
Comparative Analysis of Codon Usage Patterns in the Chloroplast Genomes of Fagopyrum Species

 ,
,  ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Data
2.2. Codon Usage Deviation Index Analysis
2.3. Calculation of Relative Synonymous Codon Usage (RSCU) and Relative Synonymous Codon Usage Frequency (RFSC)
2.4. Identification of Optimal Codons and High-Frequency Codons
2.5. Neutral Plot Analysis and ENC Analysis
2.6. PR2 Plot Analysis and Correspondence Analysis (COA)
2.7. Gene Expression Level Analysis
2.8. Comparative Analysis of Codon Usage Frequency
2.9. Cluster Analysis and Phylogenetic Analysis
3. Results
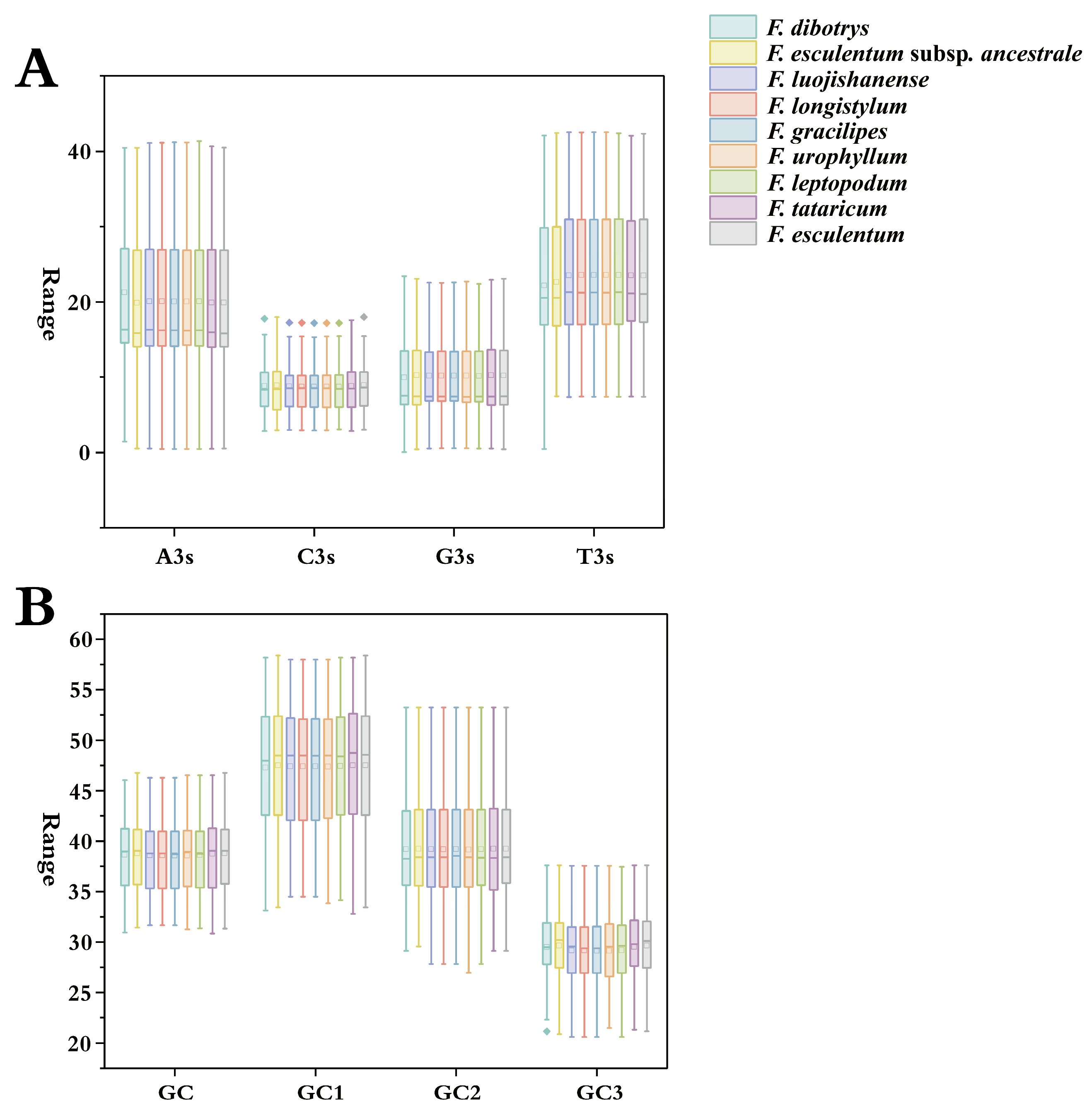
3.1. Characteristics of Codon Usage Bias
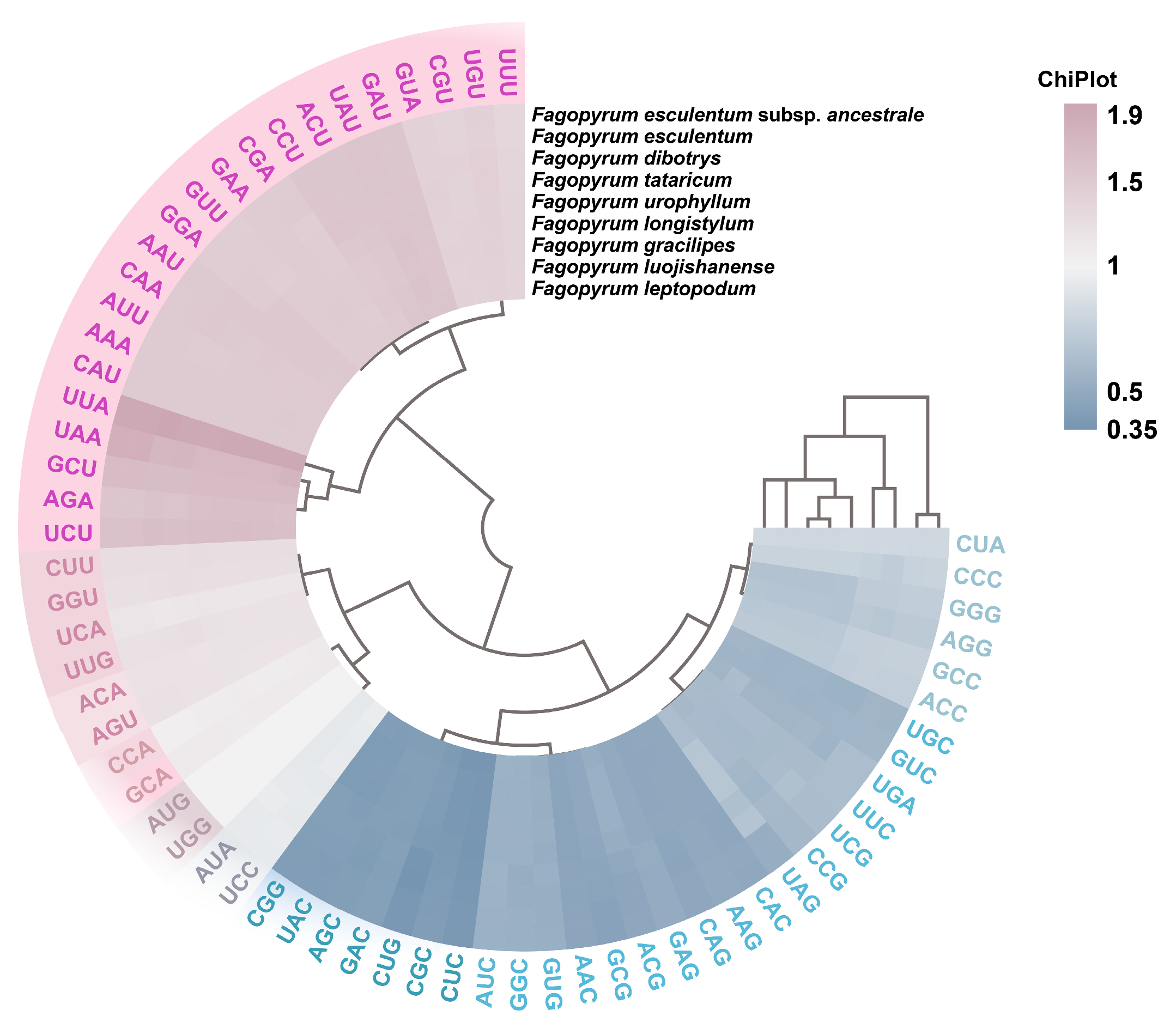
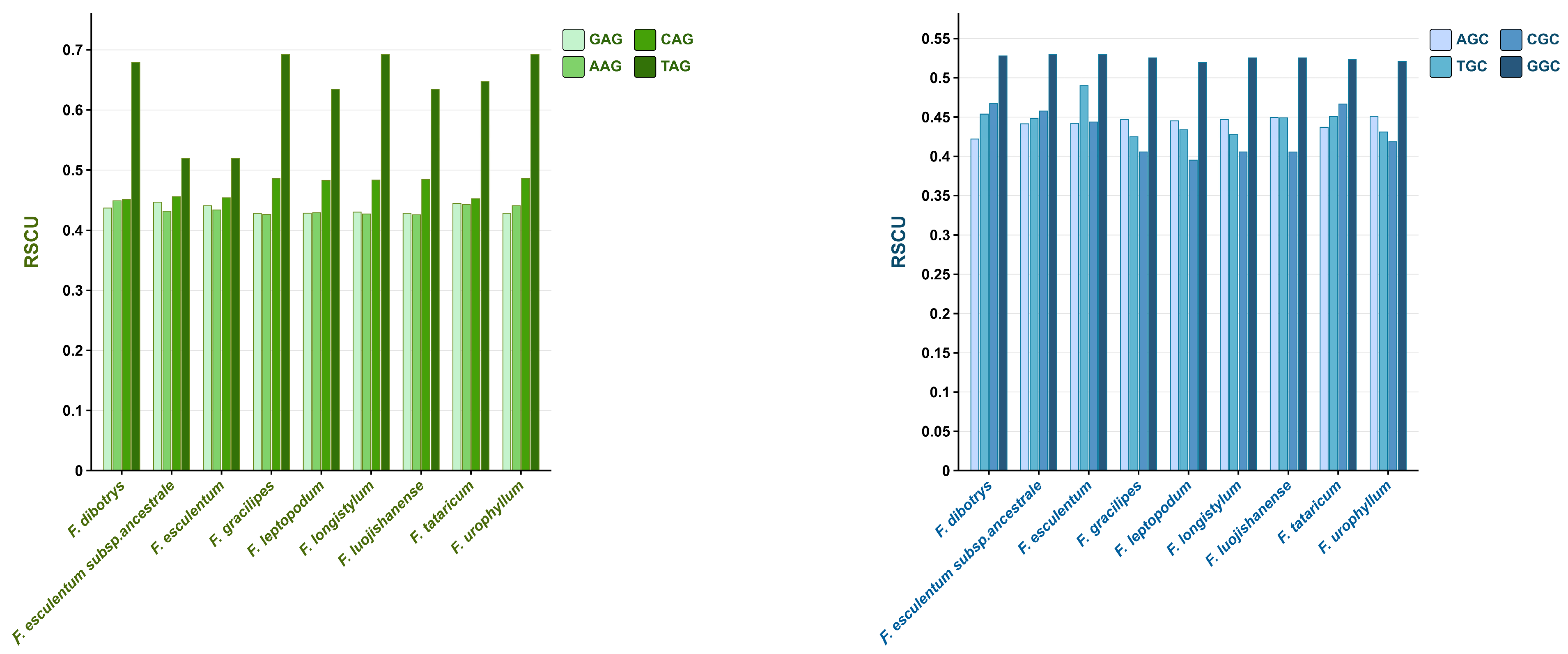
3.2. Preferred Codon Analysis
3.3. High-Frequency Codon Analysis
3.4. Neutrality Plot Analysis
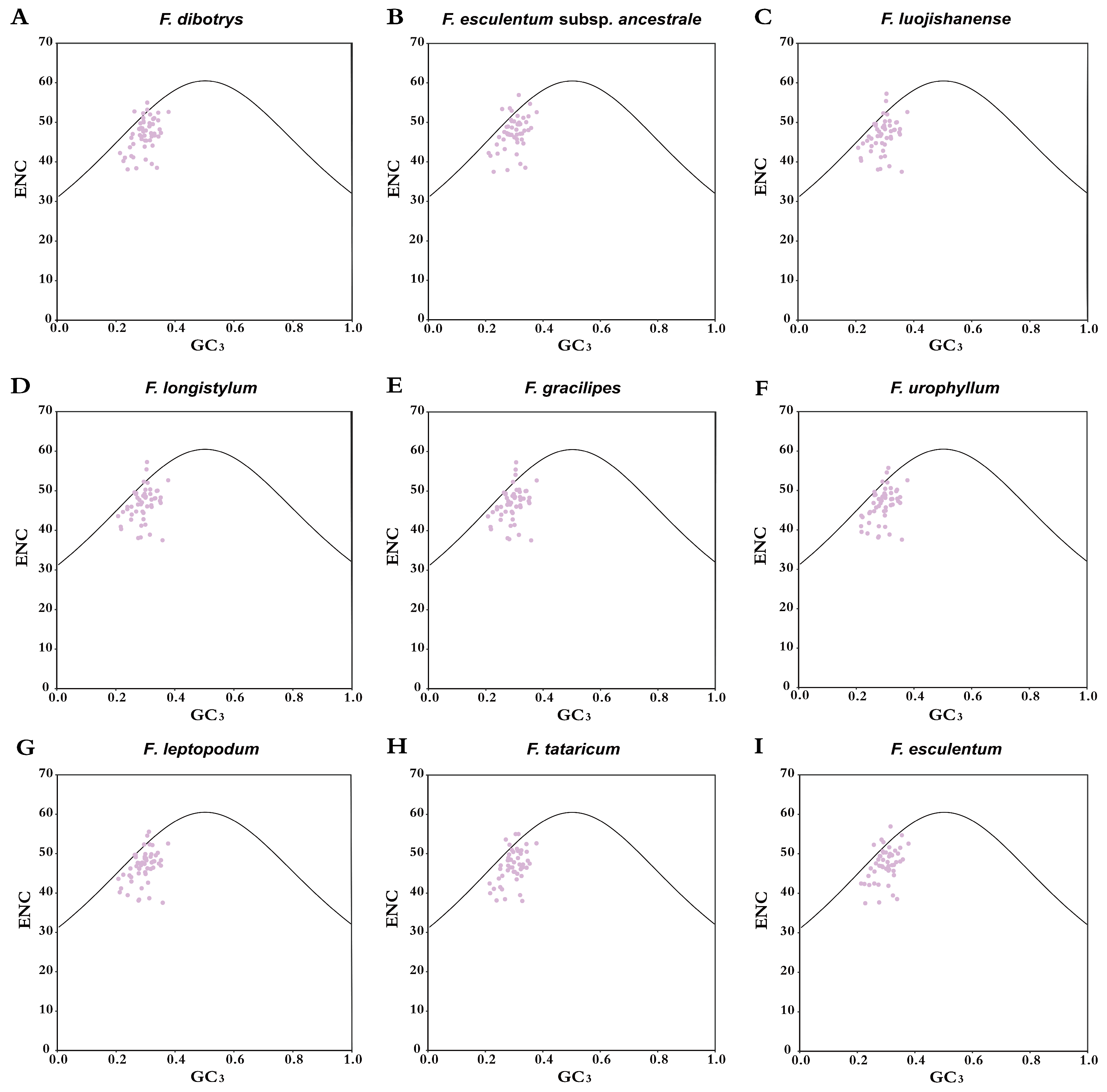
3.5. ENC Analysis
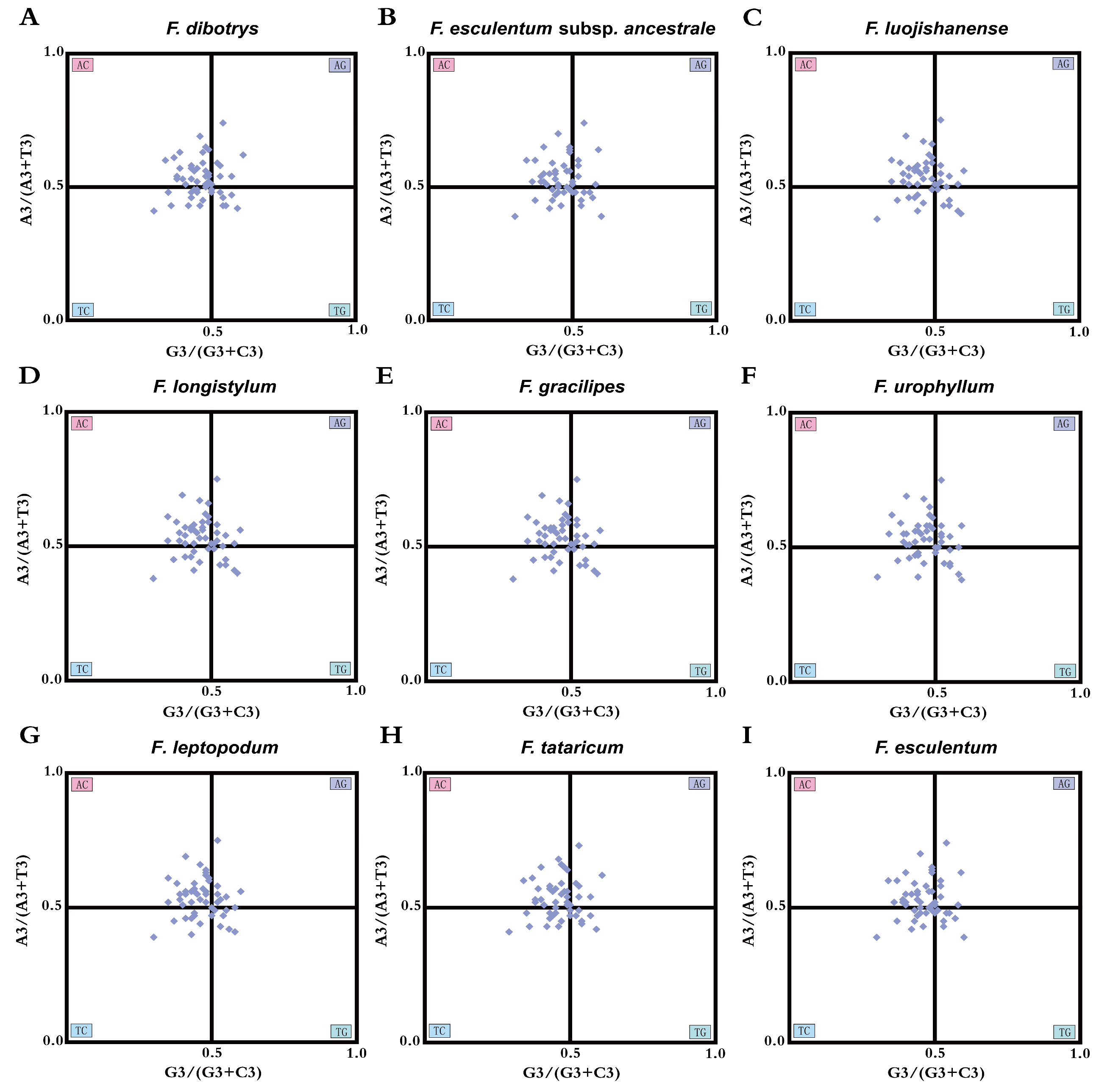
3.6. PR2 Plot Analysis
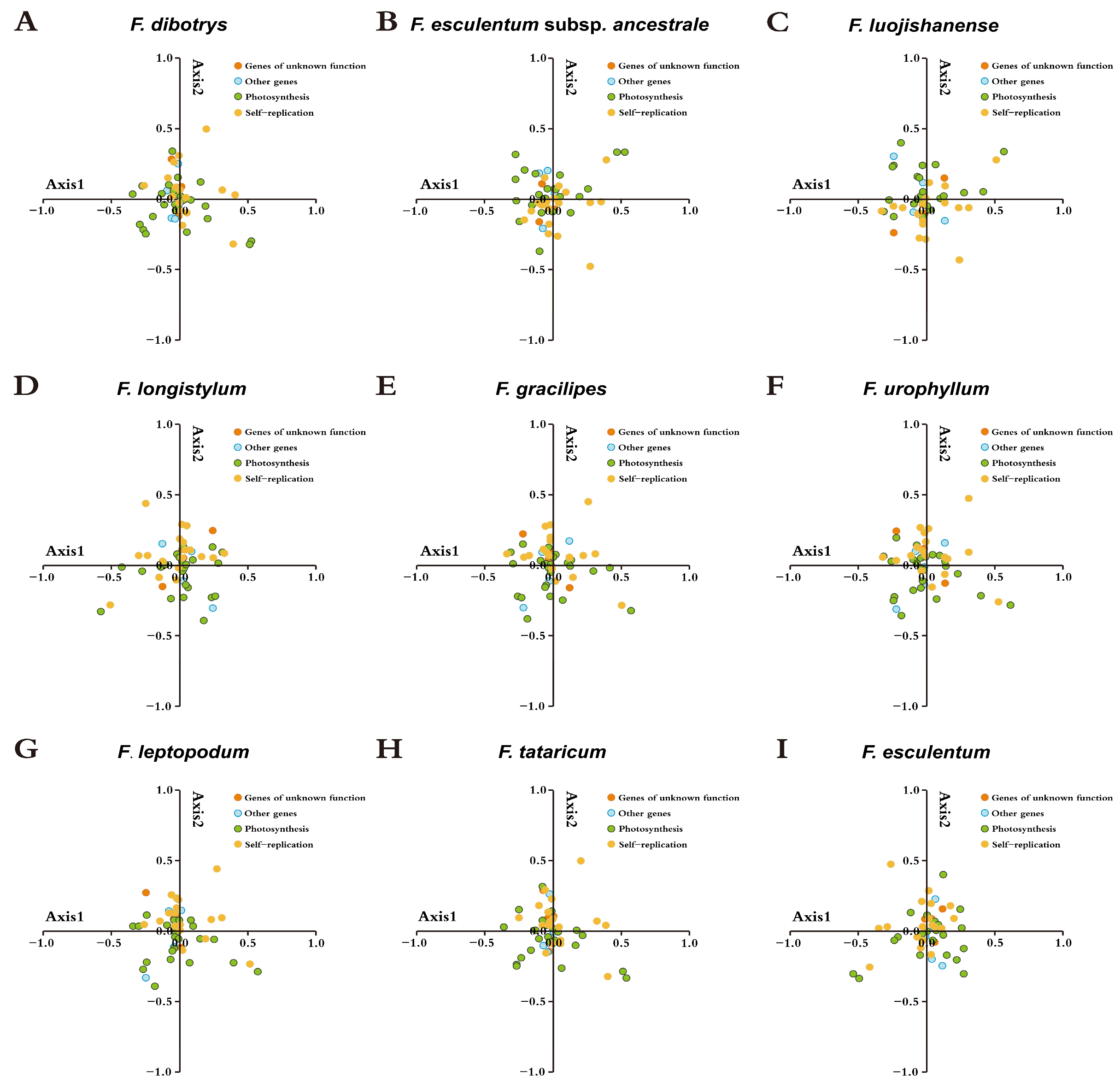
3.7. Correspondence Analysis (COA)
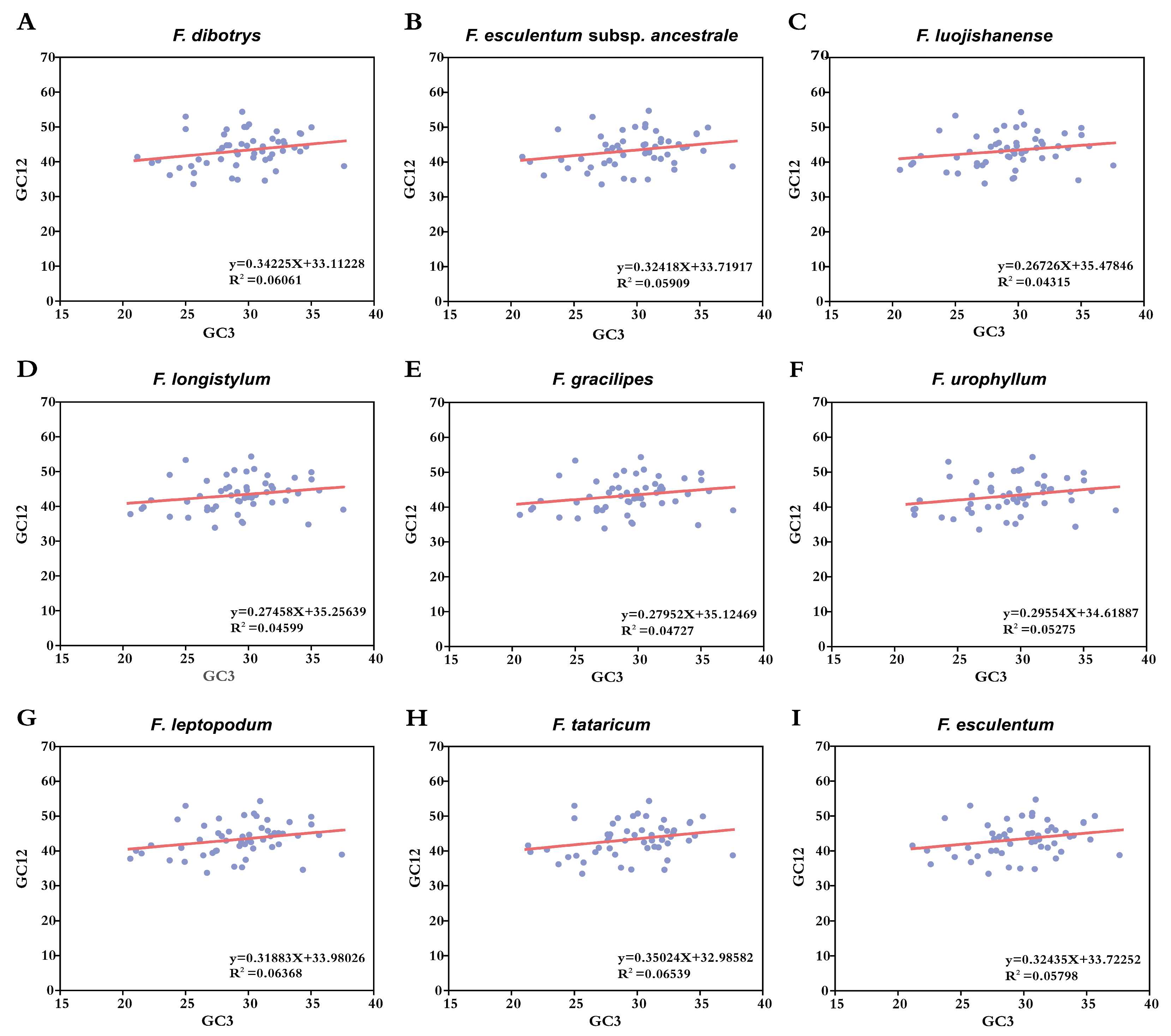
3.8. Correlation Between Codon Bias and Nucleotide Bias
3.9. Correlation Between Codon Bias and Protein Properties
3.10. Correlation Between Codon Bias and Conservative (MILC) Values
3.11. Codon Usage Frequency of Different Species
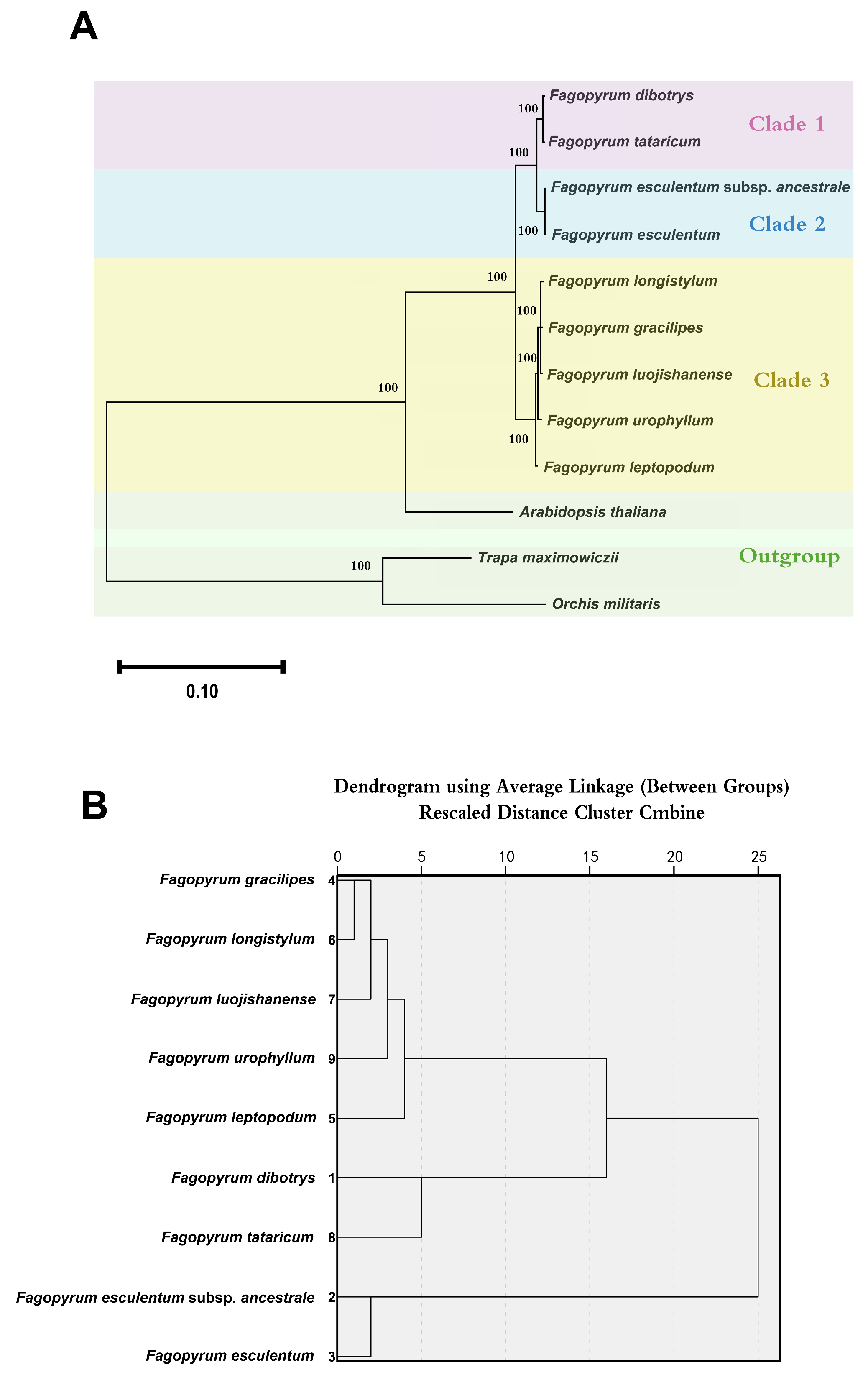
3.12. Phylogenetic Analysis and Cluster Analysis of Fagopyrum Species
4. Discussion
4.1. Factors Influencing Codon Usage Bias in Fagopyrum Chloroplast Genomes
4.2. Comparative Analysis of the Codon Usage Ratio Between Fagopyrum and Model Organisms
4.3. Phylogeny and Cluster Analysis of Fagopyrum
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chen, Y.; Hu, N.; Wu, H. Analyzing and Characterizing the Chloroplast Genome of Salix wilsonii. BioMed Res. Int. 2019, 2019, 5190425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, B.; Li, B.; Zhou, Q.; Wang, G.; Jiang, X.; Wang, C.; Xu, Z. Comparative analysis of codon usage patterns in chloroplast genomes of six Euphorbiaceae species. PeerJ 2020, 8, e8251. [Google Scholar] [CrossRef]
- Joey, S.; Edgar, B.L.; Edward, E.S.; Randall, L.S. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar]
- Wang, Z.; Cai, Q.; Wang, Y.; Li, M.; Wang, C.; Wang, Z.; Jiao, C.; Xu, C.; Wang, H.; Zhang, Z. Comparative Analysis of Codon Bias in the Chloroplast Genomes of Theaceae Species. Front. Genet. 2022, 13, 824610. [Google Scholar] [CrossRef]
- Tang, D.; Wei, F.; Cai, Z.; Wei, Y.; Khan, A.; Miao, J.; Wei, K. Analysis of codon usage bias and evolution in the chloroplast genome of Mesona chinensis Benth. Dev. Genes Evol. 2020, 231, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Zhang, Q.; Wang, C.; Li, F.; Tian, F.; Lu, Y.; Hu, Y.; Yang, H.; Cui, G. Analysis of codon usage patterns of the chloroplast genome in Delphinium grandiflorum L. reveals a preference for AT-ending codons as a result of major selection constraints. PeerJ 2021, 9, e10787. [Google Scholar] [CrossRef]
- Gun, L.; Liang, Z.; Pei, X. Codon usage pattern and genetic diversity in chloroplast genomes of Panicum species. Gene 2021, 802, 145866. [Google Scholar]
- Sheng, J.; She, X.; Liu, X.; Wang, J.; Hu, Z. Comparative analysis of codon usage patterns in chloroplast genomes of five Miscanthus species and related species. PeerJ 2021, 9, e12173. [Google Scholar] [CrossRef] [PubMed]
- Gerdol, M.; Moro, G.D.; Venier, P.; Pallavicini, A. Analysis of synonymous codon usage patterns in sixty-four different bivalve species. PeerJ 2015, 3, e1520. [Google Scholar] [CrossRef]
- Jyotika, S.; Supriyo, C.; Arif, U. Codon Usage Bias in Two Hemipteran Insect Species: Bemisia tabaci and Homalodisca coagulata. Adv. Biol. 2014, 2014, 1–7. [Google Scholar]
- Mazumdar, P.; Binti Othman, R.; Mebus, K.; Ramakrishnan, N.; Ann Harikrishna, J. Codon usage and codon pair patterns in non-grass monocot genomes. Ann. Bot. 2017, 120, 893–909. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.; Sun, Z.; Li, C.; Zhao, X.; Li, M.; Deng, R.; Huang, Y.; Zhao, H.; Chen, H.; Wu, Q. Overexpression of Fagopyrum tataricum FtbHLH2 enhances tolerance to cold stress in transgenic Arabidopsis. Plant Physiol. Biochem. 2018, 125, 85–94. [Google Scholar] [CrossRef]
- Song, Y.; Shen, M.; Cao, F.; Yang, X. Compare Analysis of Codon Usage Bias of Nuclear Genome in Eight Sapindaceae Species. Int. J. Mol. Sci. 2024, 26, 39. [Google Scholar] [CrossRef] [PubMed]
- Marta, H.; Michał, D.; Monika, K.M.; Jakub, P.; Anna, S.; Marek, S.; Agnieszka, P. Photosynthetic efficiency, growth and secondary metabolism of common buckwheat (Fagopyrum esculentum Moench) in different controlled-environment production systems. Sci. Rep. 2022, 12, 257. [Google Scholar]
- Deng, J.; Zhao, J.; Huang, J.; Damaris, R.N.; Li, H.; Shi, T.; Zhu, L.; Cai, F.; Zhang, X.; Chen, Q. Comparative proteomic analyses of Tartary buckwheat (Fagopyrum tataricum) seeds at three stages of development. Funct. Integr. Genom. 2022, 22, 1449–1458. [Google Scholar] [CrossRef]
- Jian, J.; Ikenna, C.O.; Chibuike, C.U. Buckwheat proteins: Functionality, safety, bioactivity, and prospects as alternative plant-based proteins in the food industry. Crit. Rev. Food Sci. Nutr. 2020, 62, 11–13. [Google Scholar] [CrossRef]
- Liu, F.; He, C.; Wang, L.; Wang, M. Effect of milling method on the chemical composition and antioxidant capacity of Tartary buckwheat flour. Int. J. Food Sci. Technol. 2018, 53, 2457–2464. [Google Scholar] [CrossRef]
- Jie, L.Q.; Yu, L.; Hu, W.A.; Fu, C.Q.; Mei, W.J.; Lu, P.; Yi, Y. Plastome comparison and phylogenomics of Fagopyrum (Polygonaceae): Insights into sequence differences between Fagopyrum and its related taxa. BMC Plant Biol. 2022, 22, 339. [Google Scholar]
- Li, J.; Feng, S.; Zhang, Y.; Xu, L.; Luo, Y.; Yuan, Y.; Yang, Q.; Feng, B. Genome-wide identification and expression analysis of the plant-specific PLATZ gene family in Tartary buckwheat (Fagopyrum tataricum). BMC Plant Biol. 2022, 22, 160. [Google Scholar] [CrossRef]
- Zhu, F. Chemical composition and health effects of Tartary buckwheat. Food Chem. 2016, 203, 231–245. [Google Scholar] [CrossRef]
- Kwang-Chul, K.; Hui-Ting, C.; Ileana, R.L.; Rosalind, W.-C.; Alice, B.; Henry, D. Codon Optimization to Enhance Expression Yields Insights into Chloroplast Translation. Plant Physiol. 2016, 172, 62–77. [Google Scholar]
- Wang, L.; Zheng, B.; Yuan, Y.; Xu, Q.; Chen, P. Transcriptome profiling of Fagopyrum tataricum leaves in response to lead stress. BMC Plant Biol. 2020, 20, 54. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Jia, Z.; Sun, J.; Wang, X.; Zhao, X.; Zhao, C.; Liang, F.; Song, X.; Guan, J.; Jia, X.; et al. Structural variants involved in high-altitude adaptation detected using single-molecule long-read sequencing. Nat. Commun. 2023, 14, 8282. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Yuan, H.; Cao, H.; Yazdani, M.; Tadmor, Y.; Li, L. Carotenoid Metabolism in Plants: The Role of Plastids. Mol. Plant 2018, 11, 58–74. [Google Scholar] [CrossRef]
- Song, Y.; Feng, L.; Alyafei, M.A.M.; Jaleel, A.; Ren, M. The Role of Chloroplast Gene Expression in Plant Responses to Environmental Stress. Int. J. Mol. Sci. 2020, 21, 6082. [Google Scholar]
- Geng, X.; Huang, N.; Zhu, Y.; Qin, L.; Hui, L. Codon usage bias analysis of the chloroplast genome of cassava. S. Afr. J. Bot. 2022, 151, 970–975. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Li, Y.; Ji, K.-K.; Zhu, J.; Ling, P.; Zhou, T.; Fan, L.-Y.; Xie, S.-Q. Genome-wide codon usage pattern analysis reveals the correlation between codon usage bias and gene expression in Cuscuta australis. Genomics 2020, 112, 2695–2702. [Google Scholar] [CrossRef]
- Som, A.; Sahoo, S.; Chakrabarti, J. Coding DNA sequences: Statistical distributions. Math. Biosci. 2003, 183, 49–61. [Google Scholar] [CrossRef]
- Anders, F. Estimating the “effective number of codons”: The Wright way of determining codon homozygosity leads to superior estimates. Genetics 2006, 172, 1301–1307. [Google Scholar]
- Wan, X.F.; Xu, D.; Kleinhofs, A.; Zhou, J. Quantitative relationship between synonymous codon usage bias and GC composition across unicellular genomes. BMC Evol. Biol. 2004, 4, 19. [Google Scholar] [CrossRef]
- Pere, P.; Ignacio, B.; Santiago, G.-V. E-CAI: A novel server to estimate an expected value of Codon Adaptation Index (eCAI). BMC Bioinform. 2008, 9, 65. [Google Scholar]
- Joshua, B.P.; Grzegorz, K. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Choudhury, M.N.; Uddin, A.; Chakraborty, S. Codon usage bias and its influencing factors for Y-linked genes in human. Comput. Biol. Chem. 2017, 69, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Tong, C.; Shi, J. Analysis of Codon Usage Between Different Poplar Species. J. Genet. Genom. 2007, 34, 555–561. [Google Scholar] [CrossRef]
- Liu, Q. Analysis of codon usage pattern in the radioresistant bacterium Deinococcus radiodurans. Bio Syst. 2006, 85, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Luo, Y.; Sha, A.; Xiao, W.; Xiong, Z.; Chen, X.; He, J.; Peng, L.; Zou, L. Analysis of synonymous codon usage patterns in mitochondrial genomes of nine Amanita species
. Front. Microbiol. 2023, 14, 1134228. [Google Scholar]
- Wang, Z.; Wang, G.; Cai, Q.; Jiang, Y.; Wang, C.; Xia, H.; Wu, Z.; Li, J.; Ou, Z.; Xu, Z.; et al. Genomewide comparative analysis of codon usage bias in three sequenced Jatropha curcas. J. Genet. 2021, 100, 20. [Google Scholar] [CrossRef]
- Daniel, U.; Bin, T.; Paul, G.H. The response of amino acid frequencies to directional mutation pressure in mitochondrial genome sequences is related to the physical properties of the amino acids and to the structure of the genetic code. J. Mol. Evol. 2006, 62, 340–361. [Google Scholar]
- Feng, X.; Liu, Z.; Mo, Y.; Zhang, S.; Ma, X.X. Role of nucleotide pair frequency and synonymous codon usage in the evolution of bovine viral diarrhea virus. Arch. Virol. 2025, 170, 64. [Google Scholar] [CrossRef]
- Banerjee, T.; Gupta, S.K.; Ghosh, T.C. Towards a resolution on the inherent methodological weakness of the “effective number of codons used by a gene”. Biochem. Biophys. Res. Commun. 2005, 330, 1015–1018. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, J.; Yang, W.; Li, Z.; Hai, Y.; Duan, B.; Zhang, H.; Yang, X.; Xia, C.; Conglong, X. Analysis of codon usage patterns in 48 Aconitum species. BMC Genom. 2023, 24, 703. [Google Scholar] [CrossRef]
- Tekaia, F. Genome Data Exploration Using Correspondence Analysis. Bioinform. Biol. Insights 2016, 2016, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, A.A.; Mohamed, S.; Radwa, M. vhcub: Virus-host codon usage co-adaptation analysis. F1000Research 2019, 8, 2137. [Google Scholar]
- Komi, N.; Manawa, A.; Selina, T.Y. Human genes with codon usage bias similar to that of the nonstructural protein 1 gene of influenza A viruses are conjointly involved in the infectious pathogenesis of influenza A viruses. Genetica 2022, 150, 97–115. [Google Scholar]
- Adam, H.; Joseph, S.; Catherine, P. CBDB: The codon bias database. BMC Bioinform. 2012, 13, 62. [Google Scholar]
- Wang, Z.; Li, J.; Liu, X.; Zhu, M.; Li, M.; Ye, Q.; Zhou, Z.; Yang, Y.; Yu, J.; Sun, W.; et al. Transcriptomic analysis of codon usage patterns and gene expression characteristics in leafy spurge. BMC Plant Biol. 2024, 24, 1118. [Google Scholar] [CrossRef]
- Tao, K.; Tang, L.; Luo, Y.; Li, L. Complete chloroplast genome of eight Phaius (Orchidaceae) species from China: Comparative analysis and phylogenetic relationship. BMC Plant Biol. 2025, 25, 37. [Google Scholar] [CrossRef]
- Seifert, E. OriginPro 9.1: Scientific data analysis and graphing software-software review. J. Chem. Inf. Model. 2014, 54, 1552. [Google Scholar] [CrossRef]
- Sudhir, K.; Glen, S.; Koichiro, T. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar]
- Sudhir, K.; Glen, S.; Michael, L.; Christina, K.; Koichiro, T. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar]
- Ivica, L.; Peer, B. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar]
- Torrent, M.; Chalancon, G.; Groot, N.S.d.; Wuster, A.; Babu, M.M. Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci. Signal. 2018, 11, eaat6409. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Dang, Y.; Zhou, M.; Li, L.; Yu, C.H.; Fu, J.; Chen, S.; Liu, Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA 2016, 113, E6117–E6125. [Google Scholar] [CrossRef]
- Dawei, W.; Baoling, Y. Analysis of codon usage bias of thioredoxin in apicomplexan protozoa. Parasites Vectors 2023, 16, 431. [Google Scholar]
- Gabriel, W.; Anabel, R.; Jun, L.; Tijana, M.; Scott, J.E.; Patricia, L.C. CHARMING: Harmonizing synonymous codon usage to replicate a desired codon usage pattern. Protein Sci. A Publ. Protein Soc. 2021, 31, 221–231. [Google Scholar]
- Zhang, Y.; Shen, Z.; Meng, X.; Zhang, L.; Liu, Z.; Liu, M.; Zhang, F.; Zhao, J. Codon usage patterns across seven Rosales species. BMC Plant Biol. 2022, 22, 65. [Google Scholar] [CrossRef]
- Yang, Q.; Xin, C.; Xiao, Q.S.; Lin, Y.T.; Li, L.; Zhao, J.L. Codon usage bias in chloroplast genes implicate adaptive evolution of four ginger species. Front. Plant Sci. 2023, 14, 1304264. [Google Scholar] [CrossRef]
- Näsvall, K.; Boman, J.; Talla, V.; Backström, N. Base composition, codon usage and patterns of gene sequence evolution in butterflies. Genome Biol. Evol. 2023, 15, evad150. [Google Scholar] [CrossRef]
- Sharp, P.M.; Emery, L.R.; Zeng, K. Forces that influence the evolution of codon bias. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2010, 365, 1203–1212. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Gong, W.; Li, Y. Comparative Analysis of the Codon Usage Pattern in the Chloroplast Genomes of Gnetales Species. Int. J. Mol. Sci. 2024, 25, 10622. [Google Scholar] [CrossRef] [PubMed]
- Parvin, A.B.; Arif, U.; Supriyo, C. Codon usage pattern and evolutionary forces of mitochondrial ND genes among orders of class Amphibia. J. Cell. Physiol. 2020, 236, 2850–2868. [Google Scholar]
- Parvin, A.B.; Arif, U.; Supriyo, C. Understanding the codon usage patterns of mitochondrial CO genes among Amphibians. Gene 2021, 777, 145462. [Google Scholar]
- Sau, K.; Gupta, S.K.; Sau, S.; Mandal, S.C.; Ghosh, T.C. Factors influencing synonymous codon and amino acid usage biases in Mimivirus. Bio Syst. 2006, 85, 107–113. [Google Scholar] [CrossRef]
- Liu, H.; Lu, Y.; Lan, B.; Xu, J. Codon usage by chloroplast gene is bias in Hemiptelea davidii. J. Genet. 2020, 99, 8. [Google Scholar] [CrossRef]
- Yang, C.; Zhao, Q.; Wang, Y.; Zhao, J.; Qiao, L.; Wu, B.; Yan, S.; Zheng, J.; Zheng, X. Comparative analysis of codon usage patterns of Plasmodium helical interspersed subtelomeric (PHIST) proteins. Front. Microbiol. 2023, 14, 1320060. [Google Scholar] [CrossRef]
- Chenkang, Y.; Qi, Z.; Ying, W.; Jiajia, Z.; Ling, Q.; Bangbang, W.; Suxian, Y.; Jun, Z.; Xingwei, Z. Comparative Analysis of Genomic and Transcriptome Sequences Reveals Divergent Patterns of Codon Bias in Wheat and Its Ancestor Species. Front. Genet. 2021, 12, 732432. [Google Scholar]
- L’Heureux, A.E.C.; Sterner, E.G.; Alcalá, X.X.M.; Katz, L.A. Lost in translation: Conserved amino acid usage despite extreme codon bias in foraminifera. mBio 2025, 16, e0391624. [Google Scholar]
- Ran, Z.; Li, Z.; Xiao, X.; An, M.; Yan, C. Complete chloroplast genomes of 13 species of sect. Tuberculata Chang (Camellia L.): Genomic features, comparative analysis, and phylogenetic relationships. BMC Genom. 2024, 25, 108. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Preferred Codons |
|---|---|
| F. dibotrys | GCU, CGA, CGU, UGU, GAA, GGU, AUU, CUA, UUA, AAA, UUU, CCA, CCU, AGU, UCU, ACC, ACU, GUA, GUU |
| F. esculentum subsp. ancestrale | GCU, CGU, UGU, GAA, GGU, CAU, AUU, CUA, CUU, UUG, AAA, CCA, UCU, ACC, ACU, GUA, GUU |
| F. esculentum | GCU, CGA, CGU, UGU, GAA, GGU, AUU, UUA, AAA, UUU, CCA, AGU, UCU, ACC, ACU, GUA, GUU |
| F. gracilipes | GCU, CGA, CGU, UGU, CAA, GAA, GGU, AUU, UUA, AAA, UUU, CCU, AGU, UCU, ACU, GUA, GUU |
| F. leptopodum | GCU, CGU, UGU, GAA, GGU, AUU, CUA, UUA, AAA, UUU, CCU, AGU, UCU, ACU, GUA, GUU |
| F. longistylum | GCU, CGA, CGU, UGU, CAA, GAA, GGU, AUU, UUA, AAA, UUU, CCU, AGU, UCU, ACU, GUA, GUU |
| F. urophyllum | GCU, CGU, UGU, GAA, GGU, AUU, UUA, AAA, UUU, CCU, AGU, UCU, ACU, GUA, GUU |
| F. luojishanense | GCU, CGA, CGU, UGU, CAA, GAA, GGU, AUU, UUA, AAA, UUU, CCU, AGU, UCU, ACU, GUA, GUU |
| F. tataricum | GCU, CGU, UGU, CAA, GAA, GGU, AUU, UUA, AAA, UUU, CCA, CCU, AGU, UCU, ACC, ACU, GUA, GUU |
| Species | Codon (RSCU) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| F. dibotrys | GCU (2.10) | CGU (1.76) | AAU (1.75) | UGU (1.70) | CAA (1.67) | GAA (1.60) | GGA (1.60) | AUU (1.56) | UUA (1.55) | AAA (1.55) |
| F. esculentum subsp. ancestrale | GCU (2.08) | AAU (1.85) | CGU (1.78) | UGU (1.70) | GAA (1.65) | GGA (1.59) | AAA (1.57) | CAA (1.57) | AUU (1.55) | UAA (1.55) |
| F. esculentum | GCU (2.07) | AAU (1.85) | CGU (1.78) | UGU (1.71) | GAA (1.66) | GGA (1.59) | AAA (1.57) | CAA (1.56) | AUU (1.56) | UAA (1.55) |
| F. gracilipes | GCU (2.09) | CGU (1.79) | AAU (1.73) | UGU (1.70) | CAA (1.68) | GAA (1.66) | GGA (1.60) | ACU (1.59) | UAA (1.58) | AAA (1.57) |
| F. leptopodum | GCU (2.07) | CGU (1.82) | AAU (1.79) | UGU (1.70) | GAA (1.66) | CAA (1.63) | GGA (1.59) | ACU (1.58) | AUU (1.57) | AAA (1.57) |
| F. longistylum | GCU (2.09) | CGU (1.79) | AAU (1.73) | UGU (1.70) | CAA (1.67) | GAA (1.67) | GGA (1.59) | ACU (1.59) | AAA (1.57) | UAA (1.57) |
| F. urophyllum | GCU (2.08) | CGU (1.79) | AAU (1.73) | UGU (1.70) | CAA (1.67) | GAA (1.67) | GGA (1.60) | ACU (1.59) | AAA (1.57) | AUU (1.57) |
| F. luojishanense | GCU (2.09) | CGU (1.77) | AAU (1.76) | UGU (1.69) | GAA (1.64) | CAA (1.60) | GGA (1.58) | UUA (1.58) | AAA (1.56) | AUU (1.56) |
| F. tataricum | GCU (2.07) | CGU (1.82) | AAU (1.73) | UGU (1.69) | CAA (1.67) | GAA (1.65) | GGA (1.58) | ACU (1.58) | AUU (1.57) | UAA (1.57) |
| Amino Acid | High-Frequency Codons |
|---|---|
| Leu | UUA |
| Val | GUU |
| Ser | UCU |
| Thr | ACU |
| Tyr | UAU |
| Gln | CAA |
| Lys | AAA |
| Ala | GCU |
| Glu | GAA |
| Arg | CGA, CGU |
| Gly | GGA |
| Phe | UUU |
| Ile | AUU |
| Pro | CCU |
| His | CAU |
| Asn | AAU |
| Asp | GAU |
| Cys | UGU |
| Species Name | R | p |
|---|---|---|
| F. dibotrys | 0.459 *** | 0.000 |
| F. esculentum | 0.466 *** | 0.000 |
| F. esculentum subsp. ancestrale | 0.465 *** | 0.000 |
| F. gracilipes | 0.385 ** | 0.005 |
| F. leptopodum | 0.357 ** | 0.009 |
| F. longistylum | 0.373 ** | 0.006 |
| F. luojishanense | 0.380 ** | 0.006 |
| F. tataricum | 0.415 ** | 0.002 |
| F. urophyllum | 0.422 ** | 0.002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Li, S.; He, D.; Liu, J.; He, X.; Lin, C.; Li, J.; Huang, Z.; Huang, L.; Nie, G.; et al. Comparative Analysis of Codon Usage Patterns in the Chloroplast Genomes of Fagopyrum Species. Agronomy 2025, 15, 1190. https://doi.org/10.3390/agronomy15051190
Liu Q, Li S, He D, Liu J, He X, Lin C, Li J, Huang Z, Huang L, Nie G, et al. Comparative Analysis of Codon Usage Patterns in the Chloroplast Genomes of Fagopyrum Species. Agronomy. 2025; 15(5):1190. https://doi.org/10.3390/agronomy15051190
Chicago/Turabian StyleLiu, Qilin, Shurui Li, Dinghong He, Jinyu Liu, Xiuzhi He, Chengruizhi Lin, Jinze Li, Zhixuan Huang, Linkai Huang, Gang Nie, and et al. 2025. "Comparative Analysis of Codon Usage Patterns in the Chloroplast Genomes of Fagopyrum Species" Agronomy 15, no. 5: 1190. https://doi.org/10.3390/agronomy15051190
APA StyleLiu, Q., Li, S., He, D., Liu, J., He, X., Lin, C., Li, J., Huang, Z., Huang, L., Nie, G., Zhang, X., & Feng, G. (2025). Comparative Analysis of Codon Usage Patterns in the Chloroplast Genomes of Fagopyrum Species. Agronomy, 15(5), 1190. https://doi.org/10.3390/agronomy15051190