
Metabolome and Transcriptome Analyses Revealing the Molecular Mechanisms of Precarious Fertility in Quinoa Cultivars

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation for Liquid Chromatography–Mass Spectrometry (LC/MS) Analysis
2.3. Metabolite Profiling on Phytohormones Using UPLC-MS/MS
2.4. Sample Preparation for RNA-Seq Analysis
2.5. RNA-Seq Analysis
2.6. Quantitative Real-Time PCR (qRT-PCR) Analysis
2.7. Data Analysis
3. Results
3.1. Morphological Characters of Three Ecotypes with Different Sterile Abilities
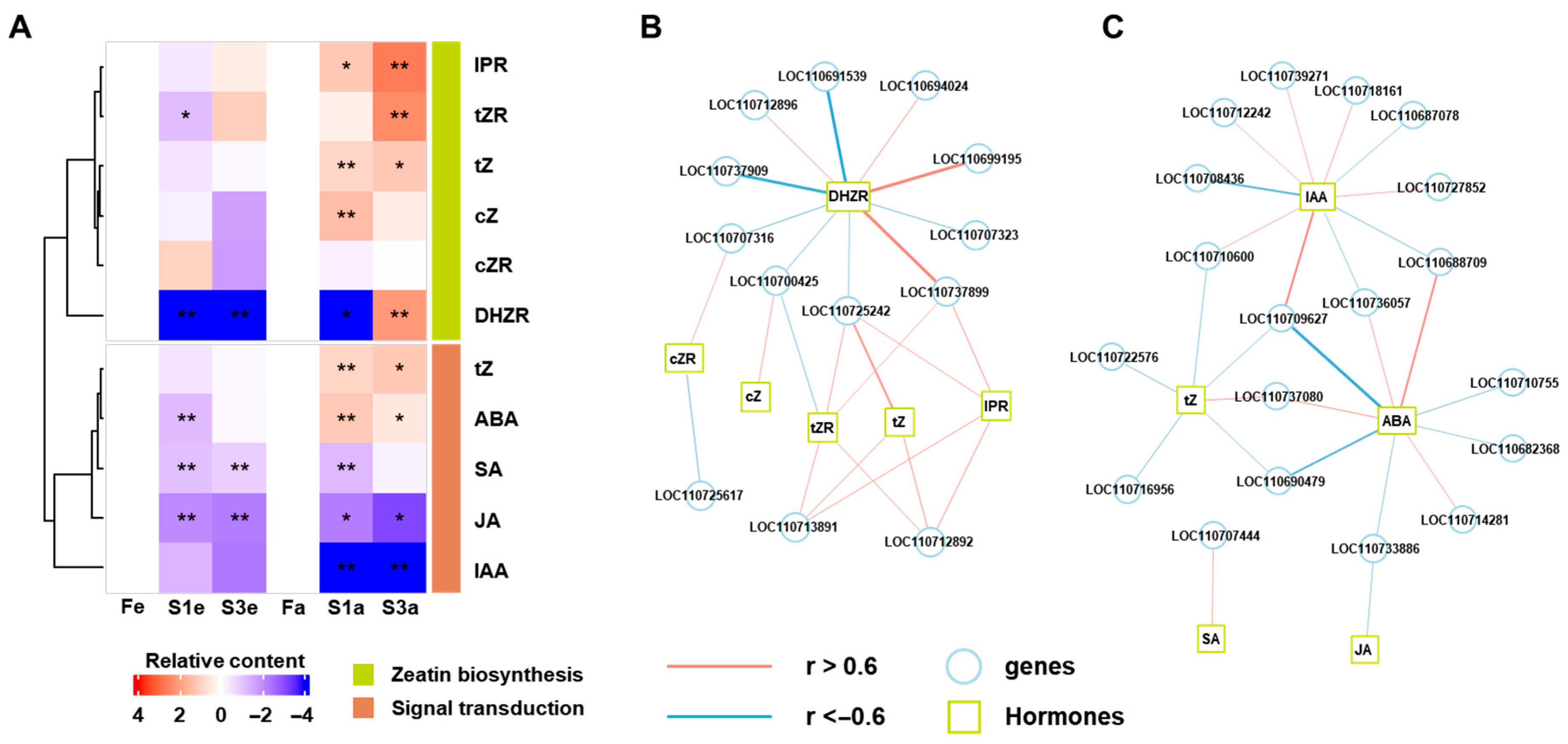
3.2. The Phytohormones in the Three Ecotypes of Quinoa Flowers
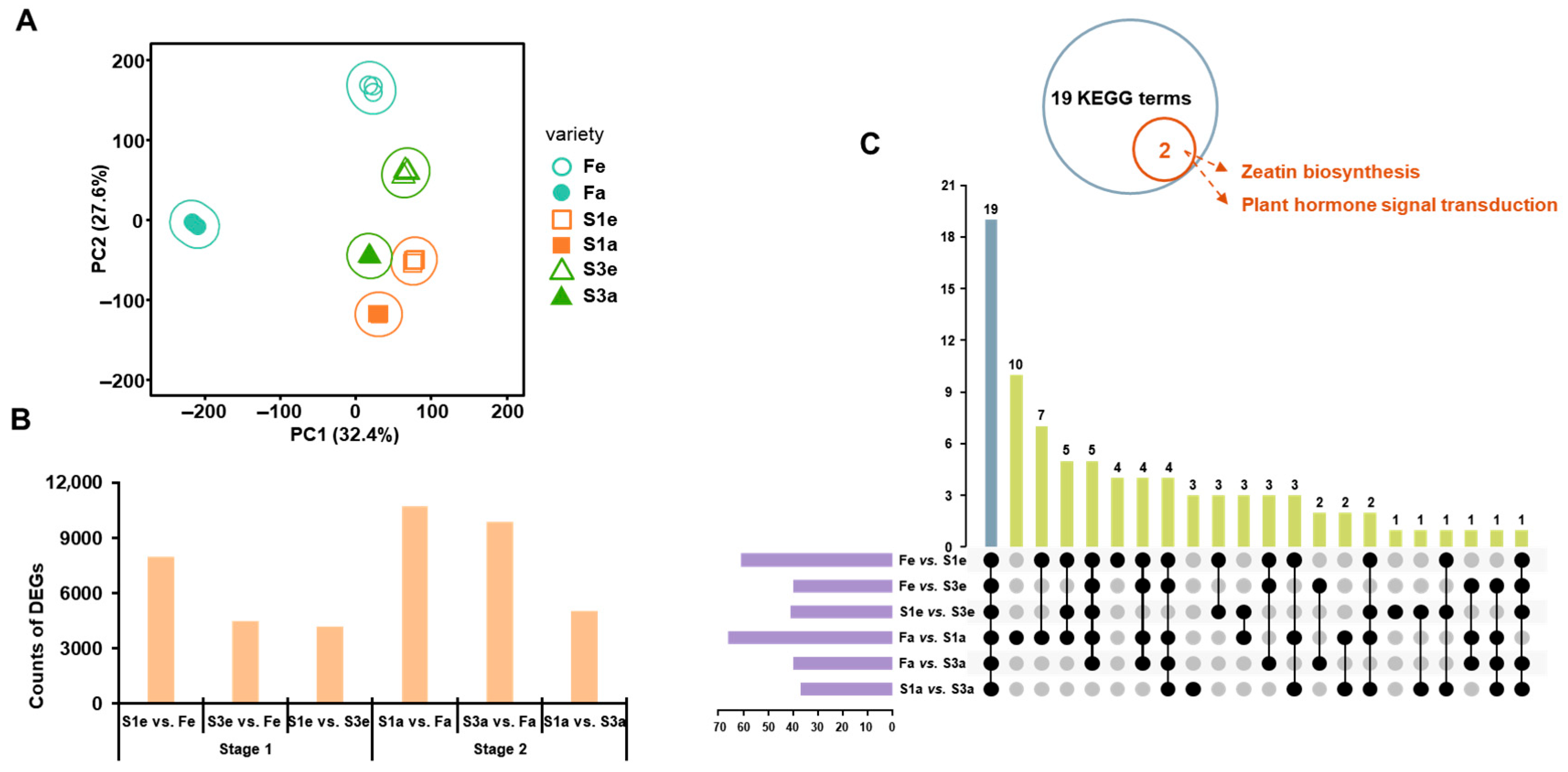
3.3. The Transcriptomic Analysis in the Three Ecotypes
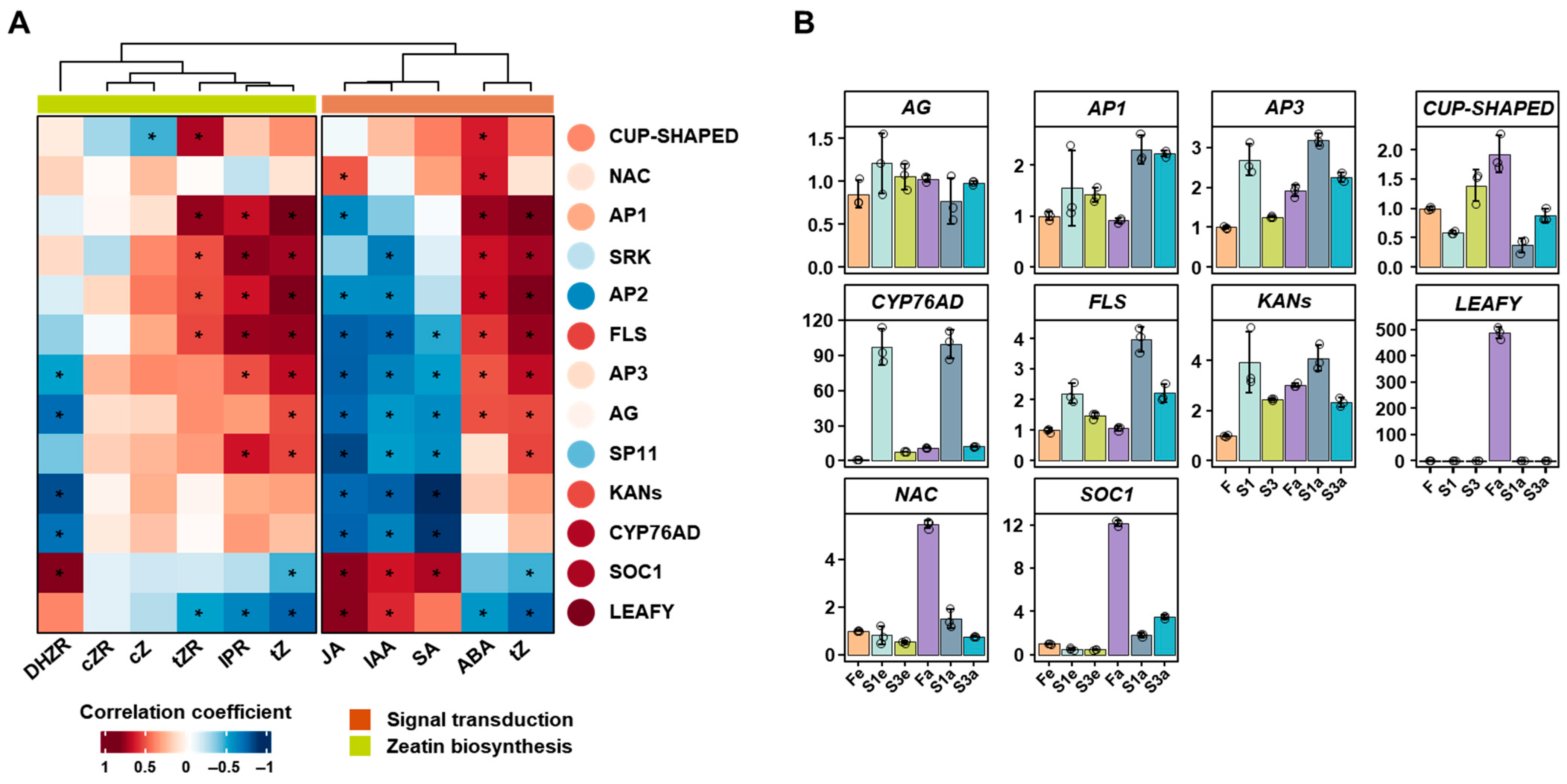
3.4. The Combined Analysis of Gene Expression and Hormone Accumulation
3.5. The Gene Expression Related to Flower Development
4. Discussion
4.1. Seed Coat Color and Inflorescence Color
4.2. Potential of Male Sterility in Quinoa Breeding
4.3. The Possible Roles of Phytohormones in Flower Fertility of Quinoa
4.4. The Transcriptome and RT-PCR Reveal the Interaction Between Phytohormones and Gene Expression in Flower Fertility of Quinoa
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Alandia, G.; Rodriguez, J.P.; Jacobsen, S.E.; Bazile, D.; Condori, B. Global expansion of quinoa and challenges for the andean region. Glob. Food Secur. 2020, 26, 100429. [Google Scholar] [CrossRef]
- Bhargava, A.; Shukla, S.; Ohri, D. Chenopodium quinoa—An indian perspective. Ind. Crops Prod. 2006, 23, 73–87. [Google Scholar] [CrossRef]
- Beloshapka, A.N.; Buff, P.R.; Fahey, G.C.; Swanson, K.S. Compositional analysis of whole grains, processed grains, grain co-products, and other carbohydrate sources with applicability to pet animal nutrition. Foods 2016, 5, 23. [Google Scholar] [CrossRef]
- Burrieza, H.P.; Rizzo, A.J.; Moura Vale, E.; Silveira, V.; Maldonado, S. Shotgun Proteomic analysis of quinoa seeds reveals novel lysine-rich seed storage globulins. Food Chem. 2019, 293, 299–306. [Google Scholar] [CrossRef]
- Wu, G.Y.; Morris, C.F.; Murphy, K.M. Evaluation of texture differences among varieties of cooked quinoa. J. Food Sci. 2014, 79, S2337–S2345. [Google Scholar] [CrossRef]
- Zevallos, V.F.; Herencia, I.L.; Chang, F.; Donnelly, S.; Ellis, J.H.; Ciclitira, P.J. Gastrointestinal effects of eating quinoa (Chenopodium quinoa Willd.) in celiac patients. Off. J. Am. Coll. Gastroenterol. ACG 2014, 109, 270. [Google Scholar] [CrossRef] [PubMed]
- Bazile, D.; Pulvento, C.; Verniau, A.; Al-Nusairi, M.S.; Ba, D.; Breidy, J.; Hassan, L.; Mohammed, M.I.; Mambetov, O.; Otambekova, M.; et al. Worldwide evaluations of quinoa: Preliminary results from post international year of quinoa FAO Projects in nine countries. Front. Plant Sci. 2016, 7, 850. [Google Scholar] [CrossRef]
- Murphy, K.M.; Bazile, D.; Kellogg, J.; Rahmanian, M. Development of a worldwide consortium on evolutionary participatory breeding in quinoa. Front. Plant Sci. 2016, 7, 608. [Google Scholar] [CrossRef]
- Ruiz, K.B.; Biondi, S.; Oses, R.; Acuña-Rodríguez, I.S.; Antognoni, F.; Martinez-Mosqueira, E.A.; Coulibaly, A.; Canahua-Murillo, A.; Pinto, M.; Zurita-Silva, A.; et al. Quinoa biodiversity and sustainability for food security under climate change. A review. Agron. Sustain. Dev. 2014, 34, 349–359. [Google Scholar] [CrossRef]
- Fuentes, F.F.; Bazile, D.; Bhargava, A.; Martínez, E.A. Implications of farmers’ seed exchanges for on-farm conservation of quinoa, as revealed by its genetic diversity in Chile. J. Agric. Sci. 2012, 150, 702–716. [Google Scholar] [CrossRef]
- Shen, Z.J.; Xu, S.X.; Huang, Q.Y.; Li, Z.Y.; Xu, Y.D.; Lin, C.S.; Huang, Y.J. TMT proteomics analysis of a pseudocereal crop, quinoa (Chenopodium Quinoa Willd.), during seed maturation. Front. Plant Sci. 2022, 13, 975073. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Lopes, C.; de Fátima Píccolo Barcelos, M.; de Goes Vieira, C.N.; de Abreu, W.C.; Ferreira, E.B.; Pereira, R.C.; de Angelis-Pereira, M.C. Effects of sprouted and fermented quinoa (Chenopodium quinoa) on glycemic index of diet and biochemical parameters of blood of wistar rats fed high carbohydrate diet. J. Food Sci. Technol. 2019, 56, 40–48. [Google Scholar] [CrossRef]
- Massawe, F.; Mayes, S.; Cheng, A. Crop Diversity: An unexploited treasure trove for food security. Trends Plant Sci. 2016, 21, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Walters, H.; Carpenter-Boggs, L.; Desta, K.; Yan, L.; Matanguihan, J.; Murphy, K. Effect of irrigation, intercrop, and cultivar on agronomic and nutritional characteristics of quinoa. Agroecol. Sustain. Food Syst. 2016, 40, 783–803. [Google Scholar] [CrossRef]
- Risi, C.J.; Galwey, N.W.; Risi, C.J.; Galwey, N.W. The Chenopodium grains of the andes: Inca crops for modern agriculture. Adv. Appl. Biol. 1984, 10, 145–216. [Google Scholar]
- Ward, S.M.; Johnson, D.L. Cytoplasmic male sterility in quinoa. Euphytica 1993, 66, 217–223. [Google Scholar] [CrossRef]
- Escribano, J.; Cabanes, J.; Jiménez-Atiénzar, M.; Ibañez-Tremolada, M.; Gómez-Pando, L.R.; García-Carmona, F.; Gandía-Herrero, F. Characterization of betalains, saponins and antioxidant power in differently colored quinoa (Chenopodium quinoa) varieties. Food Chem. 2017, 234, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.S.; Cheng, C.; Kong, J.; Li, H.J.; Hua, J.P. Plant non-coding RNAs function in pollen development and male sterility. Front. Plant Sci. 2023, 14, 1109941. [Google Scholar] [CrossRef]
- Nie, H.S.; Wang, Y.M.; Su, Y.; Hua, J.P. Exploration of miRNAs and target genes of cytoplasmic male sterility line in cotton during flower bud development. Funct. Integr. Genomics. 2018, 18, 457–476. [Google Scholar] [CrossRef]
- Bohra, A.; Gandham, P.; Rathore, A.; Thakur, V.; Saxena, R.K.; Naik, S.J.S.; Varshney, R.K.; Singh, N.P. Identification of microRNAs and their gene targets in cytoplasmic male sterile and fertile maintainer lines of pigeonpea. Planta 2021, 253, 59. [Google Scholar] [CrossRef]
- Cucinotta, M.; Cavalleri, A.; Chandler, J.W.; Colombo, L. Auxin and flower development: A blossoming field. Cold Spring Harb. Perspect. Biol. 2021, 13, a039974. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.D.; Zhu, J.X.; Gao, Q.; Luo, D.; Yuan, B.F.; Feng, Y.Q. Rapid and High-throughput determination of endogenous cytokinins in Oryza sativa by bare Fe3O4 nanoparticles-based magnetic solid-phase extraction. J. Chromatogr. A 2014, 1340, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.M.; Cai, W.J.; Ye, T.T.; Ding, J.; Feng, Y.Q. Spatio-temporal profiling of abscisic acid, indoleacetic acid and jasmonic acid in single rice seed during seed germination. Anal. Chim. Acta 2018, 1031, 119–127. [Google Scholar] [CrossRef]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Jarvis, D.E.; Ho, Y.S.; Lightfoot, D.J.; Schmöckel, S.M.; Li, B.; Borm, T.J.A.; Ohyanagi, H.; Mineta, K.; Michell, C.T.; Saber, N.; et al. The genome of Chenopodium quinoa. Nature 2017, 542, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. 13 May 2020. Available online: https://www.rdocumentation.org/packages/factoextra/versions/1.0.7 (accessed on 13 May 2024).
- Chen, C.J.; Wu, Y.; Li, J.W.; Wang, X.; Zeng, Z.H.; Xu, J.; Liu, Y.L.; Feng, J.T.; Chen, H.; He, Y.H.; et al. TBtools-II: A “One for All, All for One” Bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Gu, Z.G.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Revelle, W. Psych: Procedures for Psychological, Psychometric, and Personality Research. 13 May 2022. Available online: https://www.rdocumentation.org/packages/psych/versions/2.4.1 (accessed on 13 May 2024).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.D.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.Q.; Liu, Y.Y.; Liu, Y.; Cui, X.H.; Sun, Z.Y.; Du, Z.W.; Wu, K.; Jiang, X.L.; Mei, H.X.; Zheng, Y.Z. Genome-wide association study of seed coat color in sesame (Sesamum indicum L.). PLoS ONE 2021, 16, e0251526. [Google Scholar] [CrossRef]
- Pereira, E.; Cadavez, V.; Barros, L.; Encina-Zelada, C.; Stojković, D.; Sokovic, M.; Calhelha, R.C.; Gonzales-Barron, U.; Ferreira, I.C.F.R. Chenopodium quinoa Willd. (Quinoa) grains: A good source of phenolic compounds. Food Res. Int. 2020, 137, 109574. [Google Scholar] [CrossRef]
- Qian, G.T.; Li, X.Y.; Zhang, H.; Zhang, H.L.; Zhou, J.W.; Ma, X.H.; Sun, W.; Yang, W.; He, R.K.; Wahab, A.; et al. Metabolomics analysis reveals the accumulation patterns of flavonoids and phenolic acids in quinoa (Chenopodium quinoa Willd.) grains of different colors. Food Chem. X 2023, 17, 100594. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, X.H.; Zhang, B.; Chen, P.X.; Liu, R.H.; Tsao, R. Characterisation of phenolics, betanins and antioxidant activities in seeds of three Chenopodium quinoa Willd. genotypes. Food Chem. 2015, 166, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Calva-Estrada, S.J.; Jiménez-Fernández, M.; Lugo-Cervantes, E. Betalains and their applications in food: The current state of processing, stability and future opportunities in the industry. Food Chem. Mol. Sci. 2022, 4, 100089. [Google Scholar] [CrossRef] [PubMed]
- Calvi, P.; Terzo, S.; Amato, A. Betalains: Colours for human health. Nat. Prod. Res. 2023, 37, 1746–1765. [Google Scholar] [CrossRef]
- Timoneda, A.; Feng, T.; Sheehan, H.; Walker-Hale, N.; Pucker, B.; Lopez-Nieves, S.; Guo, R.; Brockington, S. The evolution of betalain biosynthesis in Caryophyllales. New Phytol. 2019, 224, 71–85. [Google Scholar] [CrossRef]
- Brockington, S.F.; Walker, R.H.; Glover, B.J.; Soltis, P.S.; Soltis, D.E. Complex pigment evolution in the Caryophyllales. New Phytol. 2011, 190, 854–864. [Google Scholar] [CrossRef]
- Feng, Y.; Yan, X.Z.; Guo, F.G.; Wang, S.Y.; Liu, Z.J.; Long, W.H. Identification, expression analysis of quinoa betalain biosynthesis genes and their role in seed germination and cold stress. Plant Signal. Behav. 2023, 18, e2250891. [Google Scholar] [CrossRef]
- Xu, S.X.; Huang, Q.Y.; Lin, C.S.; Lin, L.X.; Zhou, Q.; Lin, F.C.; He, E.M. Transcriptome comparison reveals candidate genes responsible for the betalain-/anthocyanidin-production in Bougainvilleas. Funct. Plant Biol. 2016, 43, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.; Jacobsen, S.-E.; Bonifacio, A.; Murphy, K. A crossing method for quinoa. Sustainability 2015, 7, 3230–3243. [Google Scholar] [CrossRef]
- Zabaleta, E.; Mouras, A.; Hernould, M.; Suharsono, N.; Araya, A. Transgenic male-sterile plant induced by an unedited Atp9 gene is restored to fertility by inhibiting its expression with antisense RNA. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 11259–11263. [Google Scholar] [CrossRef]
- Chen, L.T.; Liu, Y.G. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef]
- Kaul, M.L.H. Male Sterility in Higher Plants; Springer: Berlin, Heidelberg, 1988; Volume 10. [Google Scholar]
- Visarada, K.B.R.S.; Meena, K. Induction of male sterility: A boon for plant breeding. Int. J. Curr. Microbiol. Appl. Sci. 2021, 10, 1084–1101. [Google Scholar] [CrossRef]
- Zurita-Silva, A.; Fuentes, F.; Zamora, P.; Jacobsen, S.E.; Schwember, A.R. Breeding quinoa (Chenopodium quinoa Willd.): Potential and perspectives. Mol. Breed. 2014, 34, 13–30. [Google Scholar] [CrossRef]
- Chen, X.Y.; Li, Y.F.; Sun, H.Y.; An, X.L.; Tang, J.H. Molecular mechanisms of male sterility in maize. Plant Mol. Biol. Report. 2024, 42, 483–491. [Google Scholar] [CrossRef]
- Farinati, S.; Draga, S.; Betto, A.; Palumbo, F.; Vannozzi, A.; Lucchin, M.; Barcaccia, G. Current insights and advances into plant male sterility: New precision breeding technology based on genome editing applications. Front. Plant Sci. 2023, 14, 1223861. [Google Scholar] [CrossRef]
- Farooq, A.; Khan, U.M.; Khan, M.A.; Ali, Z.; Maqbool, R.; Sajjad, M. Male sterility systems and their applications in hybrid wheat breeding. Cereal Res. Commun. 2024, 52, 25–37. [Google Scholar] [CrossRef]
- Thomson, B.; Wellmer, F. Molecular regulation of flower development. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2019; Volume 131, pp. 185–210. [Google Scholar]
- Chandler, J.W. The hormonal regulation of flower development. J. Plant Growth Regul. 2011, 30, 242–254. [Google Scholar] [CrossRef]
- Liu, X.T.; Wu, J.; Ji, F.F.; Cao, X.Q.; Zhao, Q.C.; Cheng, C.X.; Ma, N.; Zhou, X.F.; Zhang, Z. Transcriptomic profiling of rose flower under treatment of various phytohormones and plant growth regulators. Sci. Data 2022, 9, 669. [Google Scholar] [CrossRef]
- Malik, N.U.A.; Fajer, O.; Amin, L.B.; Khalid, A.R.; Khan, N.; Bhatti, M.F.; Munir, F.; Haider, G.; Amir, R.; Gul, A. Phytohormones, plant growth and development. In Phytohormones and Stress Responsive Secondary Metabolites; Ozturk, M., Bhat, R.A., Ashraf, M., Tonelli, F.M.P., Unal, B.T., Dar, G.H., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 175–186. [Google Scholar]
- Song, S.S.; Qi, T.C.; Huang, H.; Xie, D.X. Regulation of stamen development by coordinated actions of jasmonate, auxin, and gibberellin in arabidopsis. Mol. Plant 2013, 6, 1065–1073. [Google Scholar] [CrossRef]
- Huang, H.; Chen, Y.; Wang, S.; Qi, T.; Song, S. Jasmonate action and crosstalk in flower development and fertility. J. Exp. Bot. 2023, 74, 1186–1197. [Google Scholar] [CrossRef]
- Sanders, P.M.; Lee, P.Y.; Biesgen, C.; Boone, J.D.; Beals, T.P.; Weiler, E.W.; Goldberg, R.B. The arabidopsis DELAYED DEHISCENCE1 gene encodes an enzyme in the jasmonic acid synthesis pathway. Plant Cell 2000, 12, 1041–1061. [Google Scholar] [CrossRef]
- Minato, N.; Himeno, M.; Hoshi, A.; Maejima, K.S.K.; Komatsu, K.; Takebayashi, Y.; Kasahara, H.; Yusa, A.; Yamaji, Y.; Oshima, K.; et al. The phytoplasmal virulence factor tengu causes plant sterility by downregulating of the jasmonic acid and auxin pathways. Sci. Rep. 2014, 4, 7399. [Google Scholar] [CrossRef] [PubMed]
- Żur, I.; Dubas, E.; Krzewska, M.; Waligórski, P.; Dziurka, M.; Janowiak, F. Hormonal requirements for effective induction of microspore embryogenesis in triticale (× Triticosecale Wittm.) anther cultures. Plant Cell Rep. 2015, 34, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, N.; Heisler, M.G. Self-Organizing periodicity in development: Organ positioning in plants. Development 2018, 145, dev149336. [Google Scholar] [CrossRef] [PubMed]
- Heisler, M.G.; Ohno, C.; Das, P.; Sieber, P.; Reddy, G.V.; Long, J.A.; Meyerowitz, E.M. Patterns of auxin transport and gene expression during primordium development revealed by live imaging of the arabidopsis inflorescence meristem. Curr. Biol. 2005, 15, 1899–1911. [Google Scholar] [CrossRef]
- Li, X.; Kuang, Y.; Ye, Y.; Chen, Z.; Zhang, M. Diverse function of the PISTILLATA, APETALA 3, and AGAMOUS-like MADS-Box genes involved in the floral development in Alpinia hainanensis (Zingiberaceae). Gene 2022, 839, 146732. [Google Scholar] [CrossRef]
- Wellmer, F.; Riechmann, J.L. Gene networks controlling the initiation of flower development. Trends Genet. 2010, 26, 519–527. [Google Scholar] [CrossRef]
- Lee, J.; Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 2010, 61, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xi, W.; Shen, L.; Tan, C.; Yu, H. Regulation of floral patterning by flowering time genes. Dev. Cell 2009, 16, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Yanofsky, M.F. Activation of the Arabidopsis B class homeotic genes by APETALA1. Plant Cell 2001, 13, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.M.; Austin, R.S.; Blanvillain-Baufumé, S.; Reback, M.A.; Monniaux, M.; Wu, M.-F.; Sang, Y.; Yamaguchi, A.; Yamaguchi, N.; Parker, J.E.; et al. LEAFY target genes reveal floral regulatory logic, Cis motifs, and a link to biotic stimulus response. Dev. Cell 2011, 20, 430–443. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.; Huang, Q.; Lin, C.; Li, H.; Li, Z.; Xu, Y.; Chen, F.; Zhang, X.; Wu, Z.; Xu, S. Metabolome and Transcriptome Analyses Revealing the Molecular Mechanisms of Precarious Fertility in Quinoa Cultivars. Agronomy 2025, 15, 1694. https://doi.org/10.3390/agronomy15071694
Song C, Huang Q, Lin C, Li H, Li Z, Xu Y, Chen F, Zhang X, Wu Z, Xu S. Metabolome and Transcriptome Analyses Revealing the Molecular Mechanisms of Precarious Fertility in Quinoa Cultivars. Agronomy. 2025; 15(7):1694. https://doi.org/10.3390/agronomy15071694
Chicago/Turabian StyleSong, Chao, Qingyun Huang, Chunsong Lin, Huihua Li, Ziyang Li, Yiding Xu, Fei Chen, Xueqin Zhang, Zhiqiang Wu, and Suxia Xu. 2025. "Metabolome and Transcriptome Analyses Revealing the Molecular Mechanisms of Precarious Fertility in Quinoa Cultivars" Agronomy 15, no. 7: 1694. https://doi.org/10.3390/agronomy15071694
APA StyleSong, C., Huang, Q., Lin, C., Li, H., Li, Z., Xu, Y., Chen, F., Zhang, X., Wu, Z., & Xu, S. (2025). Metabolome and Transcriptome Analyses Revealing the Molecular Mechanisms of Precarious Fertility in Quinoa Cultivars. Agronomy, 15(7), 1694. https://doi.org/10.3390/agronomy15071694