
Transcriptome-Based Analysis of the Co-Expression Network of Genes Related to Nitrogen Absorption in Rice Roots Under Nitrogen Fertilizer and Density


Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Materials and Arrangements
2.2. Sample Collection
2.3. High-Throughput Sequencing Preparation and Data Analysis
2.4. WGCNA Construction and qRT-PCR Validation
2.5. Data Analysis
3. Results
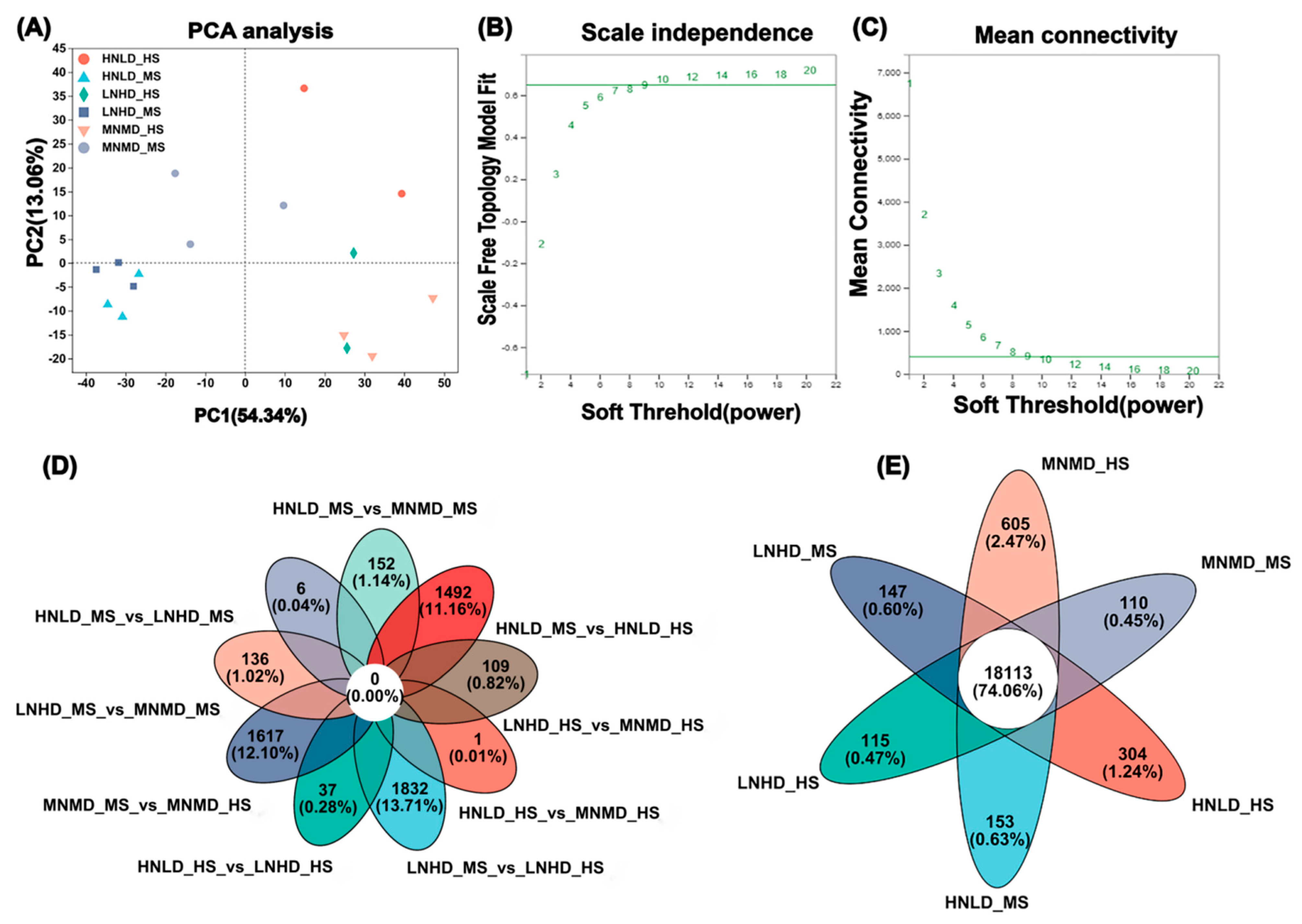
3.1. Transcriptome Data Analysis
3.2. Analysis of Root Morphological Characteristics
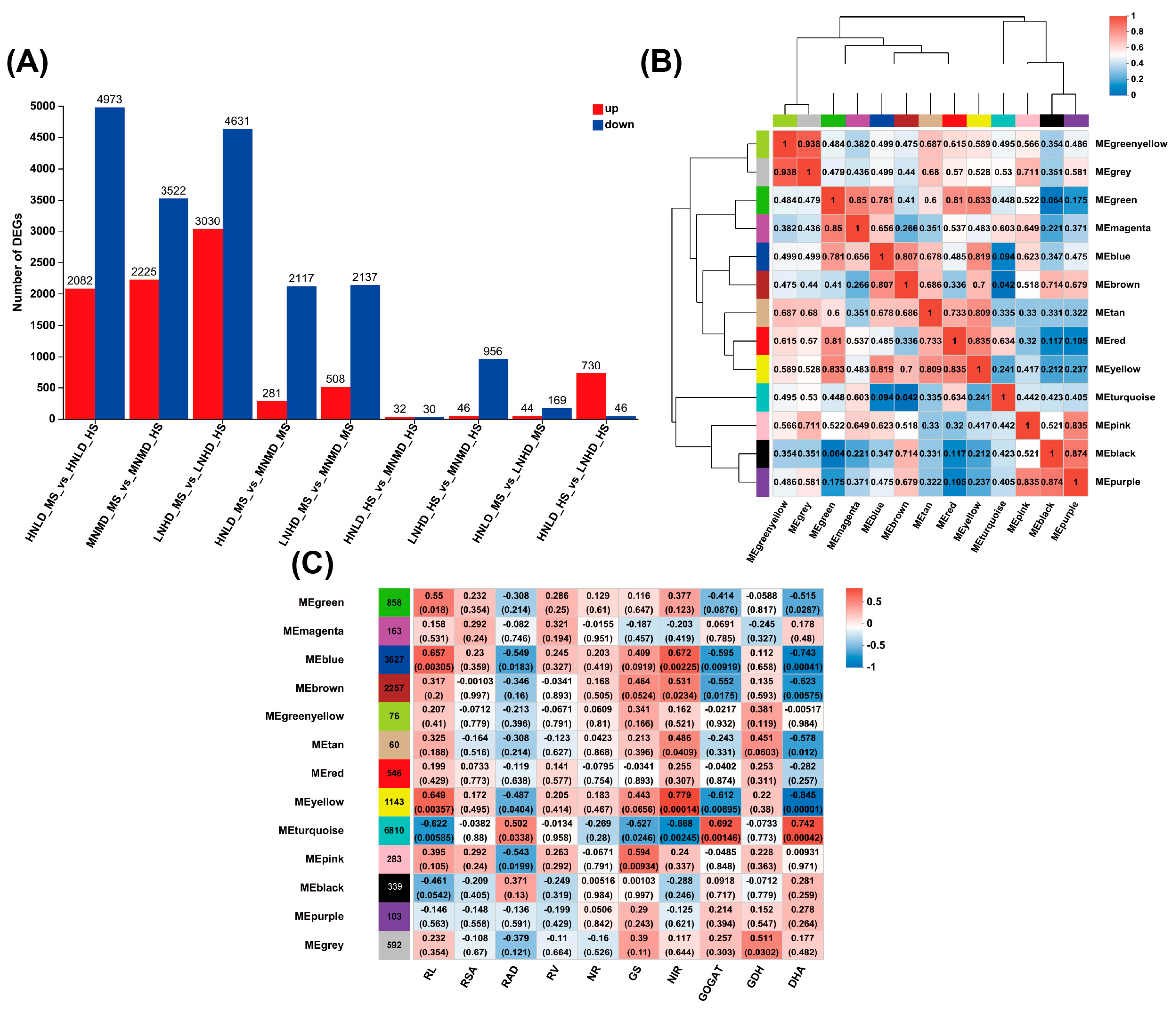
3.3. Differentially Expressed Gene (DEG) Analysis
3.4. WGCNA and Correlation Analysis
3.5. GO Enrichment Analysis
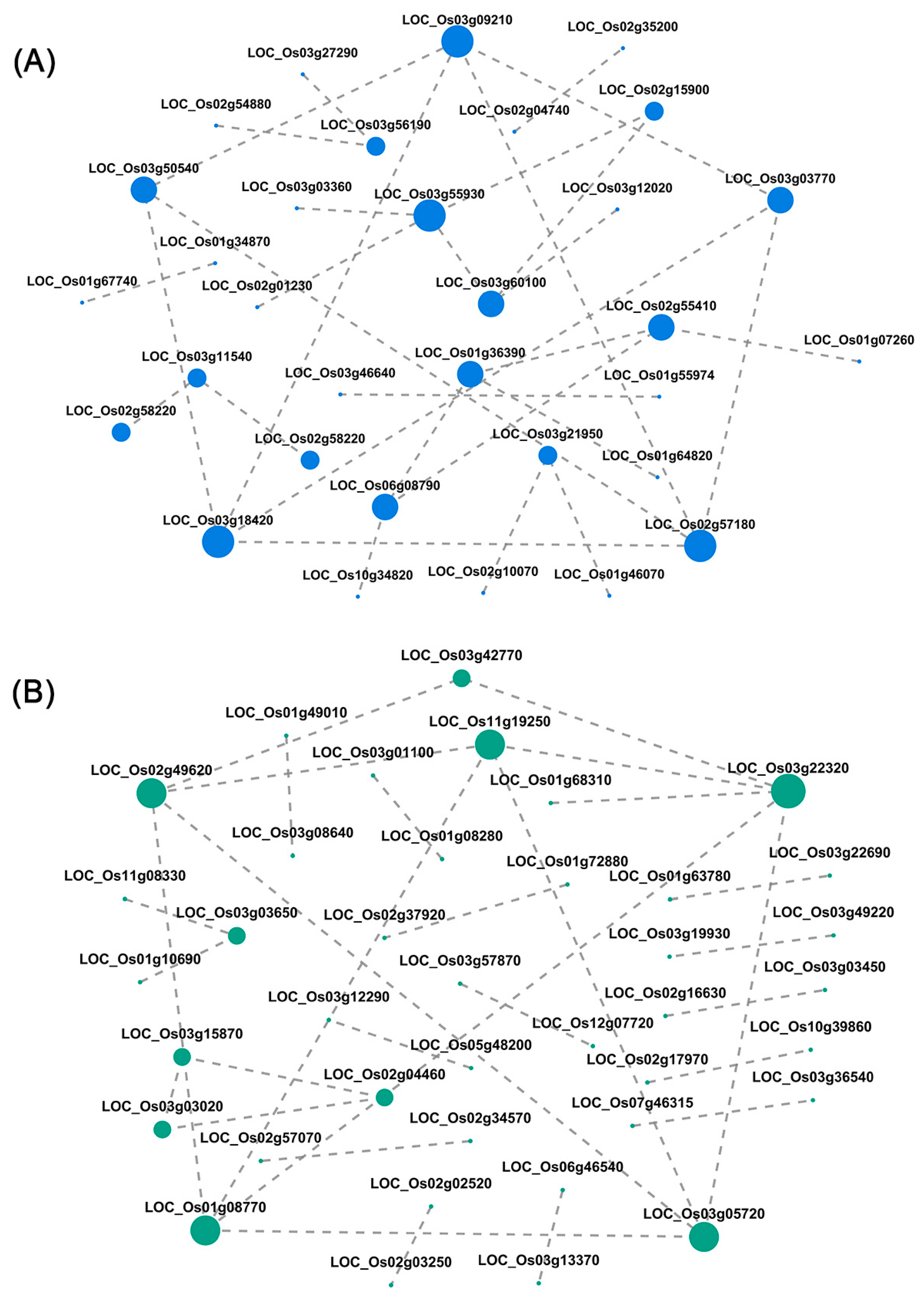
3.6. Hub Gene Network Interactions in Target Modules
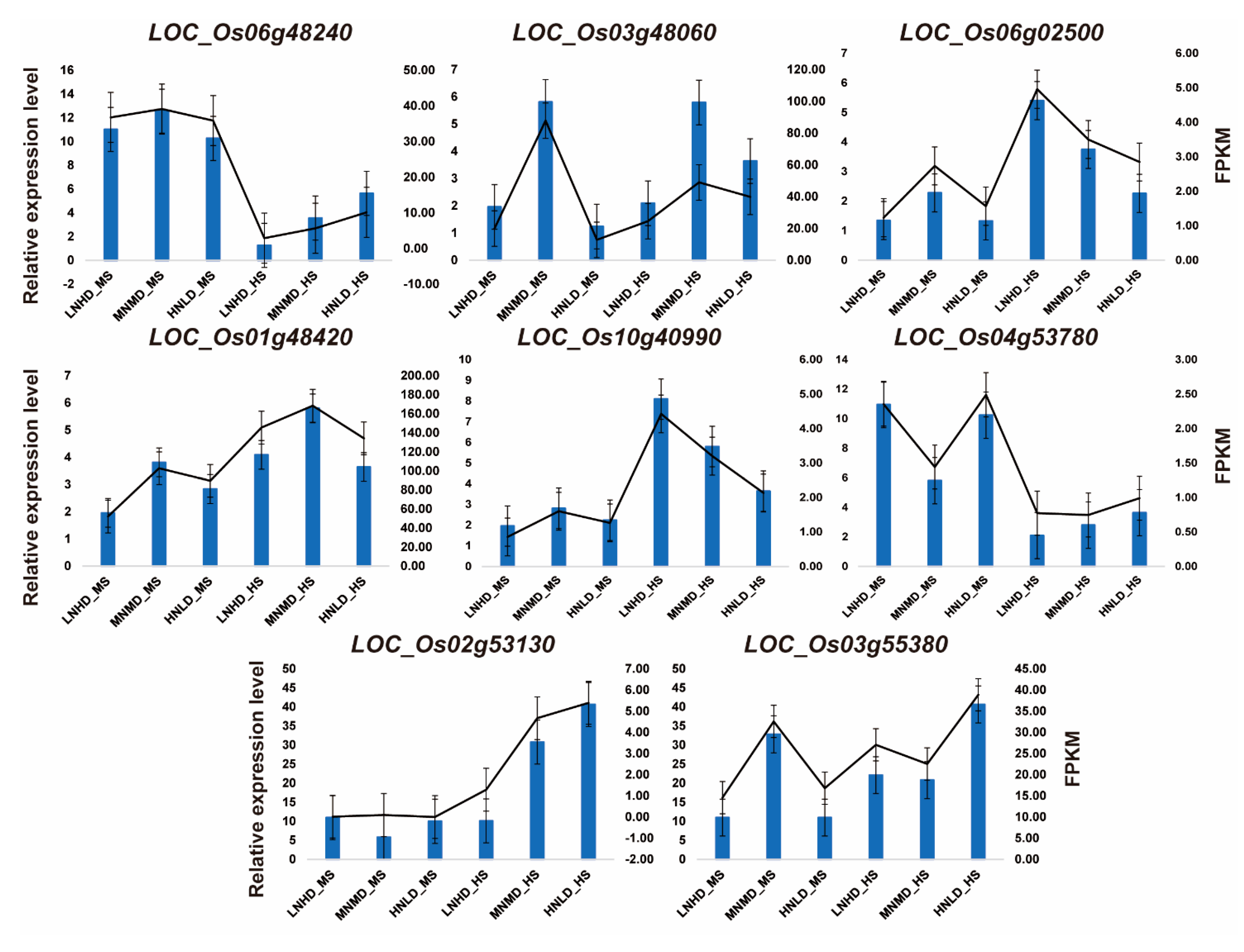
3.7. RT-qPCR Validates Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| N | Nitrogen |
| HNLD | High N/low density |
| HNLD_MS | High N/low density maturity stage |
| HNLD_HS | High N/low density heading stage |
| MNMD | Medium N/medium density |
| MNMD_MS | Medium N/medium density maturity stage |
| MNMD_HS | Medium N/medium density heading stage |
| LNHD | Low N/high density |
| LNHD_MS | Low N/high density maturity stage |
| LNHD_HS | Low N/high density heading stage |
| TSS | Total soluble solids |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| COG | Cluster of orthologous groups |
| NR | Non-redundant protein database |
| Swiss-Prot | Swiss protein database |
| Pfam | Protein families database |
| NR | Nitrate reductase activity |
| GS | Glutamine synthetase activity |
| NIR | Nitrite reductase activity |
| GOGAT | Glutamate synthase activity |
| GDH | Glutamate dehydrogenase activity |
| DHA | Dehydrogenase activity |
| RL | Root length |
| RSA | Root surface area |
| RV | Root volume |
| RAD | Root average diameter |
| Ndff | Nitrogen derived from fertilizer |
References
- Ma, B.; Karimi, M.S.; Mohammed, K.S.; Shahzadi, I.; Dai, J. Nexus between climate change, agricultural output, fertilizer use, agriculture soil emissions: Novel implications in the context of environmental management. J. Clean. Prod. 2024, 450, 141801. [Google Scholar] [CrossRef]
- Sabina, R.; Paul, J.; Sharma, S.; Hussain, N. Synthetic Nitrogen Fertilizer Pollution: Global Concerns and Sustainable Mitigating Approaches. In Agricultural Nutrient Pollution and Climate Change: Challenges and Opportunities; Springer: Berlin/Heidelberg, Germany, 2025; pp. 57–101. [Google Scholar] [CrossRef]
- Sher, A.; Zhang, L.G.; Noor, M.A.; Nadeem, M.; Ashraf, U.; Baloch, S.K.; Ameen, A.; Yuan, X.Y.; Guo, P.Y. Nitrogen use efficiency in cereals under high plant density: Manufacturing, management strategies and future prospects. Appl. Ecol. Environ. Res. 2019, 17, 10139. [Google Scholar] [CrossRef]
- Murchie, E.H.; Burgess, A.J. Casting light on the architecture of crop yield. Crop Environ. 2022, 1, 74–85. [Google Scholar] [CrossRef]
- Zhu, H.; Wen, T.; Sun, M.; Ali, I.; Sheteiwy, M.S.; Wahab, A.; Tan, W.; Wen, C.; He, X.; Wang, X. Enhancing rice yield and nitrogen utilization efficiency through optimal planting density and reduced nitrogen rates. Agronomy 2023, 13, 1387. [Google Scholar] [CrossRef]
- Wei, J.; Chai, Q.; Yin, W.; Fan, H.; Guo, Y.; Hu, F.; Fan, Z.; Wang, Q. Grain yield and N uptake of maize in response to increased plant density under reduced water and nitrogen supply conditions. J. Integr. Agric. 2024, 23, 122–140. [Google Scholar] [CrossRef]
- Ajmera, I.; Henry, A.; Radanielson, A.M.; Klein, S.P.; Ianevski, A.; Bennett, M.J.; Band, L.R.; Lynch, J.P. Integrated root phenotypes for improved rice performance under low nitrogen availability. Plant Cell Environ. 2022, 45, 805–822. [Google Scholar] [CrossRef]
- Wang, W.; Shen, C.; Xu, Q.; Zafar, S.; Du, B.; Xing, D. Grain yield, nitrogen use efficiency and antioxidant enzymes of rice under different fertilizer n inputs and planting density. Agronomy 2022, 12, 430. [Google Scholar] [CrossRef]
- Chen, L.; Xie, H.; Wang, G.; Qian, X.; Wang, W.; Xu, Y.; Zhang, W.; Zhang, H.; Liu, L.; Wang, Z. Reducing environmental risk by improving crop management practices at high crop yield levels. Field Crops Res. 2021, 265, 108123. [Google Scholar] [CrossRef]
- Satrio, R.D.; Fendiyanto, M.H.; Miftahudin, M. Tools and Techniques Used at Global Scale Through Genomics, Transcriptomics, Proteomics, and Metabolomics to Investigate Plant Stress Responses at the Molecular Level. In Molecular Dynamics of Plant Stress and Its Management; Springer: Berlin/Heidelberg, Germany, 2024; pp. 555–607. [Google Scholar] [CrossRef]
- Haq, S.A.U.; Bashir, T.; Roberts, T.H.; Husaini, A.M. Ameliorating the effects of multiple stresses on agronomic traits in crops: Modern biotechnological and omics approaches. Mol. Biol. Rep. 2024, 51, 41. [Google Scholar] [CrossRef]
- Li, Q.; Lu, X.; Wang, C.; Shen, L.; Dai, L.; He, J.; Yang, L.; Li, P.; Hong, Y.; Zhang, Q. Genome-wide association study and transcriptome analysis reveal new QTL and candidate genes for nitrogen-deficiency tolerance in rice. Crop J. 2022, 10, 942–951. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Y.; Ying, L.; Lu, H.; Liu, Y.; Liu, Y.; Xu, J.; Wu, Y.; Mo, X.; Wu, Z. Integrated transcriptomic analysis identifies coordinated responses to nitrogen and phosphate deficiency in rice. Front. Plant Sci. 2023, 14, 1164441. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, X.; Xu, L.; Li, W.; Yao, Q.; Yin, X.; Wang, Q.; Tan, W.; Xing, W.; Liu, D. Low nitrogen stress-induced transcriptome changes revealed the molecular response and tolerance characteristics in maintaining the C/N balance of sugar beet (Beta vulgaris L.). Front. Plant Sci. 2023, 14, 1164151. [Google Scholar] [CrossRef]
- Privitera, G.F.; Treccarichi, S.; Nicotra, R.; Branca, F.; Pulvirenti, A.; Piero, A.R.L.; Sicilia, A. Comparative transcriptome analysis of B. oleracea L. var. italica and B. macrocarpa Guss. genotypes under drought stress: De novo vs reference genome assembly. Plant Stress. 2024, 14, 100657. [Google Scholar] [CrossRef]
- Raghavan, V.; Kraft, L.; Mesny, F.; Rigerte, L. A simple guide to de novo transcriptome assembly and annotation. Brief. Bioinform. 2022, 23, b563. [Google Scholar] [CrossRef] [PubMed]
- Paik, H.; Yoo, S.; Nam, H.; Stevenson, M.; No, A. DTMBIO 2020: The Fourteenth International Workshop on Data and Text Mining in Biomedical Informatics. In Proceedings of the 29th ACM International Conference on Information & Knowledge Management, Virtual, 23 October 2020; pp. 3537–3538. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sun, C.; Lu, Y.; Tang, G.; Wang, R.; Wu, H.; Zhang, J.; Cai, S.; Zhu, J.; Xiong, Q. Integrated Metagenomics and 15N Isotope Tracing Reveal the Mechanisms Through which the Nitrogen-Planting Density Interaction Impacts Rice Root Nitrogen Uptake Efficiency. J. Soil Sci. Plant Nutr. 2024, 24, 1–14. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, J.; Zhang, Z.; Deng, A.; Zhang, W. Dense planting with less basal nitrogen fertilization might benefit rice cropping for high yield with less environmental impacts. Eur. J. Agron. 2016, 75, 50–59. [Google Scholar] [CrossRef]
- Fortunato, S.; Nigro, D.; Lasorella, C.; Marcotuli, I.; Gadaleta, A.; de Pinto, M.C. The role of glutamine synthetase (GS) and glutamate synthase (GOGAT) in the Improvement of nitrogen use efficiency in cereals. Biomolecules 2023, 13, 1771. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Kumar, K.; Jha, A.K.; Yadava, P.; Pal, M.; Rakshit, S.; Singh, I. Global gene expression profiling under nitrogen stress identifies key genes involved in nitrogen stress adaptation in maize (Zea mays L.). Sci. Rep. 2022, 12, 4211. [Google Scholar] [CrossRef] [PubMed]
- Platzer, M.; Kiese, S.; Asam, T.; Schneider, F.; Tybussek, T.; Herfellner, T.; Schweiggert-Weisz, U.; Eisner, P. Quantitative Structure-Property Relationship (QSPR) of Plant Phenolic Compounds in Rapeseed Oil and Comparison of Antioxidant Measurement Methods. Processes 2022, 10, 1281. [Google Scholar] [CrossRef]
- Li, Y.; Wang, M.; Teng, K.; Dong, D.; Liu, Z.; Zhang, T.; Han, L. Transcriptome profiling revealed candidate genes, pathways and transcription factors related to nitrogen utilization and excessive nitrogen stress in perennial ryegrass. Sci. Rep. 2022, 12, 3353. [Google Scholar] [CrossRef]
- Li, H.; Wang, Q.; Huang, T.; Liu, J.; Zhang, P.; Li, L.; Xie, H.; Wang, H.; Liu, C.; Qin, P. Transcriptome and Metabolome Analyses Reveal Mechanisms Underlying the Response of Quinoa Seedlings to Nitrogen Fertilizers. Int. J. Mol. Sci. 2023, 24, 11580. [Google Scholar] [CrossRef]
- Liao, G.; Yang, Y.; Xiao, W.; Mo, Z. Nitrogen modulates grain yield, nitrogen metabolism, and antioxidant response in different rice genotypes. J. Plant Growth Regul. 2023, 42, 2103–2114. [Google Scholar] [CrossRef]
- Brooks, E.G.; Elorriaga, E.; Liu, Y.; Duduit, J.R.; Yuan, G.; Tsai, C.; Tuskan, G.A.; Ranney, T.G.; Yang, X.; Liu, W. Plant promoters and terminators for high-precision bioengineering. BioDesign Res. 2023, 5, 13. [Google Scholar] [CrossRef]
- Xiong, Q.; Sun, C.; Shi, H.; Cai, S.; Xie, H.; Liu, F.; Zhu, J. Analysis of related metabolites affecting taste values in rice under different nitrogen fertilizer amounts and planting densities. Foods 2022, 11, 1508. [Google Scholar] [CrossRef]
- Wu, L.; Deng, Z.; Cao, L.; Meng, L. Effect of plant density on yield and quality of perilla sprouts. Sci. Rep. 2020, 10, 9937. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.S.; Sharma, M.; Laxmi, A. Role of sugar and auxin crosstalk in plant growth and development. Physiol. Plantarum 2022, 174, e13546. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Zhao, C.; Zhang, M.; Sun, C.; Liu, X.; Hu, J.; Zeeshan, M.; Zaid, A.; Dai, T.; Tian, Z. Nitrogen enhances the effect of pre-drought priming against post-anthesis drought stress by regulating starch and protein formation in wheat. Physiol. Plantarum 2023, 175, e13907. [Google Scholar] [CrossRef] [PubMed]
- Effah, Z.; Li, L.; Xie, J.; Liu, C.; Xu, A.; Karikari, B.; Anwar, S.; Zeng, M. Regulation of nitrogen metabolism, photosynthetic activity, and yield attributes of spring wheat by nitrogen fertilizer in the semi-arid loess plateau region. J. Plant Growth Regul. 2023, 42, 1120–1133. [Google Scholar] [CrossRef]
- Lee, S.; Na, D.; Park, C. Comparability of reference-based and reference-free transcriptome analysis approaches at the gene expression level. BMC Bioinform. 2021, 22, 310. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Expre_Gene Number (%) | Expre_Transcript Number (%) | All_Gene Number (%) | All_Transcript Number (%) |
|---|---|---|---|---|
| GO | 30,731 (0.8003) | 38,610 (0.8131) | 41,843 (0.7474) | 50,705 (0.7643) |
| KEGG | 11,132 (0.2899) | 15,482 (0.326) | 12,549 (0.2241) | 17,342 (0.2614) |
| COG | 31,543 (0.8215) | 40,291 (0.8485) | 40,390 (0.7214) | 50,157 (0.7561) |
| NR | 38,206 (0.995) | 47,297 (0.996) | 55,224 (0.9864) | 65,564 (0.9883) |
| Swiss-Prot | 25,139 (0.6547) | 32,671 (0.688) | 29,796 (0.5322) | 38,156 (0.5752) |
| Pfam | 29,373 (0.765) | 36,945 (0.778) | 36,944 (0.6599) | 45,409 (0.6845) |
| Total_anno | 38,214 (0.9952) | 47,307 (0.9962) | 55,235 (0.9866) | 65,577 (0.9885) |
| Total | 38,398 (1.0) | 47,486 (1.0) | 55,986 (1) | 66,338 (1) |
| Sample | Total Reads | Total Mapped | Multiple Mapped | Uniquely Mapped |
|---|---|---|---|---|
| LNHD_MS3 | 43,788,196.00 | 40,563,536 (92.64%) | 1,027,295 (2.35%) | 39,536,241 (90.29%) |
| LNHD_MS2 | 44,738,918.00 | 42,442,486 (94.87%) | 1,123,167 (2.51%) | 41,319,319 (92.36%) |
| LNHD_MS1 | 41,543,966.00 | 36,536,385 (87.95%) | 982,550 (2.37%) | 35,553,835 (85.58%) |
| MNMD_MS3 | 44,271,468.00 | 41,234,659 (93.14%) | 978,466 (2.21%) | 40,256,193 (90.93%) |
| MNMD_MS2 | 43,717,944.00 | 41,367,829 (94.62%) | 1,000,154 (2.29%) | 40,367,675 (92.34%) |
| MNMD_MS1 | 44,407,378.00 | 41,329,139 (93.07%) | 878,963 (1.98%) | 40,450,176 (91.09%) |
| HNLD_MS3 | 42,980,190.00 | 40,655,624 (94.59%) | 1,112,703 (2.59%) | 39,542,921 (92.0%) |
| HNLD_MS2 | 44,741,482.00 | 42,198,136 (94.32%) | 1,153,670 (2.58%) | 41,044,466 (91.74%) |
| HNLD_MS1 | 43,808,086.00 | 41,100,507 (93.82%) | 1,103,059 (2.52%) | 39,997,448 (91.3%) |
| LNHD_HS3 | 45,854,996.00 | 43,976,160 (95.9%) | 1,294,726 (2.82%) | 42,681,434 (93.08%) |
| LNHD_HS2 | 45,247,476.00 | 43,222,035 (95.52%) | 1,211,992 (2.68%) | 42,010,043 (92.85%) |
| LNHD_HS1 | 53,822,518.00 | 51,136,296 (95.01%) | 1,336,284 (2.48%) | 49,800,012 (92.53%) |
| MNMD_HS3 | 45,275,630.00 | 43,062,454 (95.11%) | 1,077,873 (2.38%) | 41,984,581 (92.73%) |
| MNMD_HS2 | 44,707,566.00 | 42,658,057 (95.42%) | 1,184,548 (2.65%) | 41,473,509 (92.77%) |
| MNMD_HS1 | 54,236,202.00 | 52,204,495 (96.25%) | 1,486,009 (2.74%) | 50,718,486 (93.51%) |
| HNLD_HS3 | 42,556,750.00 | 37,092,719 (87.16%) | 994,379 (2.34%) | 36,098,340 (84.82%) |
| HNLD_HS2 | 45,492,090.00 | 43,733,711 (96.13%) | 1,030,136 (2.26%) | 42,703,575 (93.87%) |
| HNLD_HS1 | 46,356,536.00 | 44,754,616 (96.54%) | 1,042,874 (2.25%) | 43,711,742 (94.29%) |
| Treatment | RL (cm) | RSA (cm2) | RAD (mm) | RV (cm3) |
|---|---|---|---|---|
| HNLD_HS | 301.50 ab | 4901.26 b | 0.48 cd | 60.59 b |
| MNMD_HS | 379.95 a | 4210.94 b | 0.44 d | 47.97 bc |
| LNHD_HS | 325.99 ab | 1621.40 cd | 0.51 abc | 32.38 c |
| HNLD_MS | 230.95 cd | 3139.84 a | 0.56 ab | 85.22 a |
| MNMD_MS | 264.17 c | 2199.35 c | 0.60 a | 83.52 a |
| LNHD_MS | 237.55 bcd | 881.64d | 0.54 abc | 71.9 b |
| GO ID | Description | Ratio_in_Study | Ratio_in_Pop | p-Value |
|---|---|---|---|---|
| GO enrichment pathways between HNLD_HS and LNHD_HS | ||||
| GO:0051171 | regulation of nitrogen compound metabolic process | 56/585 | 2505/42,523 | 0.000373735 |
| GO enrichment pathways between HNLD_HS and MNMD_HS | ||||
| GO:0016052 | carbohydrate catabolic process | 3/45 | 339/42,523 | 0.005563181 |
| GO:0019203 | Carbohydrate phosphatase activity | 1/45 | 46/42,523 | 0.047562811 |
| GO:0016491 | oxidoreductase activity | 7/45 | 2654/42,523 | 0.020602351 |
| GO enrichment pathways between LNHD_HS and MNMD_HS | ||||
| GO:0016209 | antioxidant activity | 15/759 | 303/42,523 | 0.000411868 |
| GO:0016052 | carbohydrate catabolic process | 16/759 | 339/42,523 | 0.000452965 |
| GO:0005504 | fatty acid binding | 2/759 | 20/42,523 | 0.048897966 |
| GO:0016491 | oxidoreductase activity | 75/759 | 2654/42,523 | 0.000101426 |
| GO:1901698 | response to nitrogen compound | 5/759 | 98/42,523 | 0.031306244 |
| GO enrichment pathways between HNLD_MS and LNHD_MS | ||||
| GO:0016667 | oxidoreductase activity, acting on a sulfur group of donors | 3/145 | 121/42,523 | 0.00834102 |
| GO:1901698 | response to nitrogen compound | 2/145 | 98/42,523 | 0.044368267 |
| GO enrichment pathways between HNLD_MS and MNMD_MS | ||||
| GO:0016209 | antioxidant activity | 26/1940 | 303/42,523 | 0.002075187 |
| GO:0005524 | ATP binding | 341/1940 | 4929/42,523 | 0.00000546 |
| GO:0016052 | carbohydrate catabolic process | 30/1940 | 339/42,523 | 0.00056884 |
| GO:0033759 | flavone synthase activity | 1/1940 | 1/42,523 | 0.045622369 |
| GO:0009812 | flavonoid metabolic process | 4/1940 | 23/42,523 | 0.019098749 |
| GO:0006541 | glutamine metabolic process | 7/1940 | 43/42,523 | 0.003098179 |
| GO:0016491 | oxidoreductase activity | 182/1940 | 2654/42,523 | 0.00000369 |
| GO enrichment pathways between LNHD_MS and MNMD_MS | ||||
| GO:0016209 | antioxidant activity | 33/2162 | 303/42,523 | 0.0000549 |
| GO:0005524 | ATP binding | 389/2162 | 4929/42,523 | 0.00000609 |
| GO:0016052 | carbohydrate catabolic process | 40/2162 | 339/42,523 | 0.00000234 |
| GO:0009812 | flavonoid metabolic process | 4/2162 | 23/42,523 | 0.02720865 |
| GO:0006541 | glutamine metabolic process | 7/2162 | 43/42,523 | 0.005604449 |
| GO:0042744 | hydrogen peroxide catabolic process | 25/2162 | 202/42,523 | 0.0000642 |
| GO:0016491 | oxidoreductase activity | 207/2162 | 2654/42,523 | 0.00000473 |
| GO enrichment pathways between HNLD_MS and HNLD_HS | ||||
| GO:0006757 | ATP generation from ADP | 19/5743 | 72/42,523 | 0.003029804 |
| GO:0016052 | carbohydrate catabolic process | 92/5743 | 339/42,523 | 0.00000196 |
| GO:0006631 | fatty acid metabolic process | 59/5743 | 216/42,523 | 0.00000194 |
| GO:0051552 | flavone metabolic process | 2/5743 | 2/42,523 | 0.018237463 |
| GO:0009812 | flavonoid metabolic process | 10/5743 | 23/42,523 | 0.000424699 |
| GO:0006541 | glutamine metabolic process | 13/5743 | 43/42,523 | 0.005375737 |
| GO:0006096 | glycolytic process | 19/5743 | 72/42,523 | 0.003029804 |
| GO:0016491 | oxidoreductase activity | 557/5743 | 2654/42,523 | 0.00000706 |
| GO enrichment pathways between MNMD_MS and MNMD_HS | ||||
| GO:0016209 | antioxidant activity | 59/4670 | 303/42,523 | 0.000012 |
| GO:0005524 | ATP binding | 782/4670 | 4929/42,523 | 0.00000914 |
| GO:0016052 | carbohydrate catabolic process | 81/4670 | 339/42,523 | 0.00000188 |
| GO:0006541 | glutamine metabolic process | 11/4670 | 43/42,523 | 0.005660847 |
| GO:0016491 | oxidoreductase activity | 429/4670 | 2654/42,523 | 0.00000697 |
| GO:0051171 | regulation of nitrogen compound metabolic process | 364/4670 | 2505/42,523 | 0.00000621 |
| GO enrichment pathways between LNHD_MS and LNHD_HS | ||||
| GO:0005524 | ATP binding | 1053/6248 | 4929/42,523 | 0.0000104 |
| GO:0016051 | carbohydrate biosynthetic process | 105/6248 | 339/42,523 | 0.00000208 |
| GO:0051552 | flavone metabolic process | 2/6248 | 2/42,523 | 0.021586138 |
| GO:0006541 | glutamine metabolic process | 14/6248 | 43/42,523 | 0.00356955 |
| GO:0006096 | glycolytic process | 24/6248 | 72/42,523 | 0.0000682 |
| GO:0016491 | oxidoreductase activity | 607/6248 | 2654/42,523 | 0.00000754 |
| Gene Module | GO Number | Functional Classification | Study Gene Ratio | Reference Gene Ratio | Enrichment p-Value |
|---|---|---|---|---|---|
| Turquoise | GO:0051173 | positive regulation of N compound metabolic process | 112/5133 | 548/42,523 | 0.00000328 |
| Turquoise | GO:0051172 | negative regulation of N compound metabolic process | 69/5133 | 317/42,523 | 0.00000362 |
| Turquoise | GO:0051171 | regulation of N compound metabolic process | 509/5133 | 2505/42,523 | 0.00000697 |
| Turquoise | GO:0005524 | ATP binding | 926/5133 | 4929/42,523 | 0.00000985 |
| Turquoise | GO:0016730 | oxidoreductase activity, acting on iron–sulfur proteins as donors | 4/5133 | 12/42,523 | 0.047268181 |
| Blue | GO:0016684 | oxidoreductase activity, acting on peroxide as acceptor | 76/2886 | 263/42,523 | 0.00000164 |
| Blue | GO:0052716 | hydroquinone–oxygen oxidoreductase activity | 12/2886 | 36/42,523 | 0.00000251 |
| Blue | GO:0016614 | oxidoreductase activity, acting on CH-OH group of donors | 46/2886 | 322/42,523 | 0.00000357 |
| Blue | GO:0016616 | oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor | 40/2886 | 268/42,523 | 0.00000368 |
| Blue | GO:0016491 | oxidoreductase activity | 352/2886 | 2654/42,523 | 0.00000527 |
| Blue | GO:0016651 | oxidoreductase activity, acting on NAD(P)H | 18/2886 | 94/42,523 | 0.0000549 |
| Blue | GO:0016682 | oxidoreductase activity, acting on diphenols and related substances as donors, oxygen as acceptor | 14/2886 | 64/42,523 | 0.0000808 |
| Blue | GO:0016679 | oxidoreductase activity, acting on diphenols and related substances as donors | 15/2886 | 75/42,523 | 0.000134451 |
| Blue | GO:0050664 | oxidoreductase activity, acting on NAD(P)H, oxygen as acceptor | 6/2886 | 14/42,523 | 0.0001814 |
| Blue | GO:0016628 | oxidoreductase activity, acting on the CH-CH group of donors, NAD or NADP as acceptor | 11/2886 | 46/42,523 | 0.000200178 |
| Blue | GO:0016620 | oxidoreductase activity, acting on the aldehyde or oxo group of donors, NAD or NADP as acceptor | 11/2886 | 70/42,523 | 0.007278342 |
| Blue | GO:0016717 | oxidoreductase activity, acting on paired donors, with oxidation of a pair of donors resulting in the reduction in molecular oxygen to two molecules of water | 5/2886 | 20/42,523 | 0.009410658 |
| Blue | GO:0016860 | intramolecular oxidoreductase activity | 9/2886 | 58/42,523 | 0.015659569 |
| Blue | GO:0016903 | oxidoreductase activity, acting on the aldehyde or oxo group of donors | 12/2886 | 90/42,523 | 0.020260485 |
| Blue | GO:0016671 | oxidoreductase activity, acting on a sulfur group of donors, disulfide as acceptor | 5/2886 | 24/42,523 | 0.020597531 |
| Blue | GO:0016655 | oxidoreductase activity, acting on NAD(P)H, quinone or similar compound as acceptor | 9/2886 | 66/42,523 | 0.043312538 |
| Blue | GO:0016209 | antioxidant activity | 79/2886 | 303/42,523 | 0.0000027 |
| Blue | GO:0016052 | carbohydrate catabolic process | 43/2886 | 339/42,523 | 0.0000779 |
| Blue | GO:0044275 | cellular carbohydrate catabolic process | 11/2886 | 80/42,523 | 0.022650601 |
| Blue | GO:0009812 | flavonoid metabolic process | 6/2886 | 23/42,523 | 0.003591583 |
| Module | Gene ID | Gene Description | GO ID | GO Description | KEGG Pathway ID | KEGG Pathway Definition |
|---|---|---|---|---|---|---|
| Blue | LOC_Os03g09210 | NADH dehydrogenase 1 alpha subcomplex subunit 13, putative, expressed | GO: 0005743; GO: 0016021; GO: 0070469; GO: 0005747; GO: 0008137 | CC: mitochondrial inner membrane; CC: integral component of membrane; CC: respiratory chain; CC: mitochondrial respiratory chain complex I; MF: NADH dehydrogenas; | map00190 | Oxidative phosphorylation |
| LOC_Os03g50540 | 2Fe-2S iron-sulfur cluster binding domain containing protein, expressed | GO: 0042773; GO: 0006979; GO: 0005743; GO: 0005747; GO: 0009507; GO: 0051539; GO: 0008137 | BP: ATP synthesis coupled electron transport; BP: response to oxidative stress; CC: mitochondrial inner membrane; CC: mitochondrial respiratory chain complex I; CC: chloroplast; MF: 4 iron, 4 sulfur cluster binding; MF: NADH dehydrogenas; | map00190 | Oxidative phosphorylation | |
| LOC_Os03g18420 | CHCH domain-containing protein, expressed | GO: 0006120; GO: 0070469; GO: 0005747; GO: 0005739 | BP: mitochondrial electron transport, NADH to ubiquinone; CC: respiratory chain; CC: mitochondrial respiratory chain complex I; CC: mitochondrion; | map00190 | Oxidative phosphorylation | |
| LOC_Os02g57180 | NADH dehydrogenase 1 alpha subcomplex subunit 9, mitochondrial precursor, putative, expressed | GO: 1901006; GO: 0016021; GO: 0005739; GO: 0005747; GO: 0003824; GO: 0044877 | BP: ubiquinone-6 biosynthetic process; CC: integral component of membrane; CC: mitochondrion; CC: mitochondrial respiratory chain complex I; MF: catalytic activity; MF: macromolecular complex binding; | map00190 | Oxidative phosphorylation | |
| LOC_Os03g03770 | expressed protein | GO: 0070469; GO: 0005758; GO: 0005747; GO: 0005743; GO: 0008137 | CC: respiratory chain; CC: mitochondrial intermembrane space; CC: mitochondrial respiratory chain complex I; CC: mitochondrial inner membrane; MF: NADH dehydrogenas; | map00190 | Oxidative phosphorylation | |
| Turquoise | LOC_Os11g19250 | AARP2CN domain-containing protein, expressed | GO: 0042254; GO: 0000462; GO: 0000479; GO: 0030688; GO: 0005730; GO: 0005634; GO: 0003924; GO: 0034511; GO: 0005525 | BP: ribosome biogenesis; BP: maturation of SSU-rRNA from tricistronic rRNA transcrip; BP: endonucleolytic cleavage of tricistronic rRNA transcrip; CC: preribosome, small subunit precursor; CC: nucleolus; CC: nucleus; MF: GTPase activity; MF: U3 snoRNA binding; MF: GTP binding; | ------ | ------ |
| LOC_Os02g49620 | expressed protein | GO: 0000028; GO: 0006364; GO: 0032545; GO: 0034456 | BP: ribosomal small subunit assembly; BP: rRNA processing; CC: CURI complex; CC: UTP-C complex; | map03008 | Ribosome biogenesis in eukaryotes | |
| LOC_Os01g08770 | WD domain, G-beta repeat domain-containing protein, expressed | GO: 0000462; GO: 0032040; GO: 0005730 | BP: maturation of SSU-rRNA from tricistronic rRNA transcrip; CC: small-subunit processome; CC: nucleolus; | ------ | ------ | |
| LOC_Os03g22320 | utp14 protein, putative, expressed | GO: 0006364; GO: 0032040; GO: 0005730 | BP: rRNA processing; CC: small-subunit processome; CC: nucleolus; | map03008 | Ribosome biogenesis in eukaryotes | |
| LOC_Os03g05720 | WD domain, G-beta repeat domain-containing protein, expressed | GO: 0030490; GO: 0034388; GO: 0032040; GO: 0005730; GO: 0030515 | BP: maturation of SSU-rRNA; CC: Pwp2p-containing subcomplex of 90S preribosome; CC: small-subunit processome; CC: nucleolus; MF: snoRNA binding; | map03008 | Ribosome biogenesis in eukaryotes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Zhu, Q.; Wang, H.; Xiong, Q. Transcriptome-Based Analysis of the Co-Expression Network of Genes Related to Nitrogen Absorption in Rice Roots Under Nitrogen Fertilizer and Density. Agronomy 2025, 15, 1429. https://doi.org/10.3390/agronomy15061429
Wang R, Zhu Q, Wang H, Xiong Q. Transcriptome-Based Analysis of the Co-Expression Network of Genes Related to Nitrogen Absorption in Rice Roots Under Nitrogen Fertilizer and Density. Agronomy. 2025; 15(6):1429. https://doi.org/10.3390/agronomy15061429
Chicago/Turabian StyleWang, Runnan, Qi Zhu, Haiyuan Wang, and Qiangqiang Xiong. 2025. "Transcriptome-Based Analysis of the Co-Expression Network of Genes Related to Nitrogen Absorption in Rice Roots Under Nitrogen Fertilizer and Density" Agronomy 15, no. 6: 1429. https://doi.org/10.3390/agronomy15061429
APA StyleWang, R., Zhu, Q., Wang, H., & Xiong, Q. (2025). Transcriptome-Based Analysis of the Co-Expression Network of Genes Related to Nitrogen Absorption in Rice Roots Under Nitrogen Fertilizer and Density. Agronomy, 15(6), 1429. https://doi.org/10.3390/agronomy15061429