
Characterisation of Faba Bean (Vicia faba L.) Transcriptome Using RNA-Seq: Sequencing, De Novo Assembly, Annotation, and Expression Analysis


Abstract
1. Introduction
2. Results
2.1. RNA-Seq and De Novo Transcriptome Assembly
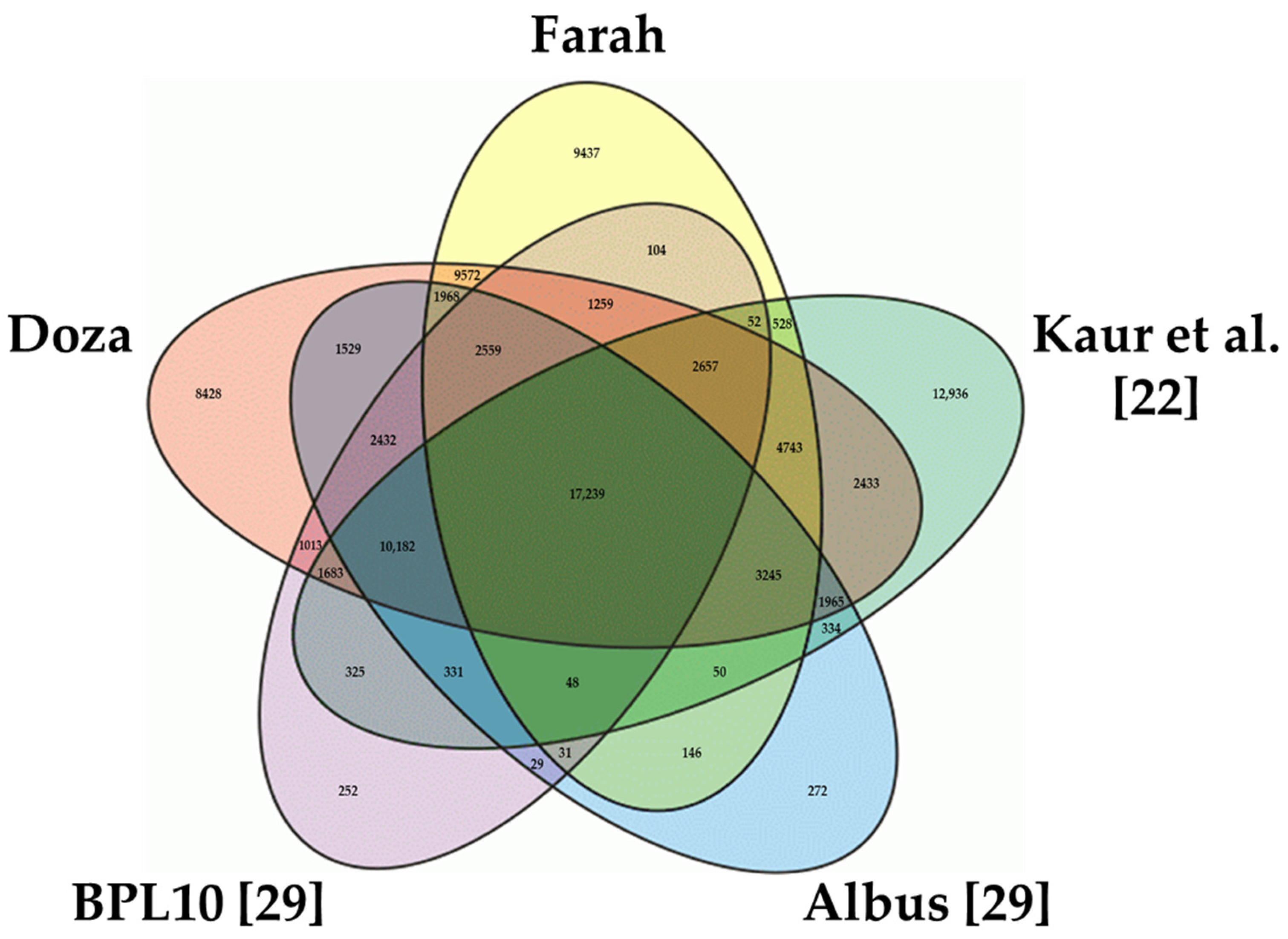
2.2. Classification and Functional Annotation of the Faba Bean Transcriptome
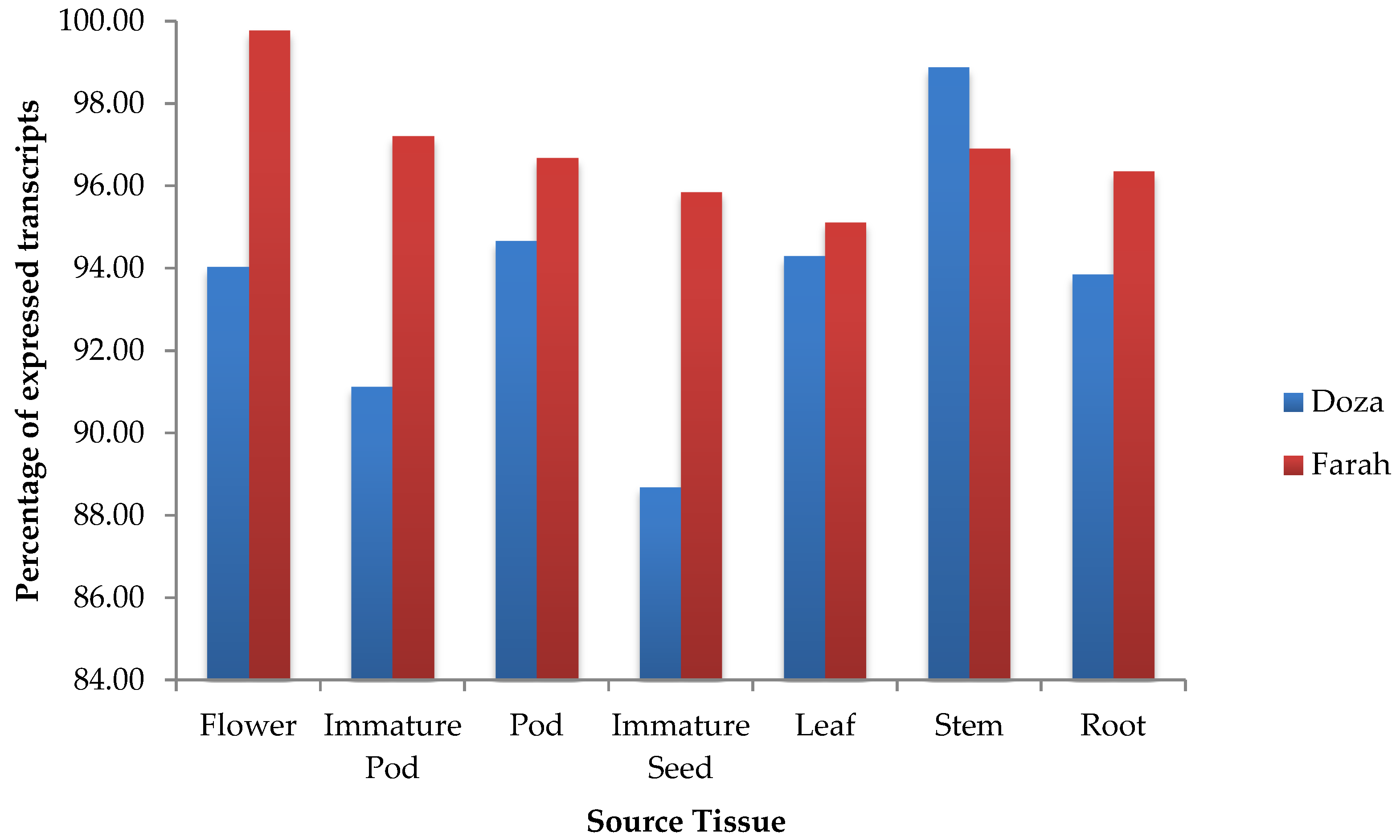
2.3. Tissue-Specific Expression Analysis
3. Discussion
3.1. De Novo Transcriptome Assembly
3.2. Annotation of the Transcriptome Assemblies
3.3. Analysis and Validation of Tissue-Specific Gene Expression Level
3.4. Applications to Genomics-Assisted Breeding
4. Materials and Methods
4.1. Plant Material
4.2. RNA Extraction
4.3. Library Preparation and Sequencing
4.4. Sequence Data Processing/Data Filtering and De Novo Assembly
4.5. Transcriptome Annotation
4.6. Tissue-Specific Expression Analysis
4.7. Validation of Tissue Expression Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bohra, A.; Pandey, M.K.; Jha, U.C.; Singh, B.; Singh, I.P.; Datta, D.; Chaturvedi, S.K.; Nadarajan, N.; Varshney, R.K. Genomics-assisted breeding in four major pulse crops of developing countries: Present status and prospects. Theor. Appl. Genet. 2014, 127, 1263–1291. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, S.S.; Migdadi, H.M.; Ammar, M.H.; Paull, J.G.; Siddique, K.H.M. Faba bean genomics: Current status and future prospects. Euphytica 2012, 186, 609–624. [Google Scholar] [CrossRef]
- Food and Agricultural Organisation of the United Nations-FAO Statistical Database. Available online: http://faostat.fao.org (accessed on 18 January 2017).
- Gnanasambandam, A.; Paull, J.; Torres, A.; Kaur, S.; Leonforte, T.; Li, H.; Zong, X.; Yang, T.; Materne, M. Impact of molecular technologies on faba bean (Vicia faba L.) breeding strategies. Agronomy 2012, 2, 132. [Google Scholar] [CrossRef]
- Sillero, J.C.; Villegas-Fernández, A.M.; Thomas, J.; Rojas-Molina, M.M.; Emeran, A.A.; Fernández-Aparicio, M.; Rubiales, D. Faba bean breeding for disease resistance. Field Crops Res. 2010, 115, 297–307. [Google Scholar] [CrossRef]
- Khan, H.R.; Paull, J.G.; Siddique, K.H.M.; Stoddard, F.L. Faba bean breeding for drought-affected environments: A physiological and agronomic perspective. Field Crops Res. 2010, 115, 279–286. [Google Scholar] [CrossRef]
- Slabu, C.; Zörb, C.; Steffens, D.; Schubert, S. Is salt stress of faba bean (Vicia faba) caused by Na+ or Cl− toxicity? JPNSS 2009, 172, 644–651. [Google Scholar] [CrossRef]
- Arbaoui, M.; Balko, C.; Link, W. Study of faba bean (Vicia faba L.) winter-hardiness and development of screening methods. Field Crops Res. 2008, 106, 60–67. [Google Scholar] [CrossRef]
- El-Sherbeeny, M.H.; Robertson, L.D. Protein content variation in a pure line faba bean (Vicia faba) collection. JSFA 1992, 58, 193–196. [Google Scholar] [CrossRef]
- Kumar, R.; Singh, M. Tannins: Their adverse role in ruminant nutrition. J. Agric. Food Chem. 1984, 32, 447–453. [Google Scholar] [CrossRef]
- Choi, H.-K.; Mun, J.-H.; Kim, D.-J.; Zhu, H.; Baek, J.-M.; Mudge, J.; Roe, B.; Ellis, N.; Doyle, J.; Kiss, G.B.; et al. Estimating genome conservation between crop and model legume species. Proc. Natl. Acad. Sci. USA 2004, 101, 15289–15294. [Google Scholar] [CrossRef] [PubMed]
- Chooi, W.Y. Variation in nuclear DNA content in the genus vicia. Genetics 1971, 68, 195–211. [Google Scholar] [PubMed]
- Bennett, M.D.; Leitch, I.J. Nuclear DNA amounts in angiosperms: Progress, problems and prospects. Ann. Bot. 2005, 95, 45–90. [Google Scholar] [CrossRef] [PubMed]
- Ellwood, S.R.; Phan, H.T.; Jordan, M.; Hane, J.; Torres, A.M.; Avila, C.M.; Cruz-Izquierdo, S.; Oliver, R.P. Construction of a comparative genetic map in faba bean (Vicia faba L.); conservation of genome structure with lens culinaris. BMC Genom. 2008, 9, 380. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Koblížková, A.; Navrátilová, A.; Macas, J. Significant expansion of Vicia pannonica genome size mediated by amplification of a single type of giant retroelement. Genetics 2006, 173, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Bond, D.A. Recent developments in breeding field beans (Vicia faba L.). Plant. Breed. 1987, 99, 1–26. [Google Scholar] [CrossRef]
- Torres, A.M.; Weeden, N.F.; Martín, A. Linkage among isozyme, rflp and rapd markers in Vicia faba. Theor. Appl. Genet. 1993, 85, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Avila, C.M.; Sillero, J.C.; Rubiales, D.; Moreno, M.T.; Torres, A.M. Identification of rapd markers linked to the Uvf-1 gene conferring hypersensitive resistance against rust (Uromyces viciae-fabae) in Vicia faba L. Theor. Appl. Genet. 2003, 107, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, N.; Avila, C.M.; Rodriguez-Suarez, C.; Moreno, M.T.; Torres, A.M. Development of scar markers linked to a gene controlling absence of tannins in faba bean. Mol. Breed. 2007, 19, 305–314. [Google Scholar] [CrossRef]
- Zeid, M.; Mitchell, S.; Link, W.; Carter, M.; Nawar, A.; Fulton, T.; Kresovich, S. Simple sequence repeats (SSRs) in faba bean: New loci from orobanche-resistant cultivar ‘giza 402’. Plant. Breed. 2009, 128, 149–155. [Google Scholar] [CrossRef]
- Gong, Y.-M.; Xu, S.-C.; Mao, W.-H.; Hu, Q.-Z.; Zhang, G.-W.; Ding, J.; Li, Z.-Y. Generation and characterization of 11 novel EST-derived microsatellites from Vicia faba (fabaceae). Am. J. Bot. 2010, 97, e69–e71. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Pembleton, L.W.; Cogan, N.O.I.; Savin, K.W.; Leonforte, T.; Paull, J.; Materne, M.; Forster, J.W. Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genom. 2012, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Kim, T.-S.; Raveendar, S.; Cho, J.-H.; Yi, J.Y.; Lee, M.C.; Lee, S.-Y.; Baek, H.-J.; Cho, G.-T.; Chung, J.-W. Transcriptome characterization and large-scale identification of SSR/SNP markers in symbiotic nitrogen fixation crop faba bean (Vicia faba L.). Turk. J. Agric. For. 2015, 39, 459–469. [Google Scholar] [CrossRef]
- Yang, T.; Bao, S.-Y.; Ford, R.; Jia, T.-J.; Guan, J.-P.; He, Y.-H.; Sun, X.-L.; Jiang, J.-Y.; Hao, J.-J.; Zhang, X.-Y.; et al. High-throughput novel microsatellite marker of faba bean via next generation sequencing. BMC Genom. 2012, 13, 602. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Izquierdo, S.; Avila, C.M.; Satovic, Z.; Palomino, C.; Gutierrez, N.; Ellwood, S.R.; Phan, H.T.; Cubero, J.I.; Torres, A.M. Comparative genomics to bridge Vicia faba with model and closely-related legume species: Stability of QTLs for flowering and yield-related traits. Theor. Appl. Genet. 2012, 125, 1767–1782. [Google Scholar] [CrossRef] [PubMed]
- Satovic, Z.; Avila, C.M.; Cruz-Izquierdo, S.; Díaz-Ruíz, R.; García-Ruíz, G.M.; Palomino, C.; Gutiérrez, N.; Vitale, S.; Ocaña-Moral, S.; Gutiérrez, M.V.; et al. A reference consensus genetic map for molecular markers and economically important traits in faba bean (Vicia faba L.). BMC Genom. 2013, 14, 932. [Google Scholar] [CrossRef] [PubMed]
- El-Rodeny, W.; Kimura, M.; Hirakawa, H.; Sabah, A.; Shirasawa, K.; Sato, S.; Tabata, S.; Sasamoto, S.; Watanabe, A.; Kawashima, K.; et al. Development of EST-SSR markers and construction of a linkage map in faba bean (Vicia faba). Breed. Sci. 2014, 64, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Kimber, R.B.; Cogan, N.O.; Materne, M.; Forster, J.W.; Paull, J.G. SNP discovery and high-density genetic mapping in faba bean (Vicia faba L.) permits identification of QTLs for ascochyta blight resistance. Plant. Sci. 2014, 217–218, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.; Cottage, A.; Wood, T.; Khamassi, K.; Hobbs, D.; Gostkiewicz, K.; White, M.; Khazaei, H.; Ali, M.; Street, D.; et al. A SNP-based consensus genetic map for synteny-based trait targeting in faba bean (Vicia faba L.). Plant. Biotechnol. J. 2016, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Avila, C.M.; Satovic, Z.; Sillero, J.C.; Nadal, S.; Rubiales, D.; Moreno, M.T.; Torres, A.M. QTL detection for agronomic traits in faba bean (Vicia faba L.). Agric. Conspec. Sci. 2005, 70, 65–73. [Google Scholar]
- Arbaoui, M.; Link, W.; Satovic, Z.; Torres, A.M. Quantitative trait loci of frost tolerance and physiologically related traits in faba bean (Vicia faba L.). Euphytica 2008, 164, 93–104. [Google Scholar] [CrossRef]
- Avila, C.M.; Satovic, Z.; Sillero, J.C.; Rubiales, D.; Moreno, M.T.; Torres, A.M. Isolate and organ-specific QTLs for ascochyta blight resistance in faba bean (Vicia faba L.). Theor. Appl. Genet. 2004, 108, 1071–1708. [Google Scholar] [CrossRef] [PubMed]
- Román, B.; Torres, A.M.; Rubiales, D.; Cubero, J.I.; Satovic, Z. Mapping of quantitative trait loci controlling broomrape (Orobanche crenata Forsk.) resistance in faba bean (Vicia faba L.). Genome 2002, 45, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Díaz, R.; Torres, A.M.; Satovic, Z.; Gutierrez, M.V.; Cubero, J.I.; Román, B. Validation of QTLs for Orobanche crenata resistance in faba bean (Vicia faba L.) across environments and generations. Theor. Appl. Genet. 2010, 120, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Atienza, S.G.; Palomino, C.; Gutiérrez, N.; Alfaro, C.M.; Rubiales, D.; Torres, A.M.; Ávila, C.M. QTLs for ascochyta blight resistance in faba bean (Vicia faba L.): Validation in field and controlled conditions. Crop. Pasture Sci. 2016, 67, 216–224. [Google Scholar] [CrossRef]
- Kumar, J.; Choudhary, A.K.; Solanki, R.K.; Pratap, A. Towards marker-assisted selection in pulses: A review. Plant. Breed. 2011, 130, 297–313. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Garg, R.; Jain, M. RNA-Seq for transcriptome analysis in non-model plants. Methods Mol. Biol. 2013, 1069, 43–58. [Google Scholar] [PubMed]
- Ray, H.; Bock, C.; Georges, F. Faba bean: Transcriptome analysis from etiolated seedling and developing seed coat of key cultivars for synthesis of proanthocyanidins, phytate, raffinose family oligosaccharides, vicine, and convicine. Plant. Genome 2015, 8, 1–11. [Google Scholar] [CrossRef]
- Arun-Chinnappa, K.S.; McCurdy, D.W. De novo assembly of a genome-wide transcriptome map of Vicia faba (L.) for transfer cell research. Front. Plant. Sci. 2015, 6, 217. [Google Scholar] [CrossRef] [PubMed]
- Madrid, E.; Horres, R.; Krezdorn, N.; Palomino, C.; Plötner, A.; Rotter, B.; Torres, A.M.; Winter, P. DeepSuperSage analysis of the Vicia faba transcriptome in response to Ascochyta fabae infection. Phytopathol. Mediterr. 2013, 52, 166–182. [Google Scholar]
- Ocaña, S.; Seoane, P.; Bautista, R.; Palomino, C.; Claros, G.M.; Torres, A.M.; Madrid, E. Large-scale transcriptome analysis in faba bean (Vicia faba L.) under Ascochyta fabae infection. PLoS ONE 2015, 10, e0135143. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.W.; Wilson, M.H.; Derks, M.F.L.; Smit, S.; Kunert, K.J.; Cullis, C.; Foyer, C.H. Enhancing faba bean (Vicia faba L.) genome resources. J. Exp. Bot. 2017, 68, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Libault, M.; Farmer, A.; Joshi, T.; Takahashi, K.; Langley, R.J.; Franklin, L.D.; He, J.; Xu, D.; May, G.; Stacey, G. An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. Plant. J. 2010, 63, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Sudheesh, S.; Sawbridge, T.I.; Cogan, N.O.I.; Kennedy, P.; Forster, J.W.; Kaur, S. De novo assembly and characterisation of the field pea transcriptome using RNA-Seq. BMC Genom. 2015, 16, 611. [Google Scholar] [CrossRef] [PubMed]
- Moreton, J.; Izquierdo, A.; Emes, R.D. Assembly, assessment, and availability of De novo generated eukaryotic transcriptomes. Front. Genet. 2015, 6, 361. [Google Scholar] [CrossRef] [PubMed]
- ABB Seeds-Doza (Faba Bean), NSW Department of Primary Industries. Available online: http://www.nvtonline.com.au/wp-content/uploads/2013/03/Fact-Sheet-Faba-Bean-Doza.pdf (accessed on 11 February 2017).
- 2017 Sowing Guide, South Australia. Available online: http://pir.sa.gov.au/__data/assets/pdf_file/0005/268862/SA_Sowing_Guide_2017.pdf (accessed on 15 April 2017).
- Plant Breeder’s Rights, IP Australia. Available online: https://www.ipaustralia.gov.au/plant-breeders-rights (accessed on 30 January 2017).
- Sudheesh, S.; Verma, P.; Forster, J.W.; Cogan, N.O.; Kaur, S. Generation and characterisation of a reference transcriptome for lentil (Lens culinaris Medik.). Int. J. Mol. Sci. 2016, 17, 1887. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’an, B.; et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Ray, H.; Georges, F. A genomic approach to nutritional, pharmacological and genetic issues of faba bean (Vicia faba): Prospects for genetic modifications. GM Crops 2010, 1, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Wheeler, S.; Xia, X.; Radchuk, R.; Weber, H.; Offler, C.E.; Patrick, J.W. Differential transcriptional networks associated with key phases of ingrowth wall construction in trans-differentiating epidermal transfer cells of Vicia faba cotyledons. BMC Plant. Biol. 2015, 15, 103. [Google Scholar] [CrossRef] [PubMed][Green Version]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant. Sci. 2014, 5, 484. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.L.; Trick, M.; Higgins, J.; Fraser, F.; Clissold, L.; Wells, R.; Hattori, C.; Werner, P.; Bancroft, I. Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat. Biotechnol. 2012, 30, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. Cap3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated megablast search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Medicago Truncatula Genome Database. Available online: http://www.medicagogenome.org/ (accessed on 15 November 2016).
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The european molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013. Available online: http://arxiv.org/abs/1303.3997 (accessed on 15 September 2016).
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008, 9, 253. [Google Scholar] [CrossRef] [PubMed]




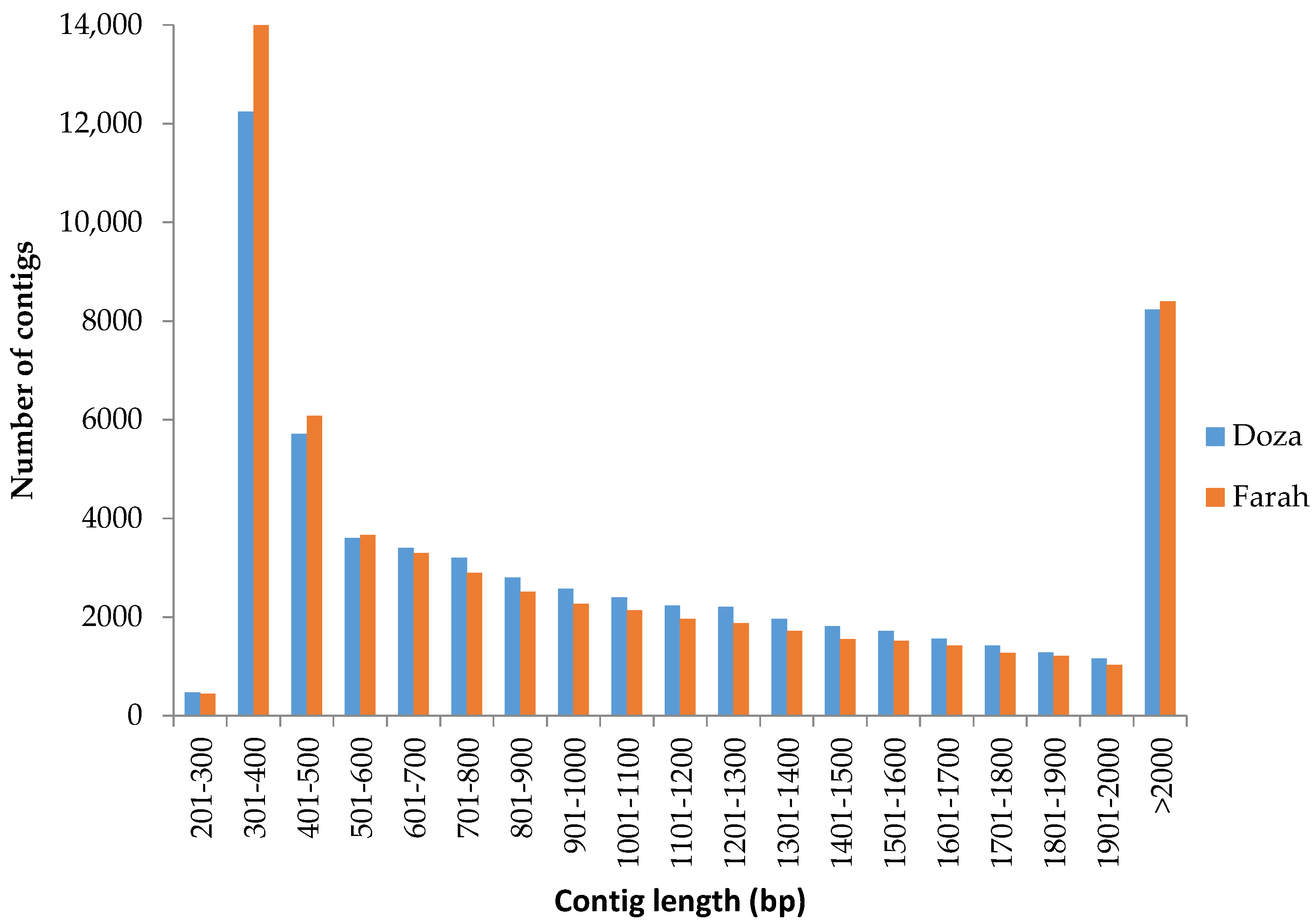{kind=link}
{kind=link}
{kind=link}
| Tissue | Doza | Farah |
|---|---|---|
| Flower | 139,572,392 | 180,879,976 |
| Immature pod | 90,270,053 | 192,279,013 |
| Pod | 42,557,479 | 159,411,032 |
| Immature seed | 65,603,441 | 163,953,461 |
| Leaf | 152,851,023 | 105,042,750 |
| Stem | 136,193,439 | 129,564,811 |
| Root | 149,339,568 | 106,690,173 |
| Total | 776,387,394 | 1,037,821,214 |
| Primary Assembly | Statistics-Doza | Statistics-Farah |
|---|---|---|
| SOAPdenovo-Trans | ||
| Total number of contigs and scaffolds | 79,782 | 73,989 |
| Total assembled (without N *) | 88.7 Mbp | 78 Mbp |
| N50 scaffold | 1751 bp | 1726 bp |
| CAP3 | ||
| Total number of contigs and scaffolds | 60,012 | 59,391 |
| Total assembled | 67.4 Mbp | 65.5 Mbp |
| N50 scaffold | 1588 bp | 1629 bp |
| Match to Comparator Legume | Doza Assembly | Farah Assembly | ||
|---|---|---|---|---|
| Transcripts with E-Value <10−50 | Transcripts with E-Value <10−10 | Transcripts with E-Value <10−50 | Transcripts with E-Value <10−10 | |
| Chickpea genome | 28,038 | 35,232 | 20,513 | 26,280 |
| Soybean CDS | 24,419 | 27,404 | 16,671 | 19,832 |
| Medicago CDS | 34,815 | 38,267 | 23,412 | 27,902 |
| Medicago genome | 32,916 | 39,968 | 24,289 | 30,265 |
| Statistics | Doza | Farah |
|---|---|---|
| Total number of sequences | 58,962 | 53,275 |
| Total length of transcripts | 66,959,534 bp | 60,943,125 bp |
| Number of transcripts with length <500 bp | 17,518 | 17,606 |
| Number of transcripts with length 500–1000 bp | 15,482 | 12,860 |
| Number of transcripts with length >1000 bp | 25,962 | 22,809 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braich, S.; Sudheesh, S.; Forster, J.W.; Kaur, S. Characterisation of Faba Bean (Vicia faba L.) Transcriptome Using RNA-Seq: Sequencing, De Novo Assembly, Annotation, and Expression Analysis. Agronomy 2017, 7, 53. https://doi.org/10.3390/agronomy7030053
Braich S, Sudheesh S, Forster JW, Kaur S. Characterisation of Faba Bean (Vicia faba L.) Transcriptome Using RNA-Seq: Sequencing, De Novo Assembly, Annotation, and Expression Analysis. Agronomy. 2017; 7(3):53. https://doi.org/10.3390/agronomy7030053
Chicago/Turabian StyleBraich, Shivraj, Shimna Sudheesh, John W. Forster, and Sukhjiwan Kaur. 2017. "Characterisation of Faba Bean (Vicia faba L.) Transcriptome Using RNA-Seq: Sequencing, De Novo Assembly, Annotation, and Expression Analysis" Agronomy 7, no. 3: 53. https://doi.org/10.3390/agronomy7030053
APA StyleBraich, S., Sudheesh, S., Forster, J. W., & Kaur, S. (2017). Characterisation of Faba Bean (Vicia faba L.) Transcriptome Using RNA-Seq: Sequencing, De Novo Assembly, Annotation, and Expression Analysis. Agronomy, 7(3), 53. https://doi.org/10.3390/agronomy7030053