
Transcription Factor Protein (TFP)-Trait Relationships During Sugarcane Internode Development



Abstract
1. Introduction
2. Materials and Methods
2.1. Material
2.2. Destructive Sampling
2.3. Biomass Composition
2.3.1. Extraction of Biomass Component
2.3.2. Cell Wall Constituents
2.3.3. Water Solubles
2.4. Calculation of Growth and Metabolic Rates
2.5. Trait-Trait Correlation
- is Spearman’s rank correlation coefficient,
- is the difference between the ranks of corresponding values,
- n is the number of observations.
2.6. Exaction of Proteins
2.7. Transcription Factor Identification and Annotation
2.8. Trait-Based Differential Expression Analysis
2.9. Protein-Protein Network Construction
2.10. Computational Analysis
3. Results and Discussion
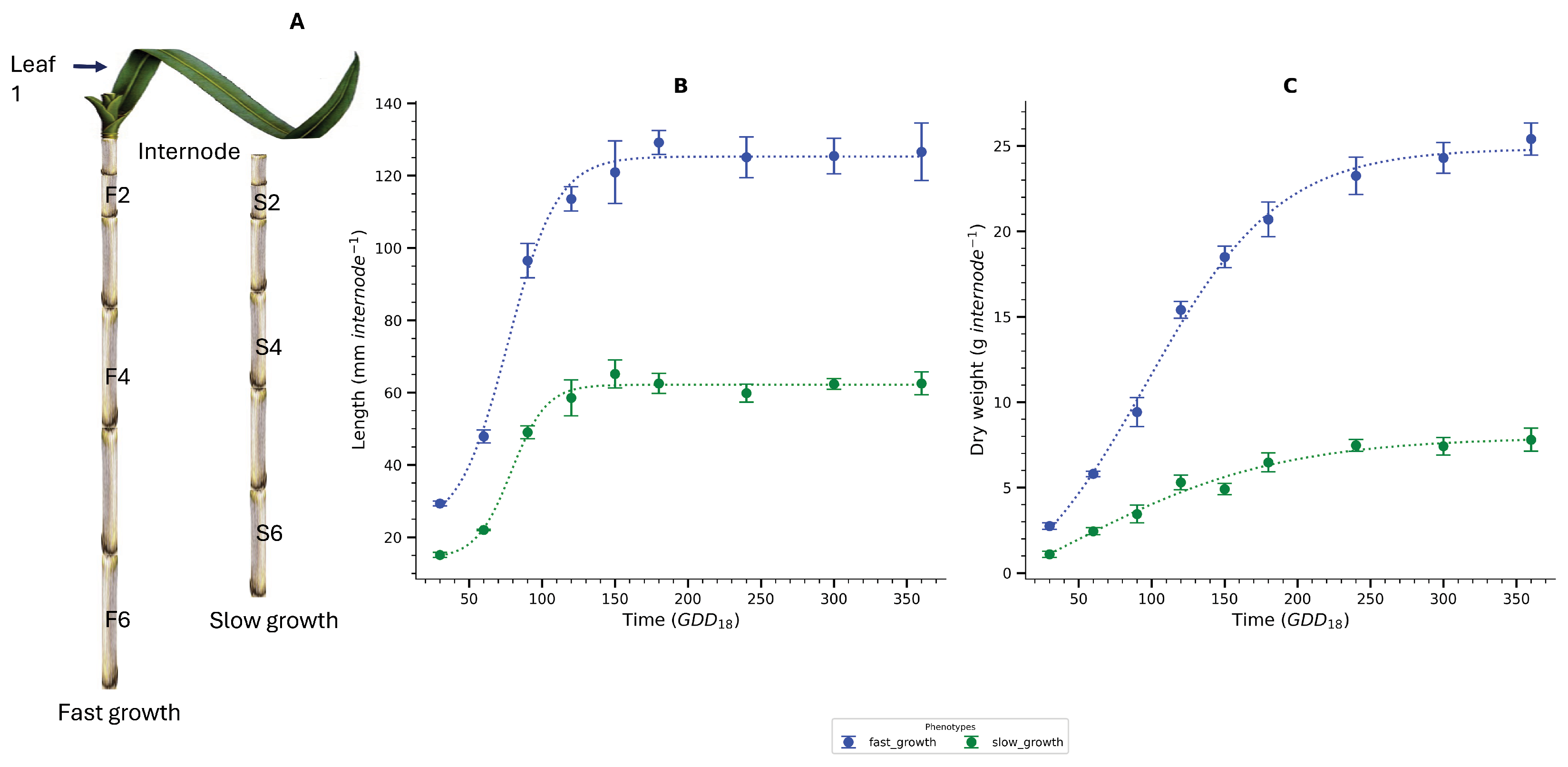
3.1. Growth and Sink Strength
3.2. Carbon Partitioning and Trait-Trait Correlations
3.3. Transcription Factors
3.4. Transcription Factor Expression
3.5. Protein-Trait Correlation
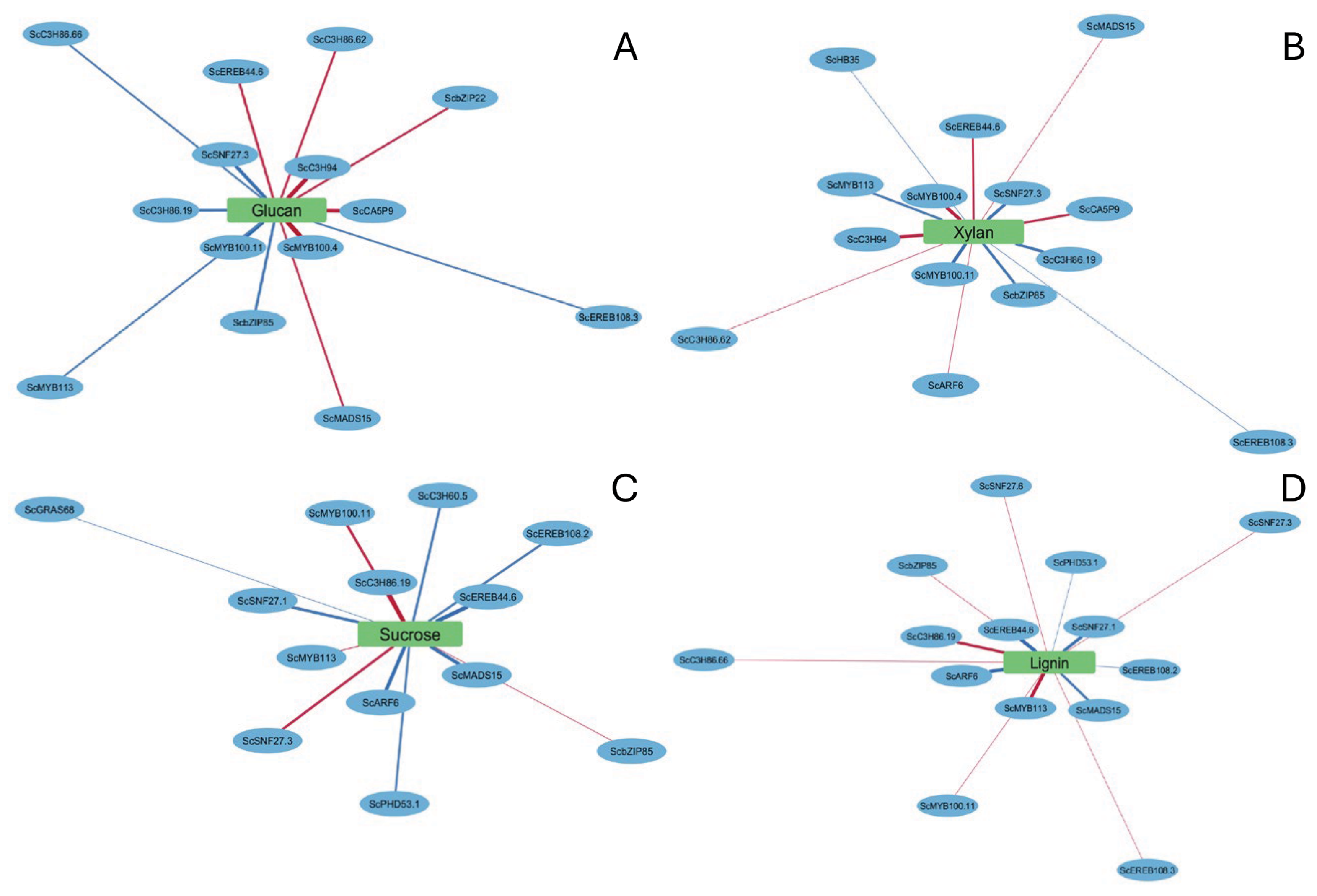
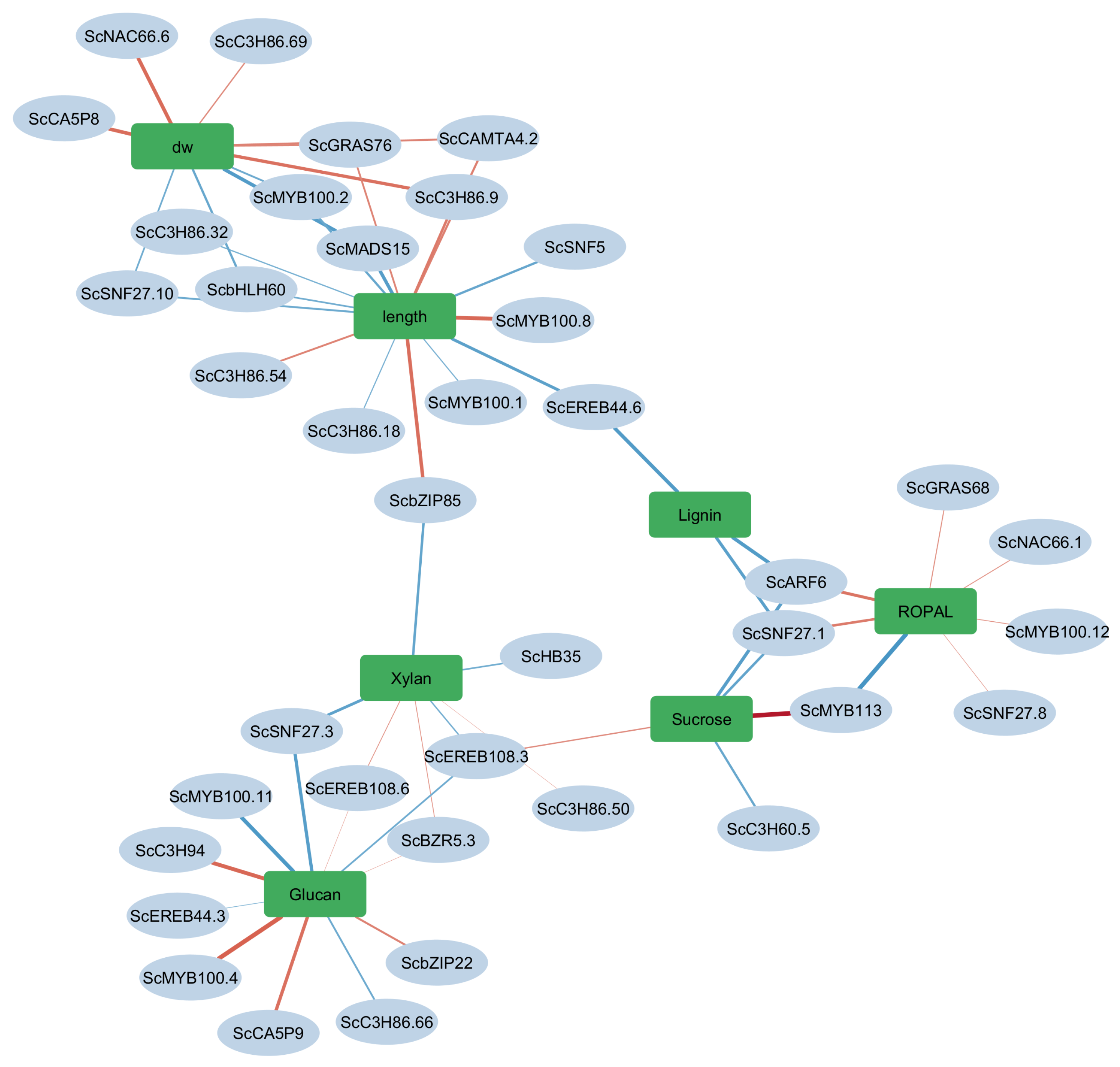
3.6. Gene-Trait Association Network Reveals Modular and Coordinated Regulation of Biomass-Related Traits
3.6.1. A Biomass Cluster: Dry Weight and Elongation
3.6.2. Cell Wall Polysaccharide Cluster: Glucan and Xylan
3.6.3. Intermediary Metabolism (ROPAL), Sucrose, and Lignin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MODDUS | Trinexapac-ethyl |
| ROPAL | Biomass-(soluble sugars+hexosans+pentosans+glucoronic-acid) |
| PCIT | Partial Correlation with Information Theory |
| GDD | = growing degree days above 37 °C base temperature |
References
- Duca, D.; Toscano, G. Biomass energy resources: Feedstock quality and bioenergy sustainability. Resources 2022, 11, 57. [Google Scholar] [CrossRef]
- Peláez-Samaniego, M.R.; Garcia-Perez, M.; Cortez, L.; Rosillo-Calle, F.; Mesa, J. Improvements of Brazilian carbonization industry as part of the creation of a global biomass economy. Renew. Sustain. Energy Rev. 2008, 12, 1063–1086. [Google Scholar] [CrossRef]
- Oh, Y.K.; Hwang, K.R.; Kim, C.; Kim, J.R.; Lee, J.S. Recent developments and key barriers to advanced biofuels: A short review. Bioresour. Technol. 2018, 257, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Fridolin, K.; Simone, G.; Nina, E.; Karl-Heinz, E.; Helmut, H.; Marina, F.-K. Growth in global materials use, GDP and population during the 20th century. Ecol. Econ. 2009, 68, 2696–2705. [Google Scholar]
- European Commission. The European Green Deal; COM (2019) 640 Final; European Commission: Brussels, Switzerland, 2019; Volume 11, p. 2019.
- Muscat, A.; de Olde, E.M.; Ripoll-Bosch, R.; Van Zanten, H.H.E.; Metze, T.A.P.; Termeer, C.J.A.M.; van Ittersum, M.K.; de Boer, I.J.M. Principles, drivers and opportunities of a circular bioeconomy. Nat. Food 2021, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Inman-Bamber, G. Sugarcane Yields and Yield-Limiting Processes. In Sugarcane: Physiology, Biochemistry, and Functional Biology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Wang, J.; Nayak, S.; Koch, K.; Ming, R. Carbon partitioning in sugarcane (Saccharum Species). Front. Plant Sci. 2013, 4, 201. [Google Scholar] [CrossRef]
- McCormick, A.J.; Cramer, M.D.; Watt, D.A. Sink strength regulates photosynthesis in sugarcane. New Phytol. 2006, 171, 759–770. [Google Scholar] [CrossRef]
- Kebrom, T.H.; McKinley, B.; Mullet, J.E. Dynamics of gene expression during development and expansion of vegetative stem internodes of bioenergy sorghum. Biotechnol. Biofuels Bioprod. 2017, 10, 159. [Google Scholar] [CrossRef]
- Lingle, S.E. Seasonal Internode Development and Sugar Metabolism in Sugarcane. Crop Sci. 1997, 37, 1222–1227. [Google Scholar] [CrossRef]
- Martin, A.; Palmer, W.; Brown, C.; Abel, C.; Lunn, J.; Furbank, R.; Grof, C.P.L. A developing Setaria viridis internode: An experimental system for the study of biomass generation in a C4 model species. Biotechnol. Biofuels 2016, 9, 45. [Google Scholar] [CrossRef]
- Botha, F.C.; Scalia, G.; Marquardt, A.; Wathen-Dunn, K. Sink Strength During Sugarcane Culm Growth: Size Matters. Sugar Tech 2023, 25, 1047–1060. [Google Scholar] [CrossRef]
- Yanagui, K.; Camargo, E.L.; de Abreu, L.G.F.; Nagamatsu, S.T.; Fiamenghi, M.B.; Silva, N.V.; Carazzolle, M.F.; Nascimento, L.C.; Franco, S.F.; Bressiani, J.A. Internode elongation in energy cane shows remarkable clues on lignocellulosic biomass biosynthesis in Saccharum hybrids. Gene 2022, 828, 146476. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fan, Y.; Yan, H.; Zhou, H.; Zhou, Z.; Weng, M.; Huang, X.; Lakshmanan, P.; Li, Y.; Qiu, L.; et al. Enhanced Activity of Genes Associated With Photosynthesis, Phytohormone Metabolism and Cell Wall Synthesis Is Involved in Gibberellin-Mediated Sugarcane Internode Growth. Front. Genet. 2020, 11, 570094. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.J.; Hoang, N.V.; Botha, F.C.; Furtado, A.; Marquardt, A.; Henry, R.J. Comparison of the root, leaf and internode transcriptomes in sugarcane (Saccharum spp. Hybrids). Curr. Res. Biotechnol. 2022, 4, 167–178. [Google Scholar] [CrossRef]
- Wang, M.; Li, A.M.; Liao, F.; Qin, C.X.; Chen, Z.L.; Zhou, L.; Li, Y.R.; Li, X.F.; Lakshmanan, P.; Huang, D.L. Control of sucrose accumulation in sugarcane (Saccharum spp. Hybrids) involves miRNA-mediated regulation of genes and transcription factors associated with sugar metabolism. GCB Bioenergy 2022, 14, 173–191. [Google Scholar] [CrossRef]
- Ferreira, S.S.; Hotta, C.T.; de Carli Poelking, V.G.; Leite, D.C.C.; Buckeridge, M.S.; Loureiro, M.E.; Barbosa, M.H.P.; Carneiro, M.S.; Souza, G.M. Co-expression network analysis reveals transcription factors associated to cell wall biosynthesis in sugarcane. Plant Mol. Biol. 2016, 91, 15–35. [Google Scholar] [CrossRef]
- Vélez-Bermúdez, I.C.; Schmidt, W. The conundrum of discordant protein and mRNA expression. Are plants special? Front. Plant Sci. 2014, 5, 619. [Google Scholar]
- Broun, P. Transcription factors as tools for metabolic engineering in plants. Curr. Opin. Plant Biol. 2004, 7, 202–209. [Google Scholar] [CrossRef]
- Grotewold, E. Transcription factors for predictive plant metabolic engineering: Are we there yet? Curr. Opin. Biotechnol. 2008, 19, 138–144. [Google Scholar] [CrossRef]
- Braun, E.L.; Dias, A.P.; Matulnik, T.J.; Grotewold, E. Chapter Five Transcription factors and metabolic engineering: Novel applications for ancient tools. In Recent Advances in Phytochemistry; Romeo, J.T., Saunders, J.A., Mattews, B.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; Volume 35, pp. 79–109. [Google Scholar] [CrossRef]
- Capell, T.; Christou, P. Progress in plant metabolic engineering. Curr. Opin. Biotechnol. 2004, 15, 148–154. [Google Scholar] [CrossRef]
- Reverter, A.; Hudson, N.J.; Nagaraj, S.H.; Pérez-Enciso, M.; Dalrymple, B.P. Regulatory impact factors: Unraveling the transcriptional regulation of complex traits from expression data. Bioinformatics 2010, 26, 896–904. [Google Scholar] [CrossRef]
- Hudson, N.J.; Reverter, A.; Dalrymple, B.P. Beyond differential expression: The quest for causal mutations and effector molecules. BMC Genom. 2009, 10, 83. [Google Scholar] [CrossRef]
- Botha, F.C.; Marquardt, A. Metabolic Control of Sugarcane Internode Elongation and Sucrose Accumulation. Agronomy 2024, 14, 1487. [Google Scholar] [CrossRef]
- Berding, N.; Marston, D.H. Operational validation of the efficacy of spectracane, a high-speed analytical system for sugarcane quality components. In Proceedings of the 2010 Conference of the Australian Society of Sugar Cane Technologists, Bundaberg, QLD, Australia, 11–14 May 2010; Australian Society of Sugar Cane Technologists: Marian, QLD, Australia, 2010; pp. 445–459. [Google Scholar]
- Pisanó, I.; Gottumukkala, L.; Hayes, D.J.; Leahy, J.J. Characterisation of Italian and Dutch forestry and agricultural residues for the applicability in the bio-based sector. Ind. Crop. Prod. 2021, 171, 113857. [Google Scholar] [CrossRef]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of structural carbohydrates and lignin in biomass. Lab. Anal. Proced. 2008, 1617, 1–16. [Google Scholar]
- Hayes, D.J. Development of near infrared spectroscopy models for the quantitative prediction of the lignocellulosic components of wet Miscanthus samples. Bioresour. Technol. 2012, 119, 393–405. [Google Scholar] [CrossRef]
- Bhagia, S.; Nunez, A.; Wyman, C.E.; Kumar, R. Robustness of two-step acid hydrolysis procedure for composition analysis of poplar. Bioresour. Technol. 2016, 216, 1077–1082. [Google Scholar] [CrossRef]
- Yang, S.; Logan, J.; Coffey, D. Mathematical formulae for calculating the base temperature for growing degree days. Agric. For. Meteorol. 1995, 74, 61–74. [Google Scholar] [CrossRef]
- Marquardt, A.; Henry, R.J.; Botha, F.C. Effect of sugar feedback regulation on major genes and proteins of photosynthesis in sugarcane leaves. Plant Physiol. Biochem. 2021, 158, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.; Chu, Y.H.; Abnave, A.; Cano, F.G.; Lee, Y.S.; Percival, S.; Jiang, N.; Grotewold, E. GRASSIUS 2.0: A gene regulatory information knowledgebase for maize and other grasses. Curr. Plant Biol. 2024, 40, 100396. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J. Educ. Behav. Stat. 2000, 25, 60–83. [Google Scholar] [CrossRef]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of ‘small-World’ Networks. Nature 1998, 393, 440–442. [Google Scholar] [CrossRef]
- Python Software Foundation. Python Language Reference, version 3.8.12. Technical Report. Python Software Foundation: Wilmington, DE, USA, 2024.
- Lingle, S.E.; Thomson, J.L. Sugarcane Internode Composition During Crop Development. BioEnergy Res. 2012, 5, 168–178. [Google Scholar] [CrossRef]
- De Souza, A.P.; Leite, D.C.; Pattathil, S.; Hahn, M.G.; Buckeridge, M.S. Composition and structure of sugarcane cell wall polysaccharides: Implications for second-generation bioethanol production. BioEnergy Res. 2013, 6, 564–579. [Google Scholar] [CrossRef]
- Chen, L.; Dou, P.; Li, L.; Chen, Y.; Yang, H. Transcriptome-wide analysis reveals core transcriptional regulators associated with culm development and variation in Dendrocalamus sinicus, the strongest woody bamboo in the world. Heliyon 2022, 8, e12600. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Xie, L.; Wen, D.; Wu, C.; Zhang, C. Transcriptome analysis reveals the mechanism of internode development affecting maize stalk strength. BMC Plant Biol. 2022, 22, 49. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Theißen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Plant Mol. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Ye, D.; Xing, J.; Duan, L.; Li, Z.; Zhang, M. Ethephon-regulated maize internode elongation associated with modulating auxin and gibberellin signal to alter cell wall biosynthesis and modification. Plant Sci. 2020, 290, 110196. [Google Scholar] [CrossRef] [PubMed]
- Ohme-Takagi, M.; Shinshi, H. Ethylene-inducible DNA binding proteins that interact with an ethylene-responsive element. Plant Cell 1995, 7, 173–182. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Phenotype 1 | Young 2 | Mature 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | p-adj | Tukey | Mean | p-adj | Tukey | Mean | p-adj | Tukey 4 | |
| Growth Traits | |||||||||
| Length (mm ) | −50.81 | <0.001 | ** 4 | 58.1 | <0.001 | ** | −1.28 | 1 | ns |
| Dry weight (g ) | −11.2 | <0.001 | ** | 7.58 | <0.001 | ** | 4.02 | 0.03 | ** |
| Water-Soluble Sugars (mg ) | |||||||||
| Total sugar | 31.59 | 0.1789 | ns | 182.46 | <0.001 | ** | −40.68 | 0.192 | ns |
| Sucrose | 40.49 | 0.2794 | ns | 241.43 | <0.001 | ** | 31.01 | 0.6912 | ns |
| Cellobiose | −0.02 | 0.9413 | ns | 2.86 | <0.001 | ** | −1.72 | <0.001 | ** |
| Glucose | −4.68 | 0.5813 | ns | −32.07 | <0.001 | ** | −38.3 | <0.001 | ** |
| Fructose | −4.03 | 0.5764 | ns | −28.49 | <0.001 | ** | −31.89 | <0.001 | ** |
| Galactose | −0.14 | 0.3249 | ns | −1.15 | <0.001 | ** | 0.23 | 0.7982 | ns |
| Arabinose | −0.02 | 0.2764 | ns | −0.13 | <0.001 | ** | −0.01 | 0.9997 | ns |
| Cell Wall Sugars (mg ) | |||||||||
| Total sugar | −23.28 | 0.0737 | ns | −83.81 | <0.001 | ** | −56.64 | <0.001 | ** |
| Hexosans | −17.02 | 0.0764 | ns | −61.57 | <0.001 | ** | −45.28 | <0.001 | ** |
| Glucan | −16.76 | 0.0759 | ns | −60.17 | <0.001 | ** | −45.42 | <0.001 | ** |
| Mannan | −0.10 | 0.1828 | ns | −0.43 | 0.0012 | ** | 0.06 | 0.9992 | ns |
| Galactan | −0.17 | 0.3208 | ns | −0.97 | 0.0046 | ** | 0.07 | 1 | ns |
| Pentosans | −6.36 | 0.0739 | ns | −21.09 | <0.001 | ** | −9.64 | 0.0556 | ns |
| Xylan | −5.28 | 0.0952 | ns | −18.54 | <0.001 | ** | −9.85 | 0.0712 | ns |
| Arabinan | −0.96 | 0.0792 | ns | −3.51 | <0.001 | ** | −1.60 | 0.1171 | ns |
| Rhamnan | −0.02 | 0.5405 | ns | −0.19 | 0.0011 | ** | 0.09 | 0.5463 | ns |
| Glucuronic acid | 0.31 | 0.4683 | ns | 1.29 | 0.5724 | ns | 1.05 | 0.7981 | ns |
| Hemicellulose 5 | −6.25 | 0.0856 | ns | −22.24 | <0.001 | ** | −11.36 | 0.0311 | ** |
| Lignin (mg ) | |||||||||
| Klason lignin | 2.85 | 0.6771 | ns | 21.86 | <0.001 | ** | 43.48 | <0.001 | ** |
| AS lignin | −0.36 | 0.4546 | ns | −3.39 | <0.001 | ** | 2.70 | 0.001 | ** |
| Other (mg ) | |||||||||
| ROPAL 6 | −22.7 | 0.4576 | ns | −170.9 | <0.001 | ** | −6.29 | 1 | ns |
| Trait | Fast_Growth | Slow_Growth | p-Value 1 | Significance |
|---|---|---|---|---|
| Dry weight | 27.15 | 21.46 | 0.03 | * |
| glucan | 14.52 | 12.86 | 0.47 | ns |
| hemicellulose | 12.24 | 8.08 | 0.06 | ns |
| lignin | 53.74 | 51.92 | 0.42 | ns |
| sucrose | 71.76 | 52.3 | 3.20 × | *** |
| Trait | Positive Correlation | Negative Correlation | ||||
|---|---|---|---|---|---|---|
| TF 1 | Correlation | p-Value | TF 1 | p-Value | ||
| Length | ScMYB100 | 0.774 | 9.07 × | ScC3H86 | −0.581 | 2.92 × |
| ScbZIP85 | 0.747 | 2.76 × | ScbHLH60 | −0.618 | 1.29 × | |
| ScC3H86 | 0.683 | 2.34 × | ScSNF27 | −0.636 | 8.30 × | |
| ScCAMTA4 | 0.653 | 5.45 × | ScSNF5 | −0.663 | 4.12 × | |
| ScGRAS76 | 0.629 | 9.84 × | ScMADS15 | −0.738 | 3.79 × | |
| – | – | – | ScEREB44 | −0.710 | 1.01 × | |
| Dry weight | ScC3H86 | 0.596 | 2.11 × | ScMADS15 | −0.750 | 2.41 × |
| ScGRAS76 | 0.624 | 1.11 × | ScbHLH60 | −0.661 | 4.43 × | |
| ScCAMTA4 | 0.637 | 8.12 × | ScMYB100 | −0.632 | 9.15 × | |
| ScC3H86 | 0.732 | 4.72 × | ScSNF27 | −0.620 | 1.24 × | |
| ScCA5P8 | 0.755 | 1.99 × | – | – | – | |
| ScNAC66 | 0.759 | 1.70 × | – | – | – | |
| Glucan | ScMYB100 | 0.811 | 1.52 × | ScEREB44 | −0.548 | 5.57 × |
| ScC3H94 | 0.778 | 7.71 × | ScEREB108 | −0.625 | 1.10 × | |
| ScCA5P9 | 0.736 | 4.09 × | ScC3H86 | −0.631 | 9.39 × | |
| ScbZIP22 | 0.644 | 6.88 × | ScSNF27 | −0.731 | 4.93 × | |
| ScEREB108 | 0.530 | 7.78 × | – | – | – | |
| ScBZR5 | 0.527 | 8.11 × | – | – | – | |
| Xylan | ScEREB108 | 0.556 | 4.76 × | ScSNF27 | −0.700 | 1.40 × |
| ScBZR5 | 0.555 | 4.87 × | ScbZIP85 | −0.679 | 2.66 × | |
| ScC3H86 | 0.526 | 8.33 × | ScHB35 | −0.613 | 1.44 × | |
| Sucrose | ScMYB113 | 0.824 | 7.58× | ScC3H60 | −0.661 | 4.35 × |
| ScEREB108 | 0.593 | 2.25 × | ScSNF27 | −0.681 | 2.49 × | |
| – | – | – | ScARF6 | −0.733 | 4.61 × | |
| Lignin | – | – | – | ScEREB44 | −0.755 | 1.99 × |
| – | – | – | ScARF6 | −0.753 | 2.17 × | |
| – | – | – | ScSNF27 | −0.718 | 7.83 × | |
| ROPAL | ScARF6 | 0.712 | 9.57 × | ScMYB113 | −0.805 | 2.10 × |
| ScSNF27 | 0.679 | 2.68 × | – | – | – | |
| ScGRAS68 | 0.570 | 3.64 × | – | – | – | |
| ScNAC66 | 0.562 | 4.29 × | – | – | – | |
| ScSNF27 | 0.539 | 6.62 × | – | – | – | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Botha, F.C.; Marquardt, A. Transcription Factor Protein (TFP)-Trait Relationships During Sugarcane Internode Development. Agronomy 2025, 15, 1475. https://doi.org/10.3390/agronomy15061475
Botha FC, Marquardt A. Transcription Factor Protein (TFP)-Trait Relationships During Sugarcane Internode Development. Agronomy. 2025; 15(6):1475. https://doi.org/10.3390/agronomy15061475
Chicago/Turabian StyleBotha, Frederik C., and Annelie Marquardt. 2025. "Transcription Factor Protein (TFP)-Trait Relationships During Sugarcane Internode Development" Agronomy 15, no. 6: 1475. https://doi.org/10.3390/agronomy15061475
APA StyleBotha, F. C., & Marquardt, A. (2025). Transcription Factor Protein (TFP)-Trait Relationships During Sugarcane Internode Development. Agronomy, 15(6), 1475. https://doi.org/10.3390/agronomy15061475