1. Introduction
The TALE (three-amino acid loop extension) protein was first identified in the genome of fruit flies [
1]. Subsequently, 70, 68, 24, 35, 17 and 46 TALEs have been identified in
T.
aestivum (
Triticum aestivum L.) [
2],
G.
max (
Glycine max L.) [
3],
S.
lycopersicum (
Solanum lycopersicum L.) [
4],
P.
tomentosa (
Populus tomentosa) [
5],
P.
granatum (
Punica granatum L.) [
6] and
G.
hirsutum (
Gossypium hirsutum L.) [
7]. The conserved domain of the TALE consists 60 amino acids and forms three helical regions. The first and second helices form a ring structure, while the second and third helices form a helix–angle–helix configuration [
8]. TALE transcription factors can be divided into two subfamilies: BELL and KNOX. The BELL subfamily contains two conserved domains, POX and homeodomain, which play important roles in plant growth and development, hormone regulation, signal transduction, meristem formation and stress response [
9,
10,
11,
12]. Conversely, in the KONX subfamily, most members contain the domains KNOX2, KNOX1, ELK and ELK-Superfamily, which play important roles in gene regulation [
9,
10,
11,
12].
TALE typically exists as heterodimers, such as OSH15-SH5, which enhances seed shedding by inhibiting lignin biosynthesis [
13]. Additionally, TALE interacts with other family proteins, including BEL1-STM complexes, which are essential for maintaining indeterminate inflorescence meristem development in
A. thaliana (
Arabidopsis thaliana L.) [
14], and GhKNL1 interacts with GhOFP4 [
15]. Moreover, AtBLE1 is crucial for ovule development [
16]. In
Z. mays (
Zea mays L.), BLH12/BLH1 regulate stem and vascular development through interaction with KN1, with double mutants exhibiting reduced plant height and fewer vascular bundles [
17]. Furthermore, BELL family members are involved in secondary cell wall formation and fruit ripening. For example, tomato SIBL4 plays a role in cell wall metabolism and carotenoid accumulation [
12], while
OsBLH6 is implicated in cell wall formation [
18]. Additionally,
TALE genes also regulate flower development [
19]. In
A. thaliana, ATH1 controls floral competency through positive regulation of
FLC, which leads to late flowering [
20]. The over-expression of
MdKNOX19 increases ABA sensitivity, up-regulates
MdABI5 gene expression and affects fruit size and seed yield [
21], revealing a complex KNOX regulatory network in plant growth and stress responses. It is worth noting that
GmSBH1 not only participates in growth and development processes but also increases tolerance to high temperatures and high humidity stresses [
22]. In
P. tomentosa, the
KNOX gene
PagKNAT2/6b enhances drought resistance by inhibiting gibberellin (GA) synthesis [
23], and another TALE gene responds to salt stress [
5].
PvKN1 is involved in regulating the lignification and cell wall development of switchgrass [
24]. During tomato fruit set, auxin regulates GA synthesis through the up-regulation of
GA20ox1 and the down-regulation of
KONX, maintaining a delicate balance between auxin and GA [
25,
26].
Gramineous crops, such as
O. sativa (
Oryza sativa L.),
Z. mays and
S. italica (
Setaria italica L.), not only hold substantial economic importance but also function as crucial model organisms for investigating gene functionality [
27,
28,
29,
30]. TALE transcription factors are widespread in both plants and animals, playing a role in numerous physiological processes. Recent research has identified
TALE family genes across numerous plant species [
2,
3,
4,
5,
6,
7]. However, a systematic bioinformatics analysis of
TALE genes in gramineous plants, like
Z. mays,
S. italica,
S. bicolor (
Sorghum bicolor L.) and
B. distachyon (
Brachypodium distachyon L.), remain absent. Consequently, we conducted a whole-genome-level identification and analysis of the
TALE gene family in five gramineous species, discovering a total of 123
TALE genes. Our research included chromosomal distribution, phylogenetic relationships, gene duplication and collinearity, evolutionary selection pressures, gene structure and cis-acting elements. Additionally, we investigated the expression variations of
OsTALE,
ZmTALE and
AtTALE genes under abiotic stress to understand the functional differentiation of the
TALE gene family in various plants. This study will provide valuable references for the further utilization of
TALE genes for the development of drought-tolerant plants in the context of global land loss, and will facilitate the functional characterization of
TALE gene responses to stress and developmental signals.
3. Results
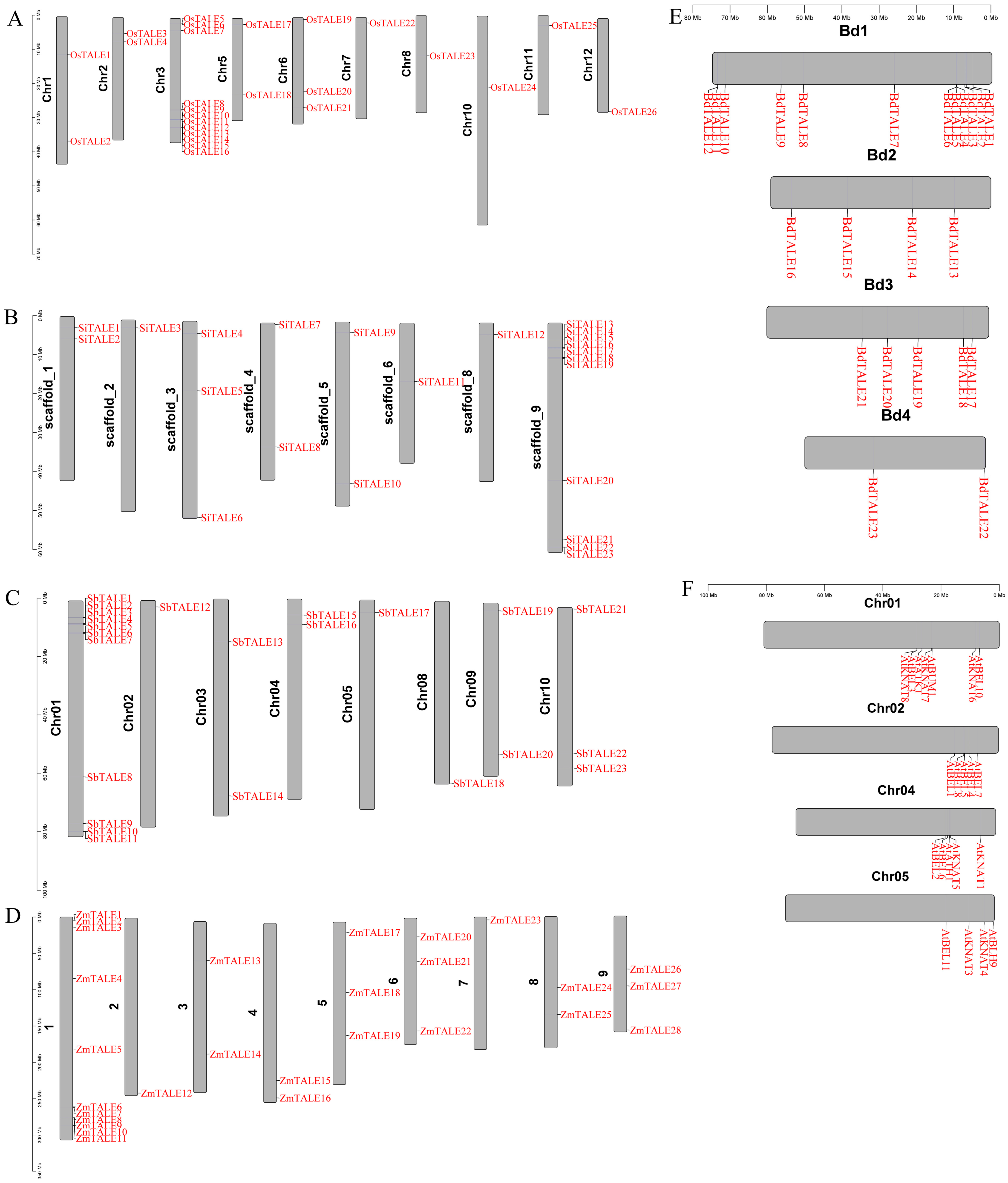
3.1. Identification of TALE Genes in Gramineae
In this research, we discovered a total of 123
TALE genes among five Gramineae species:
O. sativa (26),
Z. mays (28),
B. distachyon (23),
S. bicolor (23) and
S. italica (23) (
Figure 1 and
Supplemental Table S1). Importantly, both
O. sativa and
Z. mays displayed a greater quantity of
TALE genes relative to
B. distachyon,
S. bicolor and
S. italica, indicating a possible expansion of the
TALE gene family in these species. The identified
TALE genes were named based on their chromosomal locations, designated as
OsTALE1-
OsTALE26 for
O. sativa,
ZmTALE1-
ZmTALE28 for
Z. mays,
BdTALE1-
BdTALE23 for
B. distachyon,
SbTALE1-
SbTALE23 for
S. bicolor and
SiTALE1-
SiTALE23 for
S. italica (
Figure 1). The 26
OsTALE genes showed a wide distribution across nearly all chromosomes, with the exception of chromosomes 4, 7 and 9. Conversely, the 23
BdTALE genes were found on almost all chromosomes, except chromosome 5. The distribution of 28
ZmTALE genes were uneven across the chromosomes. Twenty-three
SbTALE genes were distributed across all chromosomes except for chromosomes 6, 7 and 8. 23
SiTALE genes were present on all chromosomes except chromosome 7. Moreover,
AtTALE genes were present on all chromosomes, apart from chromosome 3.
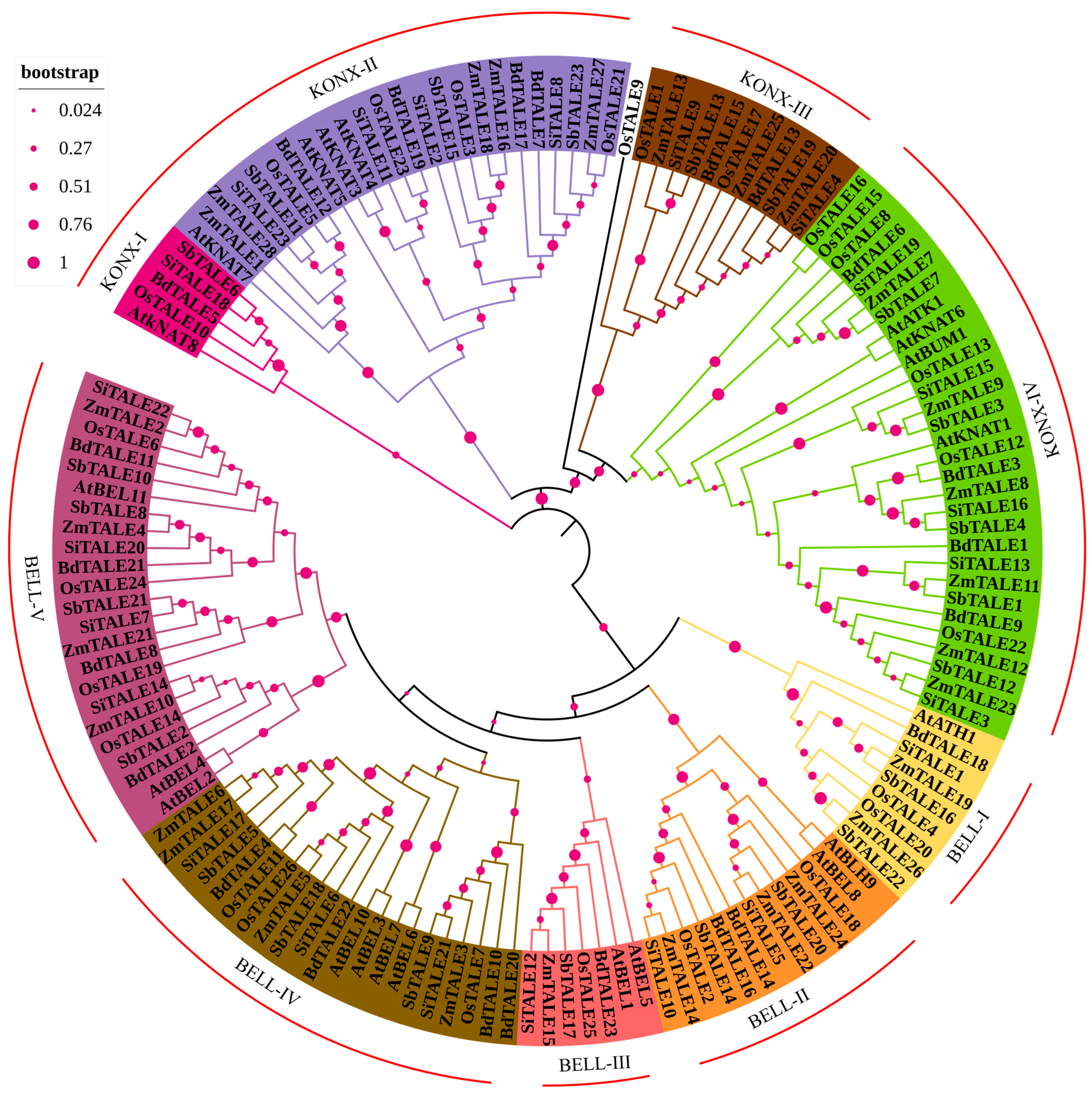
3.2. Phylogenetic Analysis of TALE Proteins in the Gramineae
To explore the evolutionary relationship for the
TALE gene family within the Gramineae, an evolutionary tree was generated utilizing the protein sequences of five Gramineae species and
A. thaliana. As illustrated in
Figure 2, these TALE proteins can be categorized into two primary subgroups: BELL and KONX. The KONX subgroup is further divided into KONX-I, KONX-II, KONX-III and KONX-IV, while the BELL subgroup consists of BELL-I, BELL-II, BELL-III, BELL-IV and BELL-V. Notably, KONX-III is uniquely present in the Gramineae. Furthermore, most proteins are classified under the KONX-IV classification, in contrast to KONX-I, which has the smallest number of TALE members.
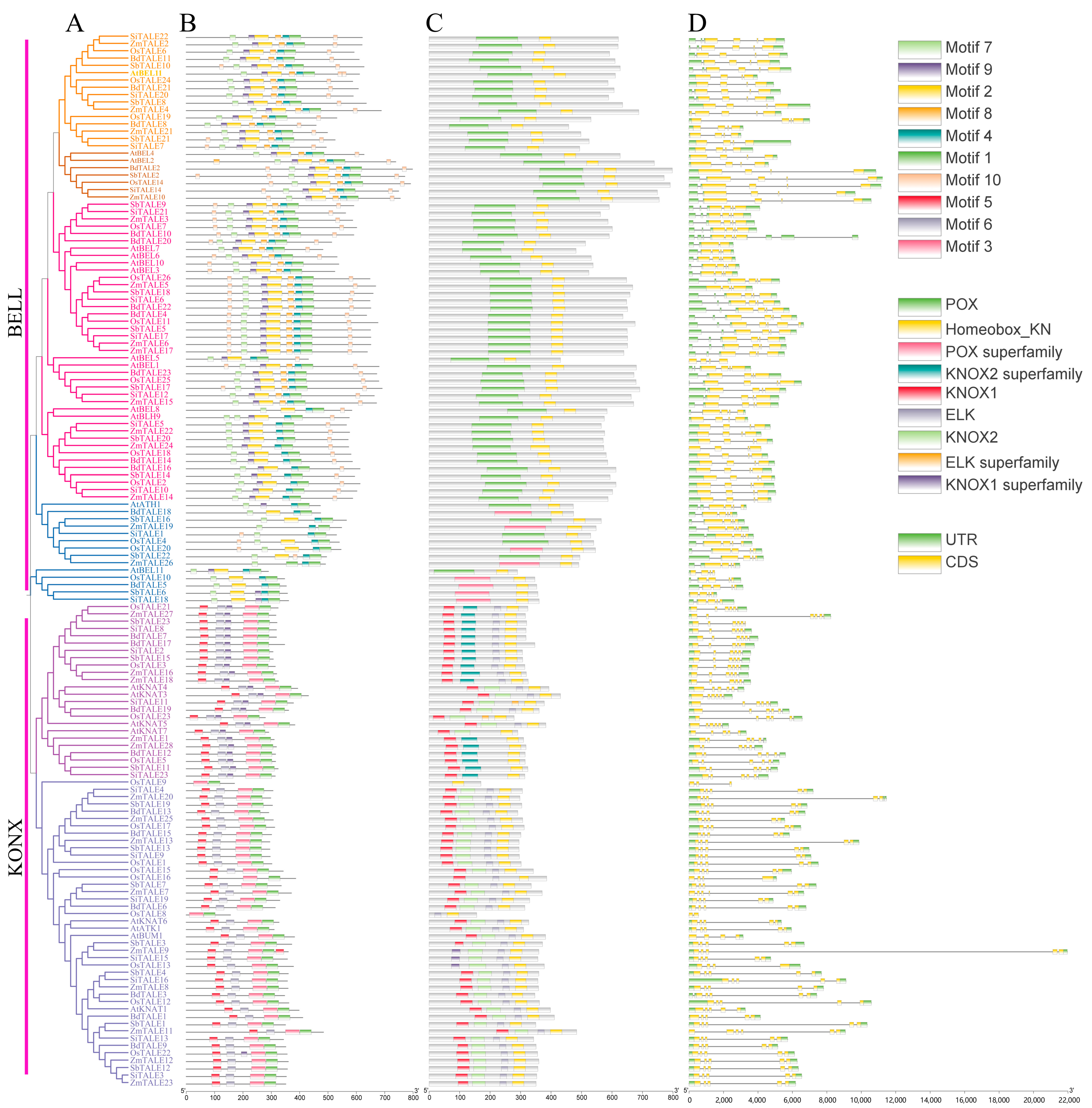
3.3. Gene Structure and Protein Conserved Motifs of TALE Members in Gramineae
To investigate the variations in gene structure and conserved motifs among members in the Gramineous TALE family, we employed MEME for the analysis of their amino acid sequences. As shown in
Figure 3, the motif count varies between two and seven. The majority of the KONX subgroup members feature Motif 1, Motif 3, Motif 4, Motif 7, Motif 8 and Motif 10, while the TELL subgroup include Motifs 1, 2, 5, 6 and 9. Notably, OsTALE8 is found to contain only Motif 1 and Motif 2, while another instance of OsTALE8 is characterized by the presence of only Motif 1 and Motif 10. Interestingly, Motif 3 is specifically associated with the KONX-I subgroup. In addition, we found that homologous genes have similar conserved motifs in different plants.
Additionally, we analyzed the conserved domains present within TALE family members in Gramineous species. Members of the TELL subgroup generally exhibit POX and Homeobox-KN domains. In contrast, BdTALE5, OsTALE10, OsTALE20 SbTALE6, SiTALE18, SbTALE16, ZmTALE19 and ZmTALE26 possess POX-superfamily and Homeobox-KN domains. On the other hand, a majority of KONX subgroup members display KNOX2, KNOX1, ELK, and ELK-superfamily domains, which play crucial roles in gene regulation. Uniquely, ZmTALE9, SiTAL15 and OsTALE13 are found to carry KNOX1-superfamily, KNOX1, ELK and ELk-Superfamily domains, suggesting they may offer unique regulatory functions or expression patterns. Other KNOX members, including ZmTALE1, ZmTALE28, SiTAL123, OsTALE5, BdTALE12 and SbTALE11, demonstrate a unique combination of domains, consisting of KNOX1, KNOX2-superfamily and ELK-superfamily domains, which may indicate their specific biological roles. On the other hand, homologous genes have similar conserved domains in different plants.
Moreover, we examined the gene structure of the TALE family in both Gramineous and
A. thaliana.
Figure 3 illustrates that the gene structure of the TALE family reveals significant variations; the quantity of introns shows patterns specific to each subfamily: members of the BELL subfamily generally have between three and six introns, whereas those in the KNOX subfamily usually have around four to five introns. This pattern could suggest a relationship between gene structure and functionality. Notably, conserved motif analysis revealed that OsTALE9 contains only Motifs 3 and 4, as well as the Homeobox-KN structural domain. In Gramineae and A. thaliana, homologous genes have similar intron–exon structures. In addition, we found considerable disparities in the untranslated region (UTR) present among various members of the Gramineae and
A. thaliana TALE genes. For instance,
AtKNAT6,
AtBEL5,
OsTALE9 and
ZmTALE24 do not possess both 5’UTR and 3’UTR, while
ZmTALE7 and
ZmTALE11 are missing 5’ UTR,
AtBEL11 is lacking 3’UTR and others show full UTR coverage. Considering the critical role that UTR plays in mRNA stability and translation, the lack of UTR in these particular genes necessitates further inquiry.
3.4. Homologous Gene Pair Analysis of TALE in Gramineae and A. thaliana
Gene duplication is crucial for plant gene family expansion [
40,
41]. To evaluate its influence on the
TALE gene family in Gramineae and
A. thaliana, we investigated potential duplication events in these species.
Figure 4 illustrates that in
O. sativa,
B. distachyon,
S. bicolor,
Z. mays,
S. italica and
A. thaliana, we found 12, 10, 14, 26, 14 and 8 genome-wide duplication (WGD) genes, as well as 2, 2, 2, 0, 2 and 0 tandem duplication (TD) genes, respectively. Noteworthy is that
Z. mays contains the highest number of WGD
TALE genes when compared to the other Gramineae and
A. thaliana species, with these genes distributed all chromosomes. Furthermore, our investigation uncovered variations in duplication patterns among species:
Z. mays and
A. thaliana showed an absence of TD genes, while
O. sativa,
B. distachyon,
S. bicolor and
S. italica each demonstrated an identical count of TD genes. These results suggest that the expansion of the
TALE gene family in Gramineae and
A. thaliana is primarily driven by WGD, especially in
Z. mays.
To evaluate the pressure of natural selection on the
TALE gene family in these species, we computed the Ka/Ks ratio, which represents the ratio of non-synonymous substitutions per non-synonymous site (Ka) to synonymous substitutions per synonymous site (Ks). The outcomes indicated that the Ka/Ks ratio for all TALE duplication genes in Gramineae and
A. thaliana were less than 1, ranging from 0.07 to 0.7 (
Supplemental Table S2), indicating that
TALE genes in these species have generally experienced purifying selection during evolution throughout their evolutionary history.
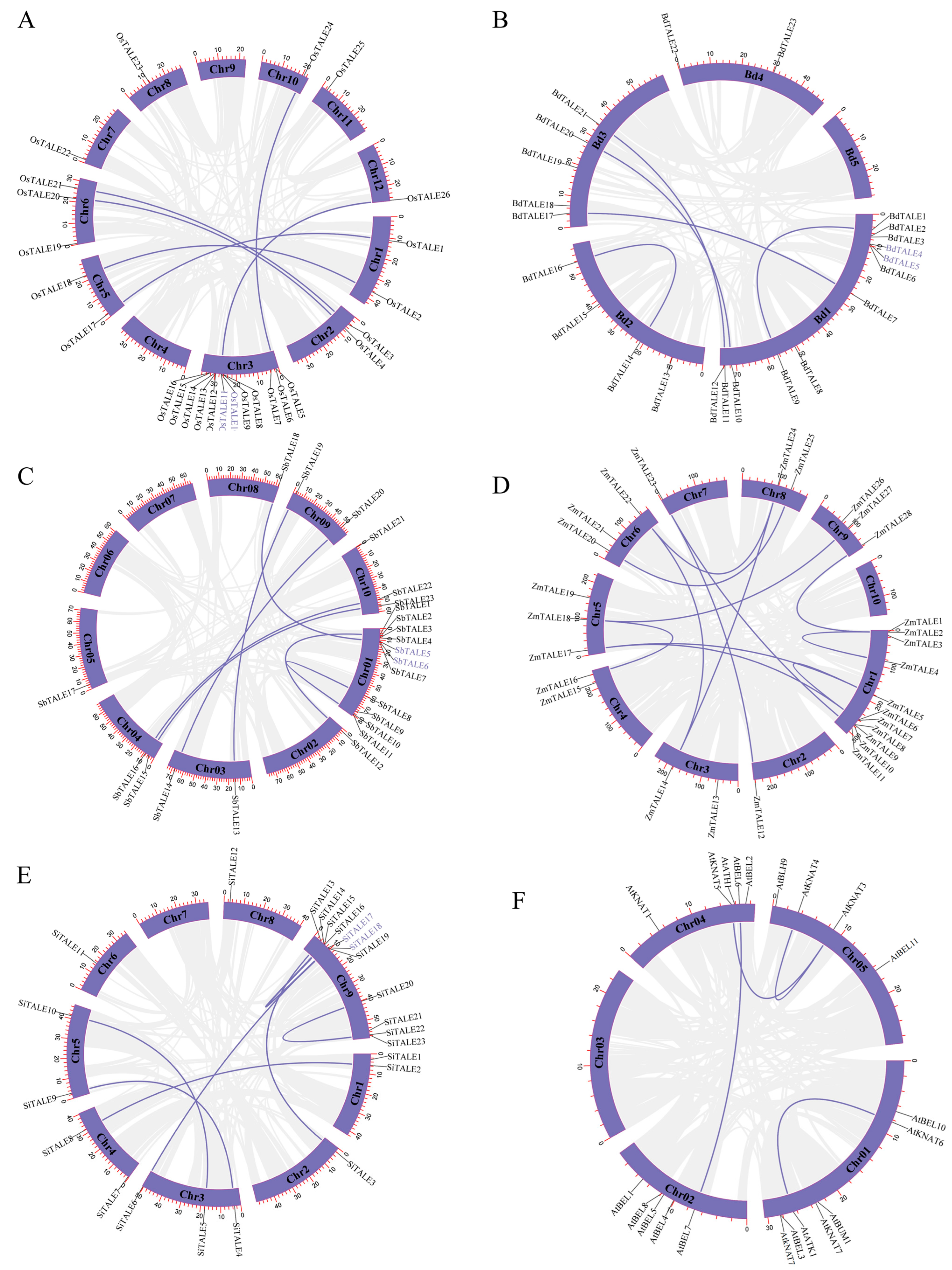
3.5. Collinearity Analysis of TALE Genes
To further elucidate the evolutionary relationship among the
TALE gene family in Gramineae, we conducted a collinearity analysis comparing
O. sativa with four Gramineae (
B. distachyon,
S. bicolor,
Z. mays and
S. italica) species and the dicotyledonous plant
A. thaliana. As shown in
Figure 5, we discovered 39, 20, 33 and 34 homologous gene pairs between
O. sativa and
Z. mays,
S. italica,
S. bicolor and
B. distachyon, respectively. In contrast, only six homologous gene pairs were identified between
O. sativa and
A. thaliana. This notable disparity indicates that the functionality of the
TALE gene family in Gramineae might be more conserved, reflecting a greater differentiation in comparison to
A. thaliana. Furthermore, the collinearity events predominantly occurred on chromosomes 1, 2, 3, 5, 6, 10, 11 and 12. These genomic regions may represent hot-spots in the evolution of the
TALE gene family within Gramineae, containing significant genetic variation and functional conservation. Importantly, the number of collinear gene pairs between
O. sativa and
Z. mays,
S. italica,
S. bicolor and
B. distachyon is relatively high, which reflects their close evolutionary relationship and suggests that the
TALE genes in these Gramineae crops may exhibit functional similarities. Specifically, we identified four genes,
OsTALE2,
OsTALE18,
OsTALE21 and
OsTALE25 that share homologous gene pairs with other Gramineae species and
A. thaliana, implying that these genes existed prior to the divergence of monocotyledons and dicotyledons.
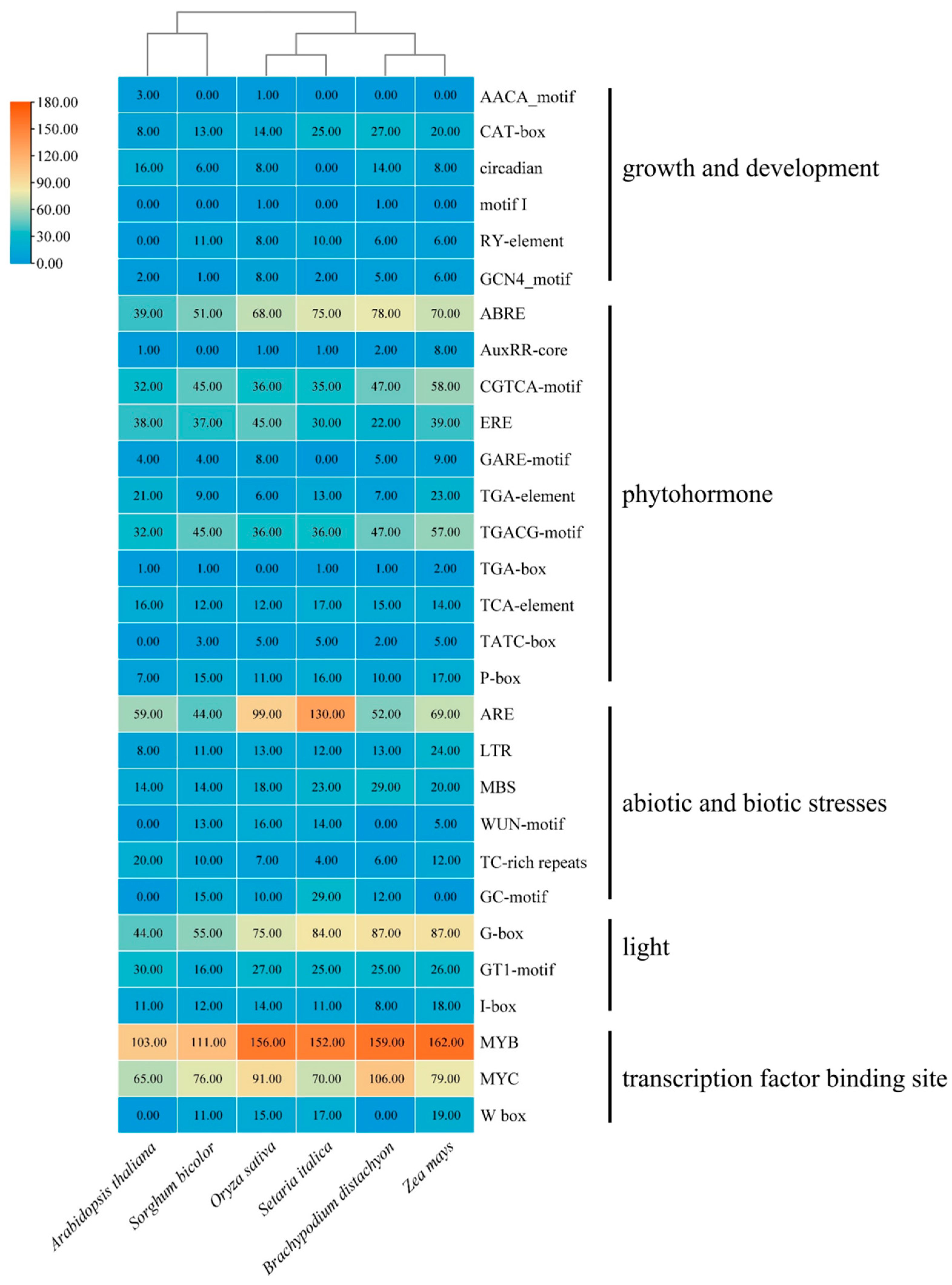
3.6. Cis-Element Analysis of TALE Genes in Gramineae
To gain deeper insights into the potential role of the
TALE gene in Gramineae, we conducted an analysis of cis-acting elements located in the 2 kb upstream region of the translation initiation site of the
TALE gene. We categorized these cis-acting elements into five separate categories: growth and development, response to plant hormone, stress response, light regulation, and binding sites for transcription factors. Our results related to growth and development revealed a strong connection between the
TALE gene and various processes, such as endosperm formation, meristem expression and circadian rhythm, as well as root-specific and seed-specific regulatory elements. Additionally, the
TALE gene displayed a wide array of responsiveness to plant hormones, indicating its involvement in a complex hormone signaling network that finely regulates plant growth, development and response to stress. The most prevalent observed elements included abscisic acid response elements (ABREs) followed by jasmonic acid response elements (CGTCA-motif and TGACG-motif), as well as ethylene response elements (EREs), implying the significant role of the
TALE gene in coping with abiotic stress (
Supplemental Table S3).
The examination of elements related to stress response provided further evidence for the possible function of the TALE gene in the adaptation of plants to stress. The occurrence of low-temperature and drought response elements in both Gramineae and A. thaliana indicates that the TALE gene may be crucial in managing these two stresses. Moreover, additional stress response elements, including those associated with anaerobic induction and mechanical injury, were also identified in some TALE genes, which enhances our comprehension of their functional roles.
The examination of light elements and binding site transcription factors has uncovered the possible involvement of the TALE gene in the transduction of light signals and the regulation of gene expression. The existence of light-responsive elements such as G-box and I-box implies that TALE genes might play a role in photosynthesis in plants. In addition, the discovery of transcription factor binding sites, including W-box, MYB and MYC, indicates that multiple transcription factors may regulate the TALE gene.
Additionally, we observed differences in the cis-acting elements of the TALE gene across various grasses and A. thaliana. For example, seed-specific regulatory elements are identified in Gramineae but are absent in A. thaliana, and circadian-related elements are observed in O. sativa, Z. mays, S. bicolor, B. distachyon and A. thaliana, yet they are missing in S. italica. In summary, our findings suggest that the TALE gene has diverse and intricate functions in Gramineae and A. thaliana, involving growth and development, hormone signaling and stress adaptation. Additionally, we have recognized both similarities and differences in the function of TALE gene among different species, providing a new perspective for understanding the evolution and functional differentiation of the TALE gene family.
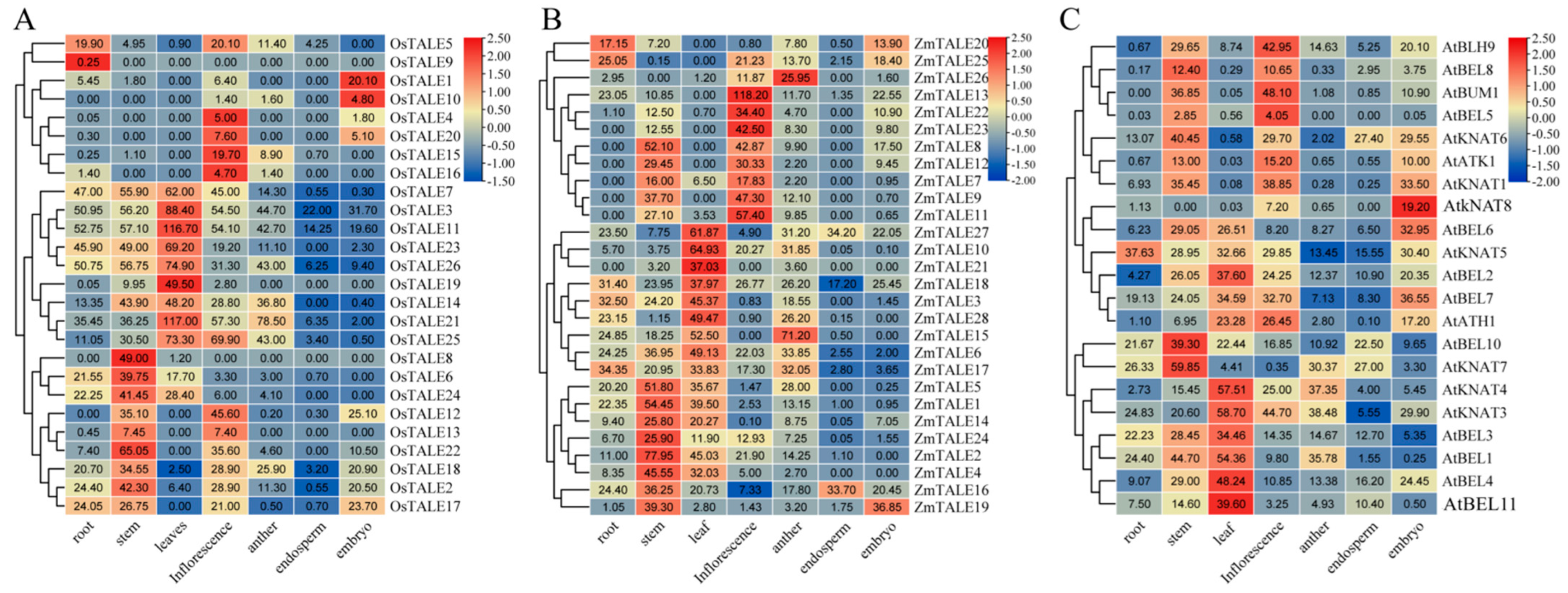
3.7. The Expression Patterns of OsTALE, ZmTALE and AtTALE Genes Across Various Tissues
Based on the published RNA-seq in the PPRD database, we mapped the
TALE gene expression heat map across the organs of
A. thaliana,
O. sativa and
Z. mays to investigate the differences in
TALE gene expression patterns between monocotyledonous plants and dicotyledonous plants. The organs included roots, stems, leaves, anthers, embryos, endosperm and inflorescence (
Supplemental Table S4). The expression levels of the
TALE gene in
A. thaliana,
O. sativa and
Z. mays were relatively high in stems, leaves and inflorescence, while the expression levels in endosperm and embryos were comparative low. Notably, the expression level of the
A.
thaliana TALE gene was significantly higher in the embryo than in
O. sativa and
Z. mays, highlighting its unique expression pattern. Furthermore, we observed that
OsTALE1,
OsTALE10,
ZmTALE20,
ZmTALE25 and
AtBEL11 were specifically highly expressed in the roots, while
OsTALE4,
OsTALE15,
OsTALE16,
OsTALE20,
ZmTALE13,
ZmTALE22 and
ZmTALE23 exhibited high expression in inflorescence. These findings suggest a differentiated regulatory role of TALE gene family members across various tissues. Additionally, we found that
OsTALE2,
ZmTALE14,
ZmTALE22,
ZmTALE24 and
AtBLH9 were orthologous genes. Importantly,
OsTALE2,
ZmTALE14 and
ZmTALE24 were highly expressed in stems, while
ZmTALE22 and
AtBLH9 showed high expression in inflorescence. This phenomenon not only reveals the functional differentiation of monocotyledonous and dicotyledonous plants throughout the evolutionary process but also indicates functional conservation and differentiation among grasses. Similar patterns were observed with
OsTALE21,
ZmTALE16,
ZmTALE18 and
AtKNAT5, as well as with
OsTALE25,
ZmTALE15 and
AtBLH1, which further confirmed the complexity and diversity of
TALE gene functions in plant evolution.
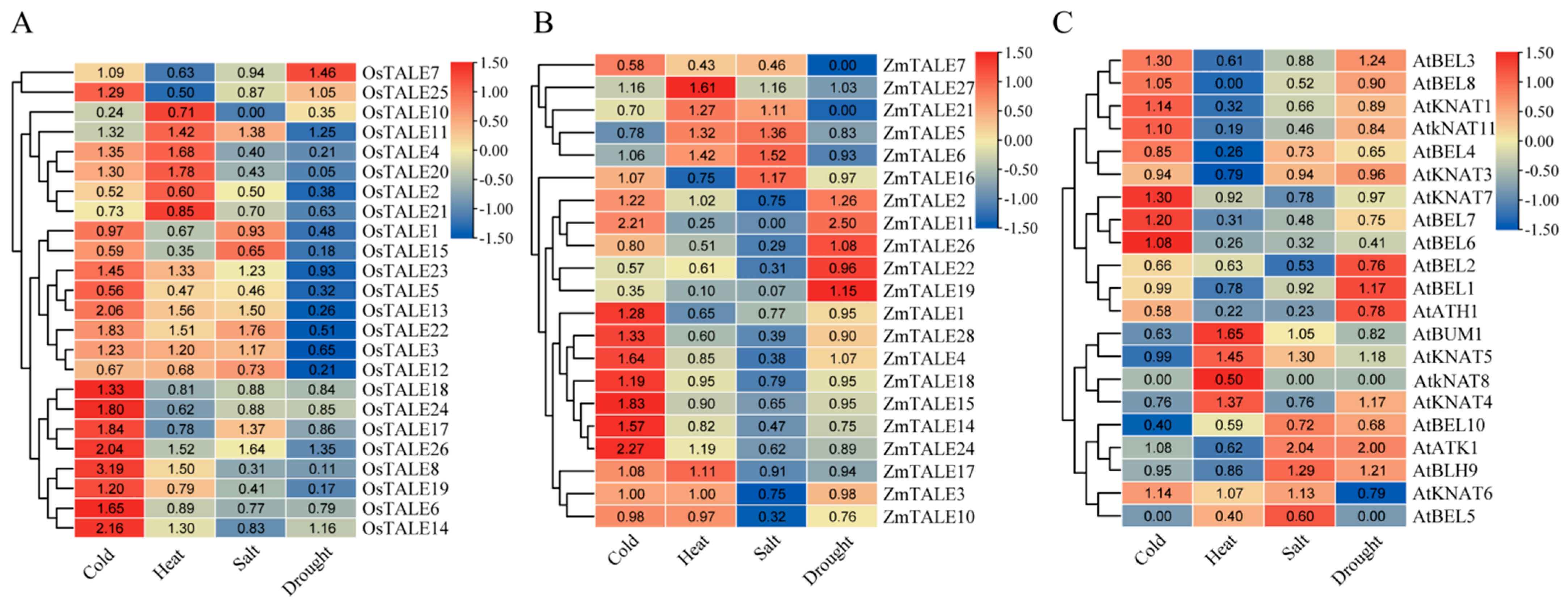
3.8. The Expression Profiles of the OsTALE, ZmTALE and AtTALE Genes Under Abiotic Stresses
The
TALE gene has been identified as playing a crucial role in plant responses to abiotic stress. In this study, we used RNA-seq data to conduct a comprehensive analysis of the expression profile changes of
ZmTALE,
OsTALE and
AtTALE genes under drought, cold, heat and salt stress (
Supplemental Table S5). The results showed that 3, 10, 6 and 16
OsTALE genes were up-regulated under drought, heat, salt and cold stress, respectively, while 14, 7, 7 and 5
OsTALE genes exhibited down-regulated under the same conditions. For the
ZmTALE gene family, the number of up-regulated genes was 2, 4, 2 and 9, under the corresponding stress conditions, with the number of down-regulated genes being 0, 7, 10 and 9. In contrast, the
AtTALE gene family showed 3 up-regulated genes under drought, heat, salt and cold stress, but the number of down-regulated genes varied, recorded as 3, 12, 8 and 4, respectively. Notably, we identified several homologous genes exhibiting distinct expression patterns in response to stress.
OsTALE18,
ZmTALE16,
ZmTALE18 and
AtKNAT4 are homologous genes;
OsTALE18 and
ZmTALE18 were up-regulated under cold stress, while
AtKNAT4 was down-regulated. In addition,
OsTALE2,
ZmTALE14,
ZmTALE22,
ZmTALE24 and
AtBLH9 also belong to homologous genes, each displaying different expression patterns. Specifically,
OsTALE2 was down-regulated under all stress conditions.
ZmTALE14 and
ZmTALE24 were up-regulated under cold stress but down-regulated under salt stress.
AtBLH9 was up-regulated specifically under drought and salt stress, showing no significant response to cold and heat stress. These findings not only enhance our understanding of the functional diversity within the
TALE gene family but also suggest that
TALE genes exhibit both functional conservation and significant differentiation among various species throughout the course of evolution. This has considerable implications for how plants adapt to complex and dynamic environments.
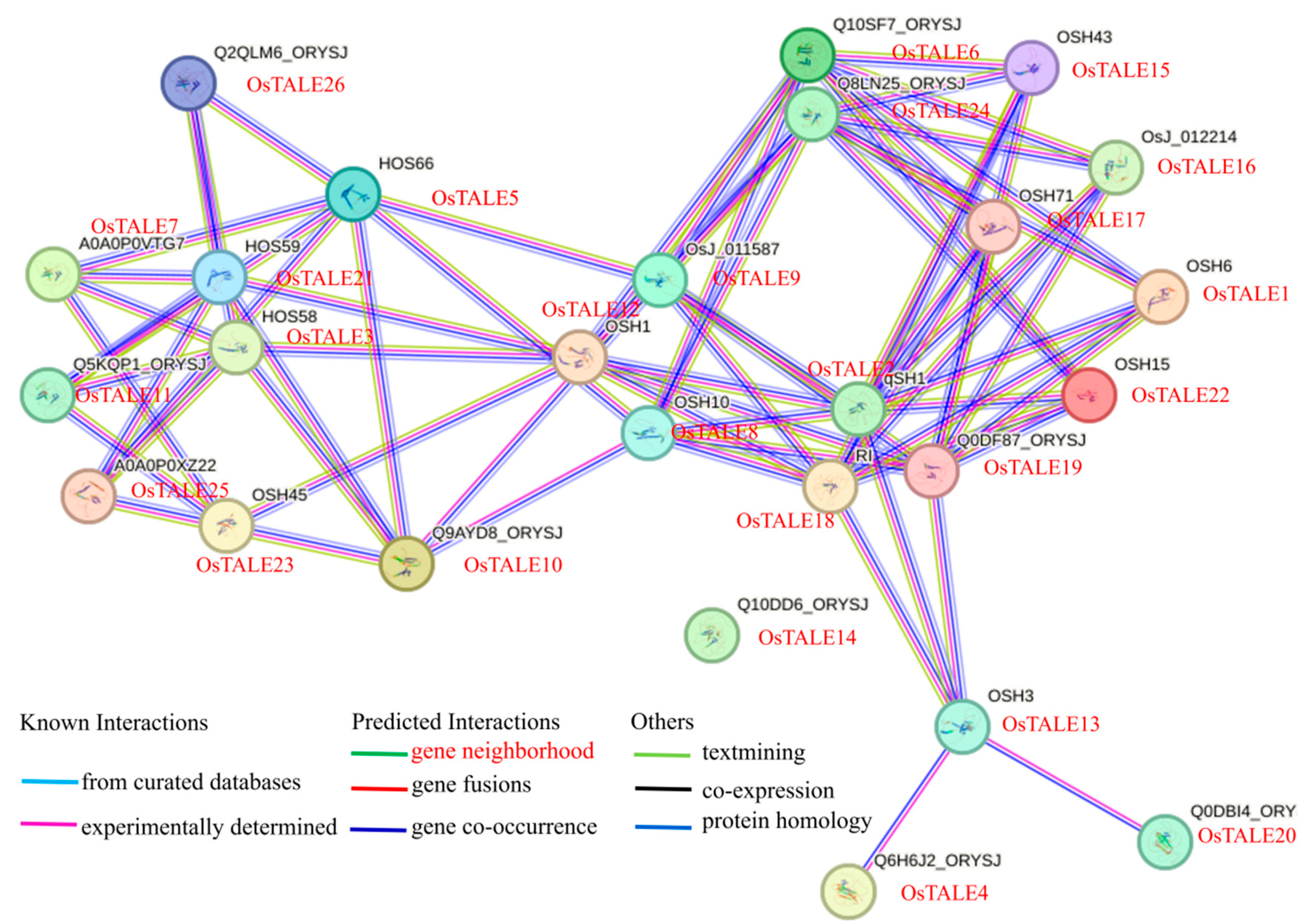
3.9. Analysis of Protein–Protein Interaction (PPI) Networks
Previous studies have shown that TALE proteins can form homologous or heterodimers. Therefore, we used the STRING database to analyze the interaction network of OsTALE proteins and found intricate interactions among family members. Notably, the (OsTALE1) OSH1, OSH15 (OsTALE22) and OSH71 (OsTALE17) genes play an important role in the formation and maintenance of undifferentiated cell meristem states. As shown in
Figure 6, OsTALE17 interacts with OsTALE2, OsTALE19, OsTALE6, OsTALE15 and OsTALE24, suggesting that they may participate in the formation and maintenance of meristem states. Additionally, OsTALE3 interacts with OsTALE5, OsTALE7, OsTALE10, OsTALE11, OsTALE12 and OsTALE21, while OItsTALE2 interacts with OsTALE8, OsTALE9, OsTALE17, OsTALE18 and OsTALE22. Previous studies have revealed that OSH15 (OsTALE22) forms heterodimers with SH5 (OsTALE18) and qSH1 (OsTALE2), repressing lignin biosynthesis genes expression and facilitating seed shedding. This suggests that OsTALE8, OsTALE9 and OsTALE17 proteins may also play a significant role in this biological process. These studies laid a solid foundation for further exploring the mechanism of TALE proteins in plant growth and development and stress response (
Figure 7 and
Figure 8).
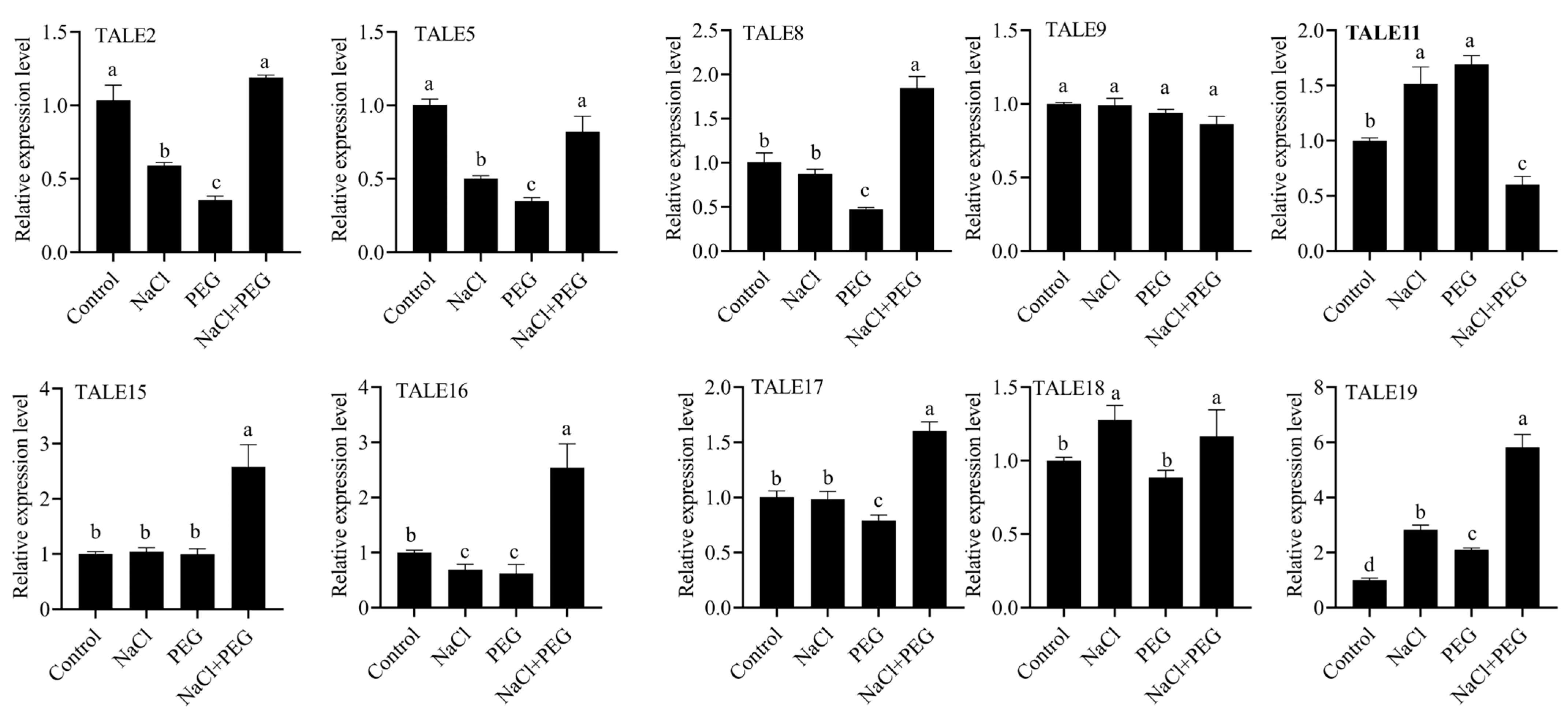
3.10. The Expression Patterns of SbTALE Genes in NaCl and PEG Stress
To further explore the function of the
TALE gene in the Gramineae family, we utilized
S. bicolor as model organism and subjected it to NaCl and PEG treatments. A random selection of 10
S. bicolor TALE genes was verified. The results are shown in
Figure 9. The expression levels of several
SbTALE genes significantly altered under 150 mM NaCl and 15% PEG6000 treatment. Under NaCl treatment, the expressions of genes such as
SbTALE11,
SbTALE18 and
SbTALE19 were up-regulated, while
SbTALE2,
SbTALE5 and
SbTALE16 were down-regulated. Conversely, during PEG treatment, the expressions levels of SbTALE18 and SbTALE46 were up-regulated, whereas
SbTALE2,
SbTALE5,
SbTALE8,
SbTALE16 and
SbTALE17 exhibited down-regulation. Notably, the
SbTALE2,
SbTALE5 and
SbTALE16 genes demonstrated down-regulation following both drought and salt stress treatment. Importantly, under the combined stress of NaCl and PEG6000, the expression levels of
TALE8,
TALE15,
TALE16,
TALE17 and
TALE19 were significantly up-regulated.
4. Discussion
TALE transcription factors are crucial in the regulation of plant growth and development, especially in meristem formation and the maintenance of organ morphogenesis. In this study, 26, 28, 23, 23 and 23
TALE genes were identified in the genomes of
O. sativa,
Z. mays,
S. italica,
S. bicolor and
B. distachyon, respectively. Given that the differentiation of crops dates back to 50 to 70 million years ago [
42], these results imply that the
TALE gene family in Gramineae might have expanded at a relatively uniform rate. Furthermore, we observed that the number of
TALE genes in Gramineae is comparable to
A. thaliana [
43], more than in
P.
granatum [
6] and less than in
G.
max [
3],
T.
aestivum [
2],
P.
tomentosa [
5] and
G.
hirsutum [
7]. This comparison highlights the significant diversity of the
TALE gene across various plant species. This study not only revealed the distribution of
TALE genes in Gramineae but also offered insights into the evolutionary and functional differentiation of
TALE gene family in plants through cross-species analysis.
Conserved domains or motifs are crucial for protein interactions. In this study, we discovered that the
TALE gene family, which consists of five Gramineae and
A. thaliana, features a conserved domain known as Homeobox-KN. Additionally, both Gramineae and
A.
thaliana TALE family members contain motif1, likely representing the most conserved protein motif. Further investigation revealed that the BELL subfamily also encompasses POX and POX-superfamily domains. The KNOX subfamily comprises KNOX1, KNOX2 and ELK domains. In
A. thaliana, BELL subfamily proteins interact with KNAT2 and KNAT5, influencing plant growth and development [
44]. Conversely, KNOX1 plays a crucial role in repressing the expression of downstream target genes, while KNOX2 is essential for the formation of homodimers [
45].
The
TALE family members of five Gramineae species and
A. thaliana were divided into two subgroups, KNOX and Bell, and the KNOX subgroup was further divided into four subgroups, while the BELL subgroup was further divided into five subgroups, which was consistent with the previous classification in
A. thaliana, cotton and poplar [
5,
7,
43]. In
A. thaliana,
O. sativa,
Z. mays,
S. italica,
S. bicolor and
B. distachyon, the BELL and KNOX subfamilies have distinct branches, which reflects the significant differences in amino acid sequences, and it is possible that these two subfamilies diverged early in the evolutionary process. In contrast, Gramineae plants show consistency in differentiation, which may be due to the fact that the five plants belong to the same family of Gramineaes and are closely related to each other. However, Gramineae and
A. thaliana show great differences in differentiation, which may be attributed to the difference in evolutionary paths between monocotyledonous and dicotyledonous plants.
Previous studies have revealed that gene replication events play a key role in the evolution of the
TALE gene family in plants [
2]. We identified 12, 10, 14, 26, 14 and 8 WGD genes and 4, 4, 4, 0, 2 and 0 TD genes in O. sativa,
B. distachyon,
S. bicolor,
Z. mays,
S. italica and
A. thaliana, respectively. These results suggest that WGD is a major factor driving the expansion of the
TALE gene family in these species. In addition, the Ka/Ks values of all homologous gene pairs were less than one, revealing that these gene pairs underwent different degrees of purification selection. The results of Huang et al. (2020) in
G. raimondii and
G. arboretum further support our conclusion [
46]. Additionally, collinearity analysis can be used to study the evolution of gene family in different species. The number of homologous gene pairs between
O. sativa and
Z. mays,
S. italica,
S. bicolor and
B. distachyon were significantly more than that between
O. sativa and
A. thaliana, and the number of
TALE homologous gene pairs with
S. italica,
S. bicolor and
B. distachyon was the most, indicating that
O. sativa and
Z. mays,
S. bicolor and
B. distachyon were closely related in evolution. It is worth noting that collinear gene pairs of
OsTALE2,
OsTALE18,
OsTALE21 and
OsTALE25 exist in dicotyledonous and monocotyledonous plants, which not only reflects gene conservation but also suggests that these homologous genes may have originated before ancestral differentiation. In addition, most of the
TALE genes of
O. sativa had a one-to-many collinearity relationship with
Z. mays,
S. italica,
S. bicolor and
B. distachyon and
A. thaliana, which further confirmed that after the differentiation of
O. sativa and these species, gene family duplication events occurred, promoting the diversity and complexity of their respective genomes.
The
TALE gene family plays a crucial role in regulating plant growth and development. For example, Wang et al. (2020) found that the
TALE gene in
P.
granatum may be involved in regulating shoot apical meristem (SAM), flower and ovule development [
6]. Similarly,
TALE genes showed different expression levels at different stages of walnut flower bud development, revealing its dynamic regulatory role in this process [
47]. The expression levels of
GhSTM3 were down-regulated and significantly affected flowering time [
48]. In poplar, most
TALE genes are highly expressed in stem tissue suggests that they may play a key regulatory role in wood formation [
5]. Consistent with this study, our results also showed that most
TALE genes in
O. sativa,
Z. mays and
A. thaliana were specifically highly expressed in stems, while some
TALE genes were highly expressed in leaves, suggesting that these
TALE genes may play an important role in different developmental stages of
O. sativa,
Z. mays and
A. thaliana. It is worth noting that TALE proteins usually form heterodimers to function [
5]. In our study, there may be interactions between OsTALE17 and OsTALE1 proteins, in addition to OsTALE3 and OsTALE5 as well as OsTALE7, OsTALE10, OsTALE11, OsTALE12 and OsTALE21. It further revealed the complex regulatory mechanism of the
TALE gene family in plant growth and development.
Previous studies have found that the function of
TALE genes is significantly related to the complex hormone network in plants. For example, the
KNOX gene promoter region in Orchidaceae contains MeJA and ABA-responsive elements [
49], while the
TALE gene promoter of pomegranates contains auxin and gibberellin-responsive elements [
6]. In this study, we identified a variety of hormone response elements, including auxin, gibberellin, methyl jasmonate, abscisic acid and salicylic acid in Gramineae and
A. thaliana. These findings suggest that hormone signaling pathways may finely regulate
TALE gene expression in Gramineae and
A. thaliana. Recent studies have shown that the
TALE gene plays an important role in plants response to environmental stress. For example, the
PagKNAT2/6b gene in poplar mediates the response to drought stress by down-regulating the expression of
PagGA20ox1 [
25]. Similarly, the expression of the
TALE gene is not only finely regulated by plant hormones but also influenced by external abiotic stress [
50,
51]. In soybean, the
TALE gene can finely regulate its expression level to adapt to adverse environments under salt and drought stress [
3]. Similarly, 11
TALE genes in poplars were found to have significant responses to salt stress [
5]. As shown in
Figure 7, the promoter region of the
TALE gene in Gramineae and
A. thaliana is rich in a variety of abiotic stress response elements, including low temperature and drought. A further study found that the expression of
OsTALE6/8/14/17/18/19/24/26 and
ZmTALE1/4/14/15/18/24/28 exhibits an increasing trend under cold stress, while
OsTALE7 and
ZmTALE2/11 show significant increases under drought stress. Moreover,
OsTALE2,
ZmTALE14,
ZmTALE22,
ZmTALE24 and
AtBLH9 are orthologous genes. However, in response to different stresses, their expression patterns showed significant differences:
OsTALE2 was down-regulated under drought and salt stress, while
ZmTALE14 and
ZmTALE24 were down-regulated under salt stress. In contrast,
AtBLH9 is up-regulated under drought and salt stress. These results suggest that
TALE genes are functionally differentiated and functionally different in different species during evolution. Different
SbTALE genes showed differential expression patterns under drought and salt stress conditions (
Figure 10). Under salt stress (NaCl),
SbTALE11,
SbTALE18 and
SbTALE19 were up-regulated, while
SbTALE2,
SbTALE5 and
SbTALE16 were down-regulated. During drought stress (PEG),
SbTALE18 and
SbTALE46 were up-regulated, whereas
SbTALE2,
SbTALE5,
SbTALE8,
SbTALE16 and
SbTALE17 were down-regulated. Notably,
SbTALE2,
SbTALE5 and
SbTALE16 were consistently down-regulated under both stresses. Under combined NaCl+PEG6000 stress,
TALE8,
TALE15,
TALE16,
TALE17 and
TALE19 showed significant up-regulation. Analysis of the cis-acting elements of the
SbTALE promoters revealed that
SbTALE2,
SbTALE5,
SbTALE8,
SbTALE17,
SbTALE18 and
SbTALE19 contain regulatory elements related to drought response. These results suggest that the
SbTALE gene family plays an important role in the
S. bicolor response to drought and salt stress. This finding is consistent with previous research indicating that
TALE genes play important roles in drought and salt tolerance in other species. These findings not only reveal the functional differentiation of
TALE genes across species but also highlight their functional differences in response to complex environmental stresses, providing valuable insights into the molecular mechanisms of plant stress resistance.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}