 1 | |
| 0.72 | −40.0 | |||
| OSINDICA_12G27600 | 0.74 | −29.1 | ABA-activated protein kinase | |
| 0.91 | −35.6 |
| H3K4me3 | PolII | ||||
|---|---|---|---|---|---|
| Enriched | Depleted | Total | Enriched | Depleted | Total |
| 26,800 | 40 | 26,840 | 21,065 | 105 | 21,170 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, M.; Byzova, M.; Martens, C.; Peeters, M.; Raj, Y.; Shukla, S.; Verwulgen, T.; De Block, M.; Van Lijsebettens, M. Methylome and Epialleles in Rice Epilines Selected for Energy Use Efficiency. Agronomy 2018, 8, 163. https://doi.org/10.3390/agronomy8090163
Schmidt M, Byzova M, Martens C, Peeters M, Raj Y, Shukla S, Verwulgen T, De Block M, Van Lijsebettens M. Methylome and Epialleles in Rice Epilines Selected for Energy Use Efficiency. Agronomy. 2018; 8(9):163. https://doi.org/10.3390/agronomy8090163
Chicago/Turabian StyleSchmidt, Martin, Marina Byzova, Cindy Martens, Marrit Peeters, Yog Raj, Shailesh Shukla, Tom Verwulgen, Marc De Block, and Mieke Van Lijsebettens. 2018. "Methylome and Epialleles in Rice Epilines Selected for Energy Use Efficiency" Agronomy 8, no. 9: 163. https://doi.org/10.3390/agronomy8090163
APA StyleSchmidt, M., Byzova, M., Martens, C., Peeters, M., Raj, Y., Shukla, S., Verwulgen, T., De Block, M., & Van Lijsebettens, M. (2018). Methylome and Epialleles in Rice Epilines Selected for Energy Use Efficiency. Agronomy, 8(9), 163. https://doi.org/10.3390/agronomy8090163