Comparative Analysis of Ratoon-Competent and Ratoon-Deficient Sugarcane by Hormonal and Transcriptome Profiling

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.3. Determination of Hormone Content
2.4. RNA Extraction, Transcriptome Sequencing, and Annotation
2.5. DEGs Screening and Enrichment Analysis
2.6. Real-Time Quantitative PCR Analysis
2.7. Statistical Analysis of the Data
3. Results
3.1. Characteristic Analysis of Hormone Content
3.1.1. Characteristics of CTK Content
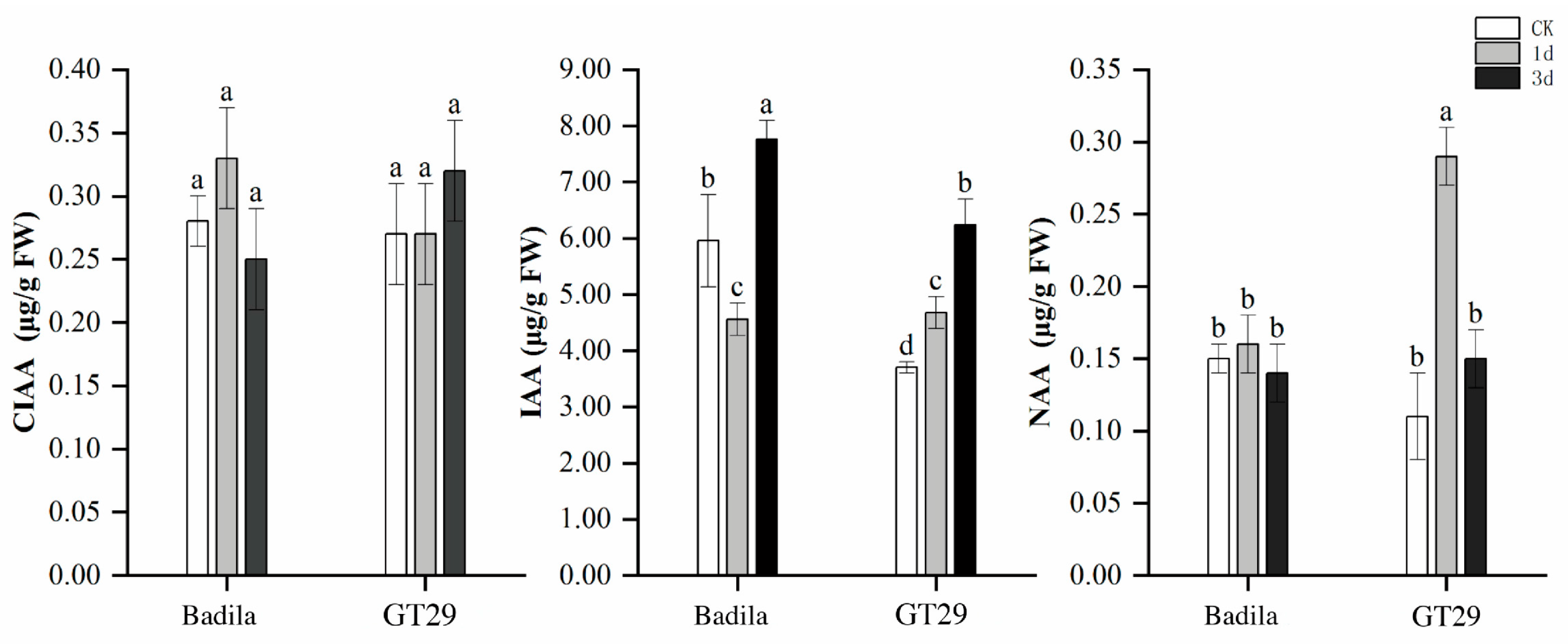
3.1.2. Characteristics of IAA Content
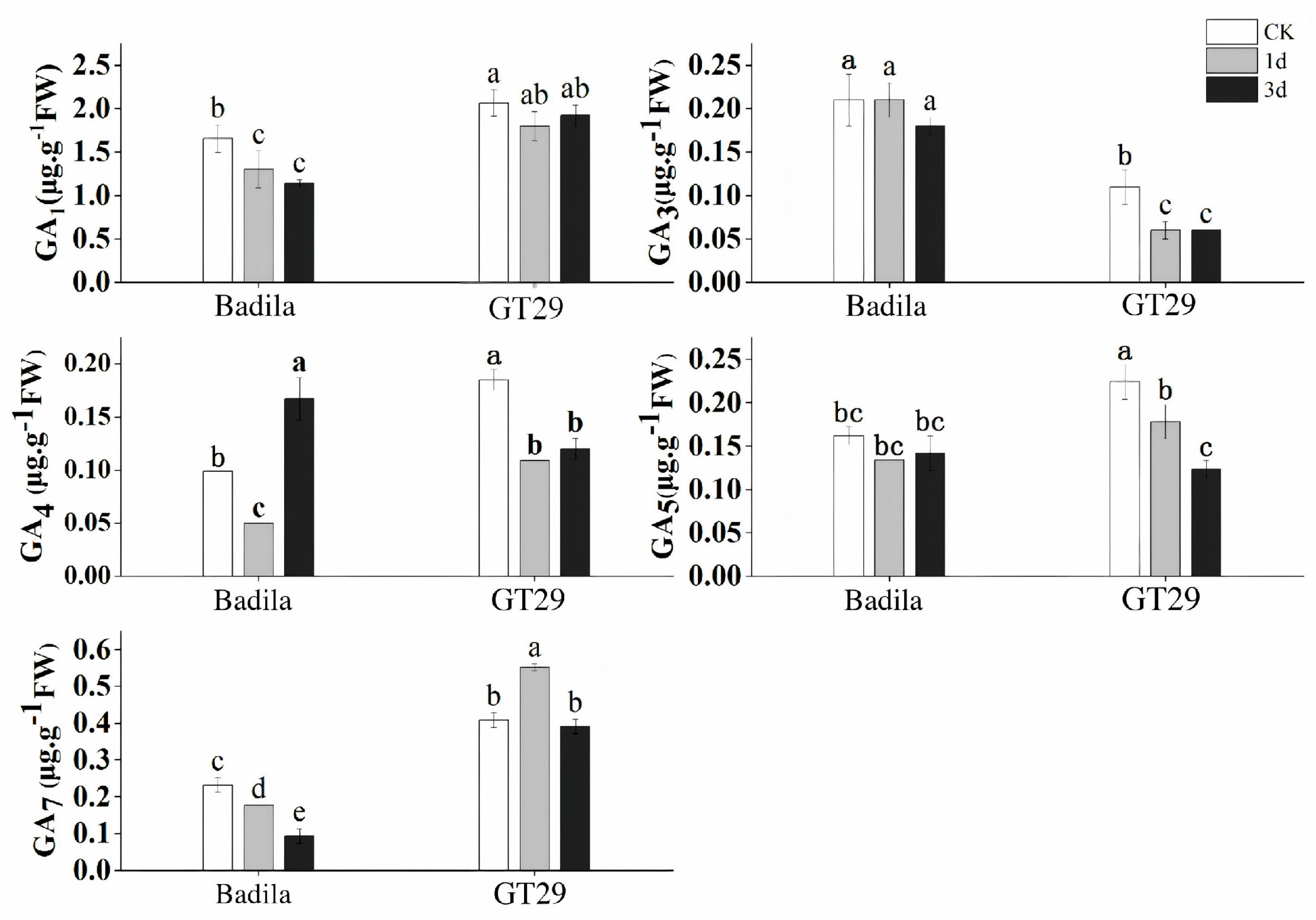
3.1.3. Characteristics of GA Content
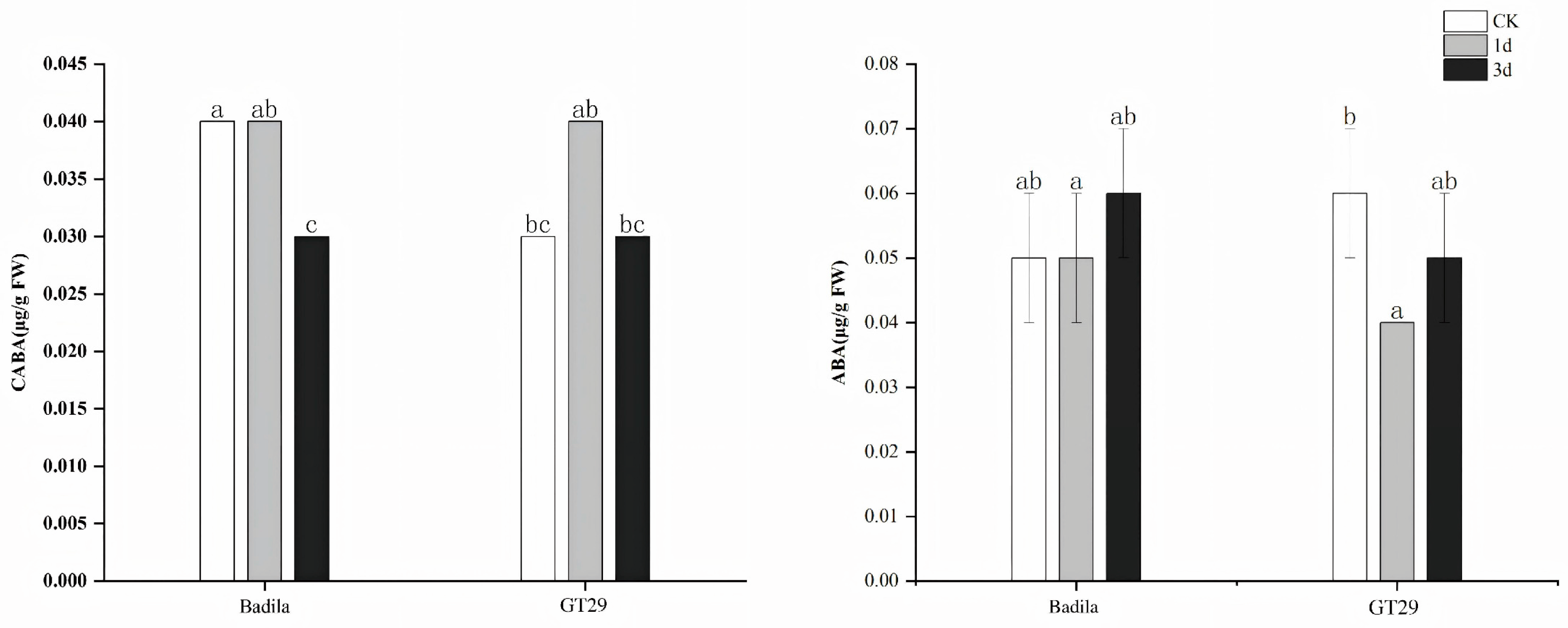
3.1.4. Characteristics of ABA Content
3.1.5. Characteristics of 5-DS Content
3.2. Transcriptome Analysis
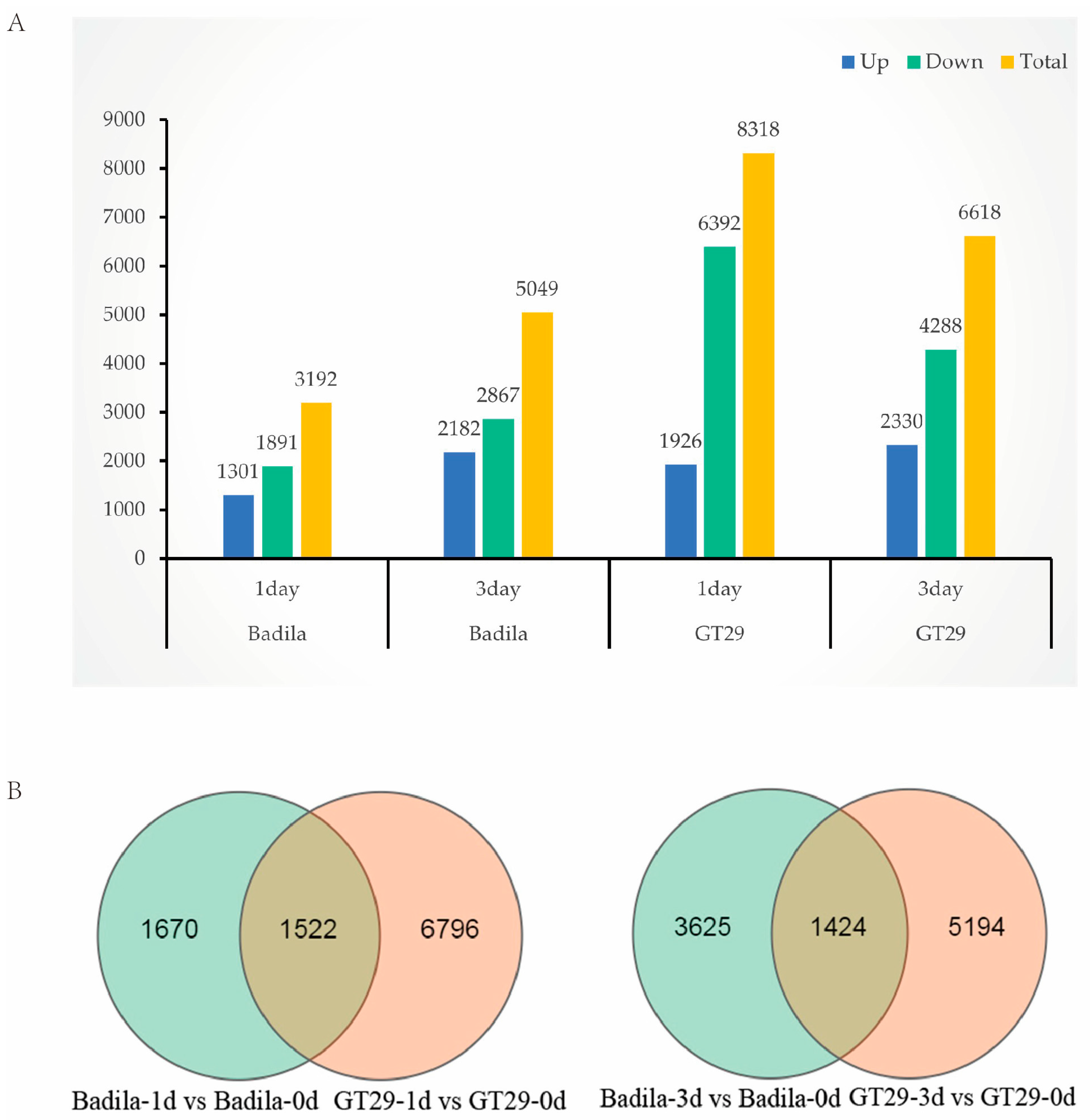
3.3. DEGs Screening
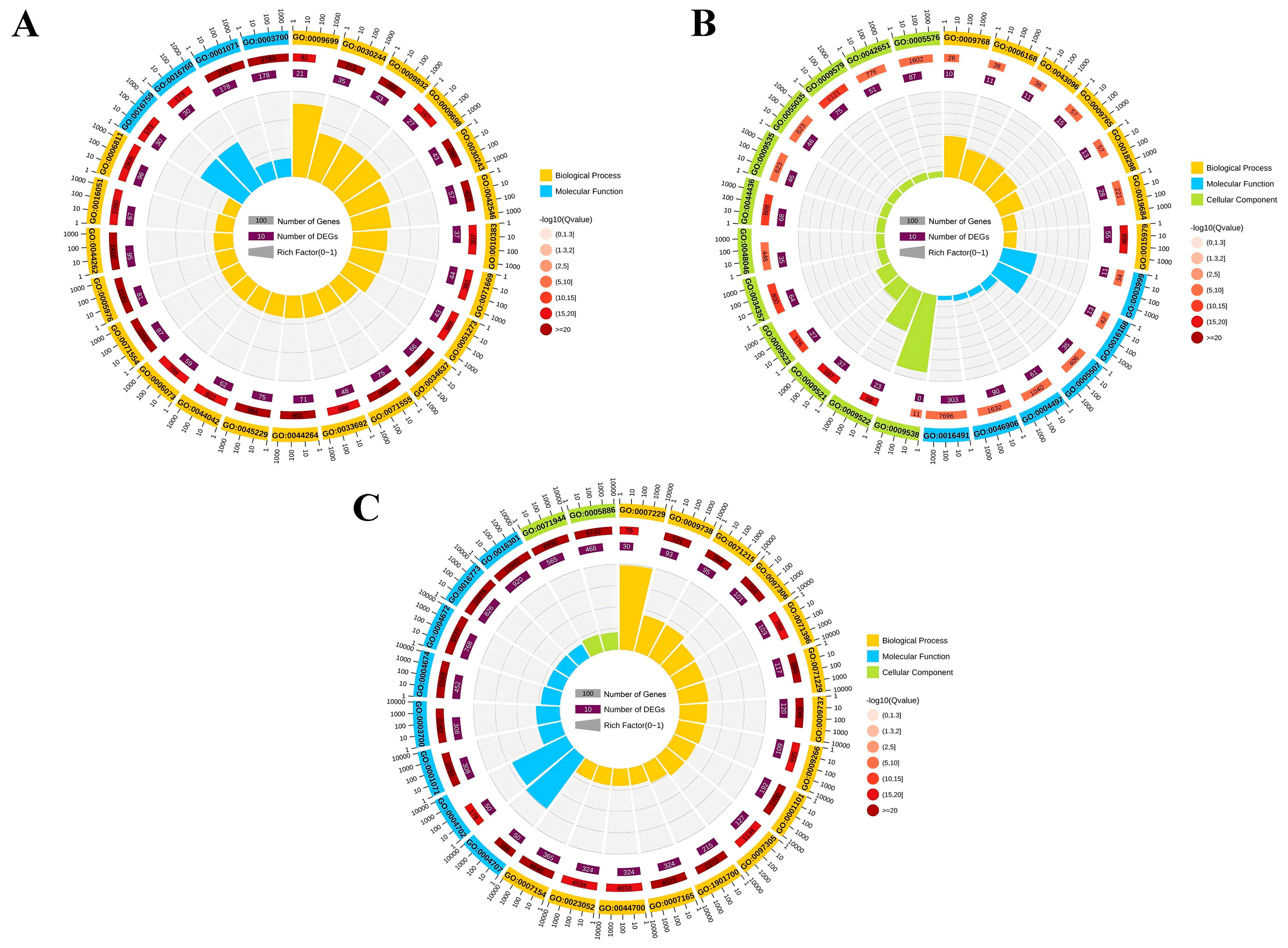
3.4. GO Analysis of DEGs
3.5. KEGG Pathway of DEGs
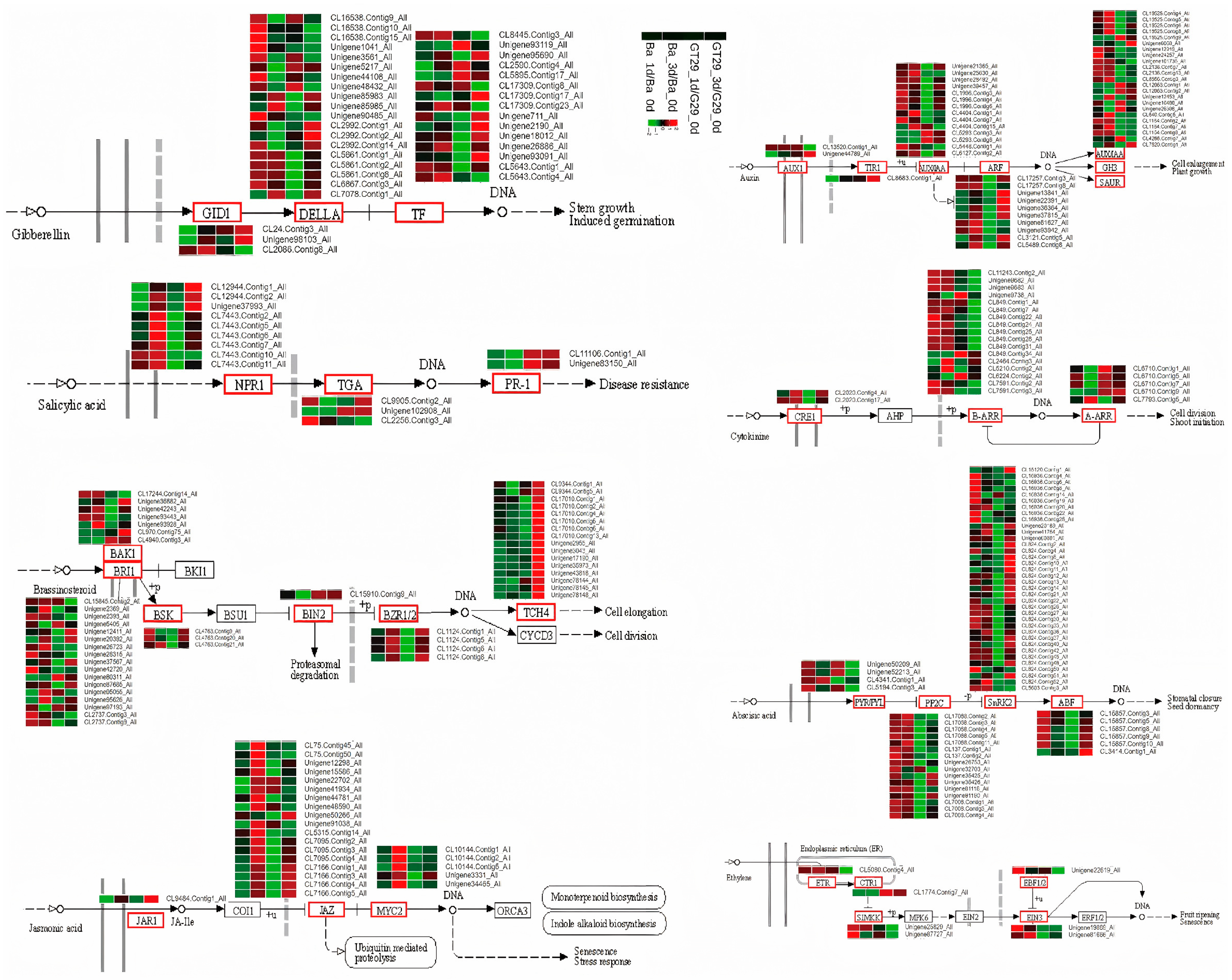
3.6. Characterization of Key Genes in Phytohormone Signal Transduction Pathways
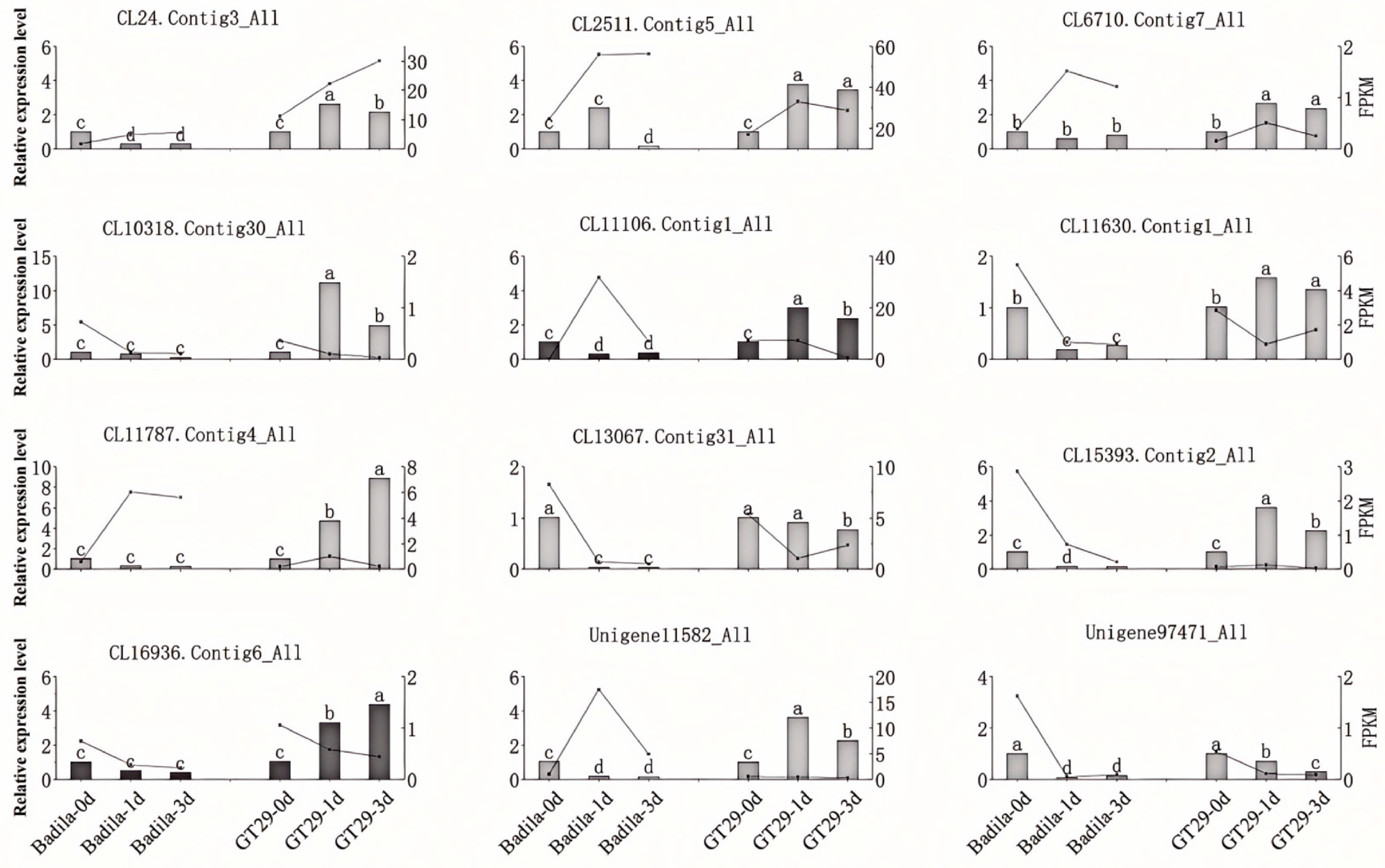
3.7. qRT-PCR Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ran, M.; Yu, B.; Cheng, C.; Li, X.; Guo, Y.; Zhao, L.; Zan, F.; Lin, X.; Hou, X.; Zhao, Y.; et al. Oligo-FISH-based analysis of the mechanisms underlying chromosome number variation in Saccharum spontaneum. Int. J. Mol. Sci. 2025, 26, 1958. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Lin, P.; Yu, F.; Deng, Z. Research progress of sugarcane rationing ability. Sugar Crops China 2020, 42, 12–18. [Google Scholar]
- Xu, F.; Wang, Z.; Lu, G.; Zeng, R.; Que, Y. Sugarcane ratooning ability: Research status, shortcomings, and prospects. Biology 2021, 10, 1052. [Google Scholar] [CrossRef] [PubMed]
- Dlamini, N.E.; Zhou, M. Predicting ratooning ability of sugarcane varieties in selection trials. Sugar Tech. 2024, 26, 52–62. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, D.; Wu, Q.; Fang, J.; Ao, J. Effects of tillage practices on yield, carbon emissions and economic benefits of ratoon sugarcane. Chin. J. Eco-Agric. 2024, 32, 1481–1491. [Google Scholar]
- Kumar, D.; Singh, P.; Verma, L.K.; Nag, N.K. An economic analysis of production of sugarcane under various methods of irrigation in Sonipat District of Haryana, India. Int. J. Curr. Microbiol. App. Sci. 2019, 8, 808–816. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Y.; Deng, J.; Li, R.; Fan, X.; Dao, J.; Quan, Y.; Hussain Bukhari, S.A. Effect of cutting depth during sugarcane (Saccharum spp. hybrid) harvest on root characteristics and yield. PLoS ONE 2021, 16, e0238085. [Google Scholar]
- Yan, H.; Zhou, H.; Luo, H.; Fan, Y.; Zhou, Z.; Chen, R.; Luo, T.; Li, X.; Liu, X.; Li, Y.; et al. Characterization of full-length transcriptome in Saccharum officinarum and molecular insights into tiller development. BMC Plant Biol. 2021, 21, 228. [Google Scholar] [CrossRef]
- Zhao, L.; Yang, K.; Zhao, P.; Qin, W.; Zhao, Y.; Zhu, J.; Zan, F.; Zhao, J.; Lu, X.; Wu, C.; et al. Sugarcane root distribution and growth as affected by genotype and crop cycle. Bragantia 2020, 79, 192–202. [Google Scholar] [CrossRef]
- Chumphu, S.; Jongrungklang, N.; Songsri, P. Association of physiological responses and root distribution patterns of ratooning ability and yield of the second ratoon cane in sugarcane elite clones. Agronomy 2019, 9, 200. [Google Scholar] [CrossRef]
- Wang, X.; Liang, Y.; Li, L.; Gong, C.; Wang, H.; Huang, X.; Li, S.; Deng, Q.; Zhu, J.; Zheng, A.; et al. Identification and cloning of tillering-related genes OsMAX1 in rice. Rice Sci. 2015, 22, 255–263. [Google Scholar]
- Wang, T.; Xu, F.; Wang, Z.; Wu, Q.; Cheng, W.; Que, Y.; Xu, L. Mapping of QTLs and screening candidate genes associated with the ability of sugarcane tillering and ratooning. Int. J. Mol. Sci. 2023, 24, 2793. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Wu, Q.; Yang, S.; Liu, J.; Zhao, Y.; Zhao, P.; Wang, L.; Lu, W.; Huang, D.; Zhang, Y.; et al. Dissection of genetic architecture for desirable traits in sugarcane by integrated transcriptomics and metabolomics. Int. J. Biol. Macromol. 2024, 280, 136009. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Corak, K.; McCord, P.; Hulse-Kemp, A.M.; Lipka, A.E. A first look at the ability to use genomic prediction for improving the ratooning ability of sugarcane. Front. Plant Sci. 2023, 14, 1205999. [Google Scholar] [CrossRef]
- Henry, A. A step forward in breeding for ratooning ability in rice. Mol. Plant 2024, 17, 368–369. [Google Scholar] [CrossRef]
- Liang, Q.; Liu, X.; Zhou, H.; Lei, J.; Lin, S.; Yan, M.; Verma, K.K.; Wei, K.; Wei, H.; Li, W. Breeding and regional production capacity performance of new sugarcane cultivar GT 66. Sci. Rep. 2025, 15, 12963. [Google Scholar] [CrossRef]
- Yang, S.; Chu, N.; Feng, N.; Zhou, B.; Zhou, H.; Deng, Z.; Shen, X.; Zheng, D. Global responses of autopolyploid sugarcane Badila (Saccharum officinarum L.) to drought stress based on comparative transcriptome and metabolome profiling. Int. J. Mol. Sci. 2023, 24, 3856. [Google Scholar] [CrossRef]
- Yao, W.; Ruan, M.; Qin, L.; Yang, C.; Chen, R.; Chen, B.; Zhang, M. Field performance of transgenic sugarcane lines resistant to sugarcane mosaic virus. Front. Plant Sci. 2017, 8, 104. [Google Scholar] [CrossRef]
- Hussain, S.; Wang, W.; Ahmed, S.; Wang, X.; Adnan; Cheng, Y.; Wang, C.; Wang, Y.; Zhang, N.; Tian, H. PIP2, an auxin induced plant peptide hormone regulates root and hypocotyl elongation in Arabidopsis. Front. Plant Sci. 2021, 12, 646736. [Google Scholar] [CrossRef]
- Bai, Y.; Cai, M.; Mu, C.; Zheng, H.; Cheng, Z.; Xie, Y.; Gao, J. Integrative analysis of exogenous auxin mediated plant height regulation in Moso bamboo (Phyllostachys edulis). Ind. Crops Prod. 2023, 200, 116852. [Google Scholar] [CrossRef]
- Kubalová, M.; Müller, K.; Dobrev, P.I.; Rizza, A.; Jones, A.M.; Fendrych, M. Auxin co-receptor IAA17/AXR3 controls cell elongation in Arabidopsis thaliana root solely by modulation of nuclear auxin pathway. New Phytol. 2024, 241, 2448–2463. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Liu, L.; Li, M.; Fang, S.; Shen, X.; Chu, J.; Zhang, Z. UNBRANCHED3 regulates branching by modulating cytokinin biosynthesis and signaling in maize and rice. New Phytol. 2017, 214, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, H.; Zhu, Q.; Liu, D.; Li, Z.; Chen, H.; Luo, J.; Gong, P.; Ismail, A.M.; Zhang, Z. Knockout of the sugar transporter OsSTP15 enhances grain yield by improving tiller number due to increased sugar content in the shoot base of rice (Oryza sativa L.). New Phytol. 2024, 241, 1250–1265. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, Y.; Qiao, Y.; Sun, H.; Liu, W.; Qiao, W.; Li, W.; Liu, M.; Dong, B. Low light stress promotes new tiller regeneration by changing source–sink relationship and activating expression of expansin genes in wheat. Plant Cell Environ. 2023, 46, 1562–1581. [Google Scholar] [CrossRef]
- Koprna, R.; Humplík, J.F.; Špíšek, Z.; Bryksová, M.; Zatloukal, M.; Mik, V.; Novák, O.; Nisler, J.; Doležal, K. Improvement of tillering and grain yield by application of cytokinin derivatives in wheat and barley. Agronomy 2020, 11, 67. [Google Scholar] [CrossRef]
- Lei, K.; Tan, Q.; Zhu, L.; Xu, L.; Yang, S.; Hu, J.; Gao, L.; Hou, P.; Shao, Y.; Jiang, D.; et al. Low red/far-red ratio can induce cytokinin degradation resulting in the inhibition of tillering in wheat (Triticum aestivum L.). Front. Plant Sci. 2022, 13, 971003. [Google Scholar] [CrossRef]
- Ji, Y.; Huang, W.; Wu, B.; Fang, Z.; Wang, X. The amino acid transporter AAP1 mediates growth and grain yield by regulating neutral amino acid uptake and reallocation in Oryza sativa. J. Exp. Bot. 2020, 71, 4763–4777. [Google Scholar] [CrossRef]
- Ferreira, L.G.; de Alencar Dusi, D.M.; Irsigler, A.S.T.; Gomes, A.C.M.M.; Mendes, M.A.; Colombo, L.; de Campos Carneiro, V.T. GID1 expression is associated with ovule development of sexual and apomictic plants. Plant Cell Rep. 2018, 37, 293–306. [Google Scholar] [CrossRef]
- Voegele, A.; Linkies, A.; Müller, K.; Leubner-Metzger, G. Members of the gibberellin receptor gene family GID1 (GIBBERELLIN INSENSITIVE DWARF1) play distinct roles during Lepidium sativum and Arabidopsis thaliana seed germination. J. Exp. Bot. 2011, 62, 5131–5147. [Google Scholar] [CrossRef]
- Ueguchi-Tanaka, M.; Nakajima, M.; Motoyuki, A.; Matsuoka, M. Gibberellin receptor and its role in gibberellin signaling in plants. Annu. Rev. Plant Biol. 2007, 58, 183–198. [Google Scholar] [CrossRef]
- Zhong, M.; Zeng, B.; Tang, D.; Yang, J.; Qu, L.; Yan, J.; Wang, X.; Li, X.; Liu, X.; Zhao, X. The blue light receptor CRY1 interacts with GID1 and DELLA proteins to repress GA signaling during photomorphogenesis in Arabidopsis. Mol. Plant 2021, 14, 1328–1342. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Sun, S.; Song, C.-P.; Zhou, J.-M.; Li, J.; Zuo, J. Nitric oxide negatively regulates gibberellin signaling to coordinate growth and salt tolerance in Arabidopsis. J. Genet. Genomics 2022, 49, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Gao, X.; He, P.; Xiao, G. Origin, evolution, and molecular function of DELLA proteins in plants. Crop J. 2022, 10, 287–299. [Google Scholar] [CrossRef]
- Li, K.; Yu, R.; Fan, L.-M.; Wei, N.; Chen, H.; Deng, X.W. DELLA-mediated PIF degradation contributes to coordination of light and gibberellin signalling in Arabidopsis. Nat. Commun. 2016, 7, 11868. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xie, Z.; Qin, T.; Zhan, C.; Jin, L.; Huang, J. The SLR 1-OsMADS 23-D14 module mediates the crosstalk between strigolactone and gibberellin signaling to control rice tillering. New Phytol. 2025, 246, 2137–2154. [Google Scholar] [CrossRef]
- Sun, H.; Guo, X.; Zhu, X.; Gu, P.; Zhang, W.; Tao, W.; Wang, D.; Wu, Y.; Zhao, Q.; Xu, G.; et al. Strigolactone and gibberellin signaling coordinately regulate metabolic adaptations to changes in nitrogen availability in rice. Mol. Plant 2023, 16, 588–598. [Google Scholar] [CrossRef]
- Zhuang, H.; Li, Y.F. Strigolactone and gibberellin crosstalk: The role of the SLR1-OsMADS23-D14 module in regulating rice tiller development. New Phytol. 2025, 246, 1893–1895. [Google Scholar] [CrossRef]
- van Mourik, H.; van Dijk, A.D.; Stortenbeker, N.; Angenent, G.C.; Bemer, M. Divergent regulation of Arabidopsis SAUR genes: A focus on the SAUR10-clade. BMC Plant Biol. 2017, 17, 245. [Google Scholar] [CrossRef]
- Yin, H.; Li, M.; Lv, M.; Hepworth, S.R.; Li, D.; Ma, C.; Li, J.; Wang, S.-M. SAUR15 promotes lateral and adventitious root development via activating H+-ATPases and auxin biosynthesis. Plant Physiol. 2020, 184, 837–851. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Forward Sequence (5′–3′) | Reverse Sequence (5′–3′) |
|---|---|---|
| 18S rRNA | CAACCATAAACGATGCCGA | AGCCTTGCGACCATACTCC |
| CL11787.Contig4 | CTTGGTCTTCCAGCAGGA | ACGGAGGTGTTCCTGAAG |
| CL15393.Contig2 | GCAAGAGGAAATACGACCC | CCCATCAGGCAAGTAACG |
| Unigene11582 | TGGTTTGGGATTGAGCAA | CGGCACAGTAGTATGGTC |
| CL2511.Contig5 | GAAGCCTCTGGACTACGA | ATCTTCTTGCCCTCCTTCT |
| Unigene97471 | ATGCTGGCTTAC CTTCTC | TTCCGAATCCCTGTATGAC |
| CL24.Contig3 | TCCTCTCCTCCTCGTACT | TCGGCTCCTCCAATACAA |
| CL13067.Contig31 | TTCAGACAAGCGTAGCCA | TGAAGGTGTGGTCAGCAT |
| CL11106.Contig1 | TGTCTTGCTGGTGTCGTT | TCTTGAGGCGAGTTCTGAG |
| CL6710.Contig7 | TGGATACACCAGGGCAAA | CACACTTCTCGGCAGTTG |
| CL16936.Contig6 | CGGATTGACGAGAATGTGTA | AAGATGCGTTGGTGTAAGTAT |
| CL10318.Contig30 | TCCAGACATCCAGTTCAG | CTTGTTGTTCACCACCAA |
| CL11630.Contig1 | AGAAGACCCGTAGCAAAG | GCTTTCCAACTCCACTCT |
| Sample ID | Total Clean Bases (Gb) | Q20 Content (%) | Q30 Content (%) | Mean Length (bp) | N50 (bp) | GC Content (%) |
|---|---|---|---|---|---|---|
| GT29-0 d1 | 10.48 | 97.04 | 92.34 | 1099 | 1678 | 48.43 |
| GT29-0 d2 | 10.56 | 97.02 | 92.23 | 1104 | 1695 | 48.32 |
| GT29-0 d3 | 10.60 | 97.05 | 92.47 | 1069 | 1643 | 49.02 |
| GT29-1 d1 | 10.50 | 97.19 | 92.72 | 1083 | 1653 | 48.47 |
| GT29-1 d2 | 10.54 | 97.16 | 92.59 | 1093 | 1687 | 48.35 |
| GT29-1 d3 | 10.53 | 97.20 | 92.74 | 1062 | 1624 | 48.69 |
| GT29-3 d1 | 10.47 | 97.25 | 92.85 | 1084 | 1683 | 48.36 |
| GT29-3 d2 | 10.53 | 97.26 | 92.83 | 1062 | 1625 | 48.71 |
| GT29-3 d3 | 10.51 | 97.20 | 92.69 | 1100 | 1694 | 48.43 |
| Badila-0 d1 | 10.55 | 97.05 | 92.37 | 1203 | 1848 | 48.57 |
| Badila-0 d2 | 9.59 | 97.09 | 92.45 | 1203 | 1825 | 48.68 |
| Badila-0 d3 | 10.54 | 97.09 | 92.42 | 1203 | 1823 | 48.59 |
| Badila-1 d1 | 10.56 | 97.16 | 92.57 | 1198 | 1828 | 48.34 |
| Badila-1 d2 | 10.46 | 97.00 | 92.30 | 1204 | 1825 | 48.33 |
| Badila-1 d3 | 10.51 | 97.00 | 92.28 | 1143 | 1758 | 48.52 |
| Badila-3 d1 | 10.47 | 97.00 | 92.28 | 1178 | 1799 | 48.55 |
| Badila-3 d2 | 10.52 | 97.00 | 92.27 | 1172 | 1780 | 48.73 |
| Badila-3 d3 | 10.50 | 97.00 | 92.26 | 1200 | 1837 | 48.36 |
| Mean | 10.47 | 97.10 | 92.48 | 1137 | 1739 | 48.53 |
| Pathway ID | Pathway Name | DEGs | Rich Ratio | Q Value |
|---|---|---|---|---|
| ko04016 | MAPK signaling pathway-plant | 122 | 0.044 | 1.28 × 10−13 |
| ko00940 | Phenylpropanoid biosynthesis | 91 | 0.048 | 2.14 × 10−12 |
| ko00941 | Flavonoid biosynthesis | 39 | 0.076 | 1.01 × 10−10 |
| ko04712 | Circadian rhythm-plant | 47 | 0.064 | 1.66 × 10−10 |
| ko04626 | Plant–pathogen interaction | 155 | 0.034 | 4.55 × 10−9 |
| ko00945 | Stilbenoid, diarylheptanoid, and gingerol biosynthesis | 21 | 0.110 | 8.23 × 10−9 |
| ko01110 | Biosynthesis of secondary metabolites | 341 | 0.026 | 6.94 × 10−6 |
| ko04075 | Plant hormone signal transduction | 91 | 0.033 | 9.66 × 10−5 |
| ko00944 | Flavone and flavonol biosynthesis | 9 | 0.129 | 1.53 × 10−4 |
| ko00500 | Starch and sucrose metabolism | 57 | 0.036 | 3.36 × 10−4 |
| ko00904 | Diterpenoid biosynthesis | 14 | 0.062 | 2.26 × 10−3 |
| ko00360 | Phenylalanine metabolism | 16 | 0.053 | 5.23 × 10−3 |
| ko03015 | mRNA surveillance pathway | 92 | 0.028 | 9.82 × 10−3 |
| ko00908 | Zeatin biosynthesis | 9 | 0.062 | 2.30 × 10−2 |
| ko00430 | Taurine and hypotaurine metabolism | 10 | 0.057 | 2.56 × 10−2 |
| Pathway ID | Pathway Name | DEGs | Rich Ration | Q Value |
|---|---|---|---|---|
| ko00196 | Photosynthesis antenna proteins | 14 | 0.350 | 7.62 × 10−11 |
| ko00195 | Photosynthesis | 28 | 0.118 | 1.61 × 10−9 |
| ko04712 | Circadian rhythm-plant | 46 | 0.063 | 1.27 × 10−6 |
| ko00941 | Flavonoid biosynthesis | 35 | 0.068 | 6.57 × 10−6 |
| ko01110 | Biosynthesis of secondary metabolites | 421 | 0.032 | 7.81 × 10−6 |
| ko01100 | Metabolic pathways | 733 | 0.029 | 3.26 × 10−5 |
| ko00280 | Valine, leucine, and isoleucine degradation | 33 | 0.051 | 3.55 × 10−3 |
| ko00640 | Propanoate metabolism | 21 | 0.062 | 3.55 × 10−3 |
| ko03010 | Ribosome | 82 | 0.037 | 5.66 × 10−3 |
| ko00940 | Phenylpropanoid biosynthesis | 72 | 0.038 | 7.15 × 10−3 |
| ko00500 | Starch and sucrose metabolism | 61 | 0.038 | 1.45 × 10−2 |
| ko00620 | Pyruvate metabolism | 42 | 0.041 | 2.09 × 10−2 |
| ko00514 | Other types of O-glycan biosynthesis | 14 | 0.062 | 2.28 × 10−2 |
| ko00945 | Stilbenoid, diarylheptanoid, and gingerol biosynthesis | 12 | 0.063 | 3.65 × 10−2 |
| ko00905 | Brassinosteroid biosynthesis | 7 | 0.085 | 4.41 × 10−2 |
| Pathway ID | Pathway Name | DEGs | Rich Ratio | Q Value |
|---|---|---|---|---|
| ko04626 | Plant–pathogen interaction | 463 | 0.101 | 1.46 × 10−36 |
| ko04016 | MAPK signaling pathway-plant | 293 | 0.106 | 1.83 × 10−25 |
| ko04075 | Plant hormone signal transduction | 268 | 0.096 | 1.57 × 10−17 |
| ko00940 | Phenylpropanoid biosynthesis | 171 | 0.091 | 3.92 × 10−9 |
| ko00500 | Starch and sucrose metabolism | 149 | 0.094 | 4.90 ×10−9 |
| ko00906 | Carotenoid biosynthesis | 54 | 0.124 | 5.84 × 10−7 |
| ko00945 | Stilbenoid, diarylheptanoid, and gingerol biosynthesis | 28 | 0.147 | 4.26 × 10−5 |
| ko00460 | Cyanoamino acid metabolism | 61 | 0.097 | 2.26 × 10−4 |
| ko01110 | Biosynthesis of secondary metabolites | 827 | 0.062 | 2.54 × 10−4 |
| ko00999 | Biosynthesis of various plant secondary metabolites | 55 | 0.099 | 2.54 × 10−4 |
| ko00591 | Linoleic acid metabolism | 21 | 0.152 | 2.72 × 10−4 |
| ko00515 | Mannose type O-glycan biosynthesis | 14 | 0.192 | 4.32 × 10−4 |
| ko00250 | Alanine, aspartate, and glutamate metabolism | 54 | 0.094 | 1.11 × 10−3 |
| ko00410 | beta-Alanine metabolism | 43 | 0.095 | 3.61 × 10−3 |
| ko00941 | Flavonoid biosynthesis | 46 | 0.09 | 7.67 × 10−3 |
| ko00520 | Amino sugar and nucleotide sugar metabolism | 113 | 0.074 | 7.67 × 10−3 |
| ko04070 | Phosphatidylinositol signaling system | 66 | 0.082 | 7.67 × 10−3 |
| ko00220 | Arginine biosynthesis | 35 | 0.095 | 9.78 × 10−3 |
| ko04712 | Circadian rhythm-plant | 59 | 0.081 | 1.56 × 10−2 |
| ko00360 | Phenylalanine metabolism | 29 | 0.095 | 2.00 × 10−2 |
| ko00524 | Neomycin, kanamycin, and gentamicin biosynthesis | 10 | 0.149 | 2.25 × 10−2 |
| ko00130 | Ubiquinone and other terpenoid-quinone biosynthesis | 34 | 0.089 | 2.37 × 10−2 |
| ko00910 | Nitrogen metabolism | 36 | 0.088 | 2.50 × 10−2 |
| ko00908 | Zeatin biosynthesis | 16 | 0.111 | 3.31 × 10−2 |
| ko00901 | Indole alkaloid biosynthesis | 3 | 0.375 | 4.12 × 10−2 |
| ko00965 | Betalain biosynthesis | 12 | 0.12 | 4.59 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Ran, M.; Zhang, J.; Zhao, P.; Zan, F.; Zhao, J.; Qin, W.; Wu, Q.; Liu, J.; Liu, X. Comparative Analysis of Ratoon-Competent and Ratoon-Deficient Sugarcane by Hormonal and Transcriptome Profiling. Agronomy 2025, 15, 1669. https://doi.org/10.3390/agronomy15071669
Zhao L, Ran M, Zhang J, Zhao P, Zan F, Zhao J, Qin W, Wu Q, Liu J, Liu X. Comparative Analysis of Ratoon-Competent and Ratoon-Deficient Sugarcane by Hormonal and Transcriptome Profiling. Agronomy. 2025; 15(7):1669. https://doi.org/10.3390/agronomy15071669
Chicago/Turabian StyleZhao, Liping, Maoyong Ran, Jing Zhang, Peifang Zhao, Fenggang Zan, Jun Zhao, Wei Qin, Qibin Wu, Jiayong Liu, and Xinlong Liu. 2025. "Comparative Analysis of Ratoon-Competent and Ratoon-Deficient Sugarcane by Hormonal and Transcriptome Profiling" Agronomy 15, no. 7: 1669. https://doi.org/10.3390/agronomy15071669
APA StyleZhao, L., Ran, M., Zhang, J., Zhao, P., Zan, F., Zhao, J., Qin, W., Wu, Q., Liu, J., & Liu, X. (2025). Comparative Analysis of Ratoon-Competent and Ratoon-Deficient Sugarcane by Hormonal and Transcriptome Profiling. Agronomy, 15(7), 1669. https://doi.org/10.3390/agronomy15071669