
Transcriptomic Regulatory Mechanisms of Isoflavone Biosynthesis in Trifolium pratense

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experimental Design
2.2. Isoflavone Content Measurement and Sample Grouping
2.3. RNA Extraction and Transcriptome Sequencing (RNA-Seq)
2.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.5. Functional Enrichment Analysis of Differentially Expressed Genes (DEGs)
2.6. qRT-PCR Validation of Differentially Expressed Genes
3. Results
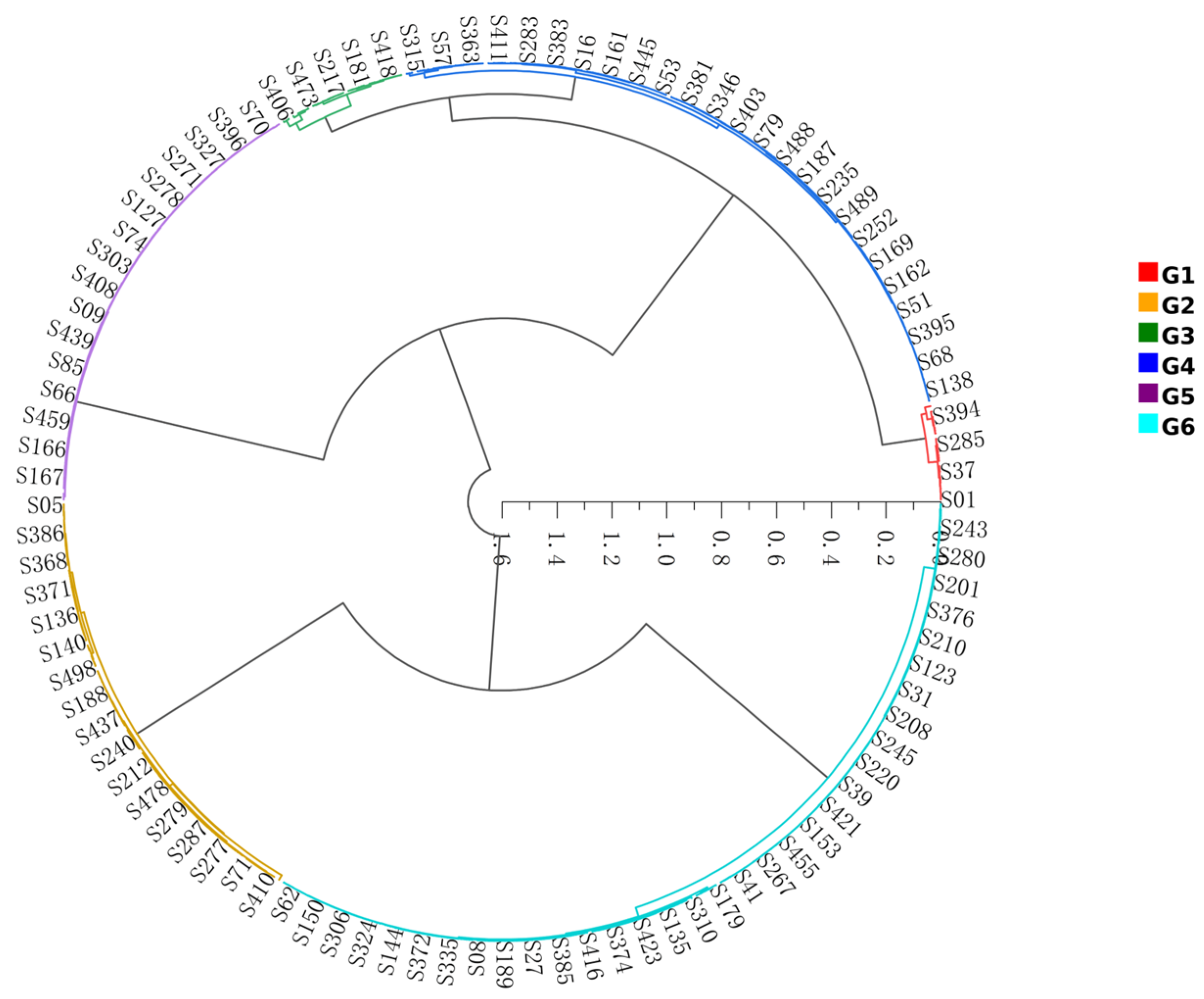
3.1. Clustering Analysis and Selection of Red Clover Isoflavone Accumulation Groups
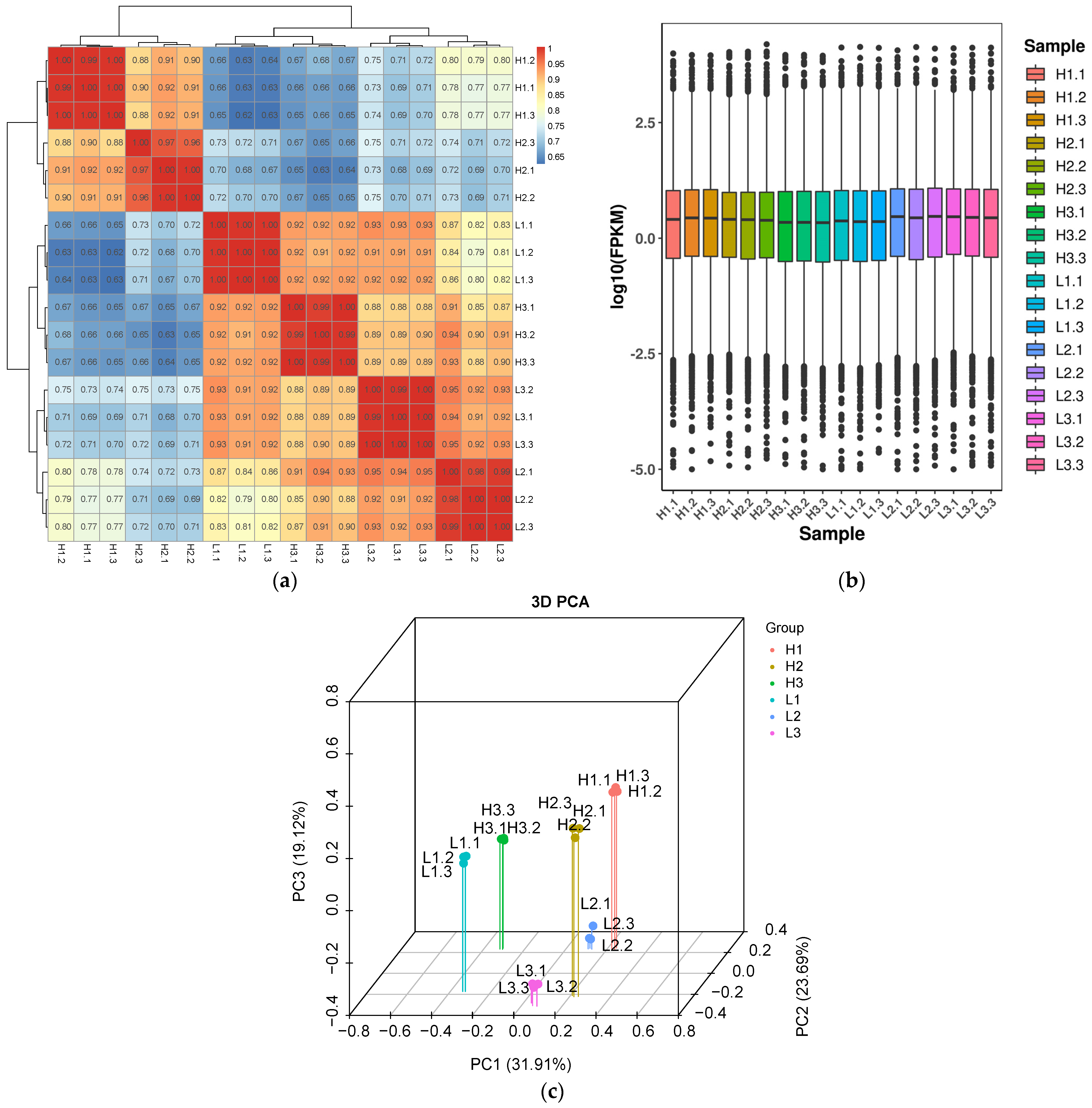
3.2. Quality Assessment of Transcriptome Sequencing Data
3.3. Correlation Analysis and Sample Clustering Evaluation of Transcriptome Data
3.4. Differentially Expressed Gene (DEG) Analysis
3.5. Analysis of Differentially Expressed Genes (DEGs)
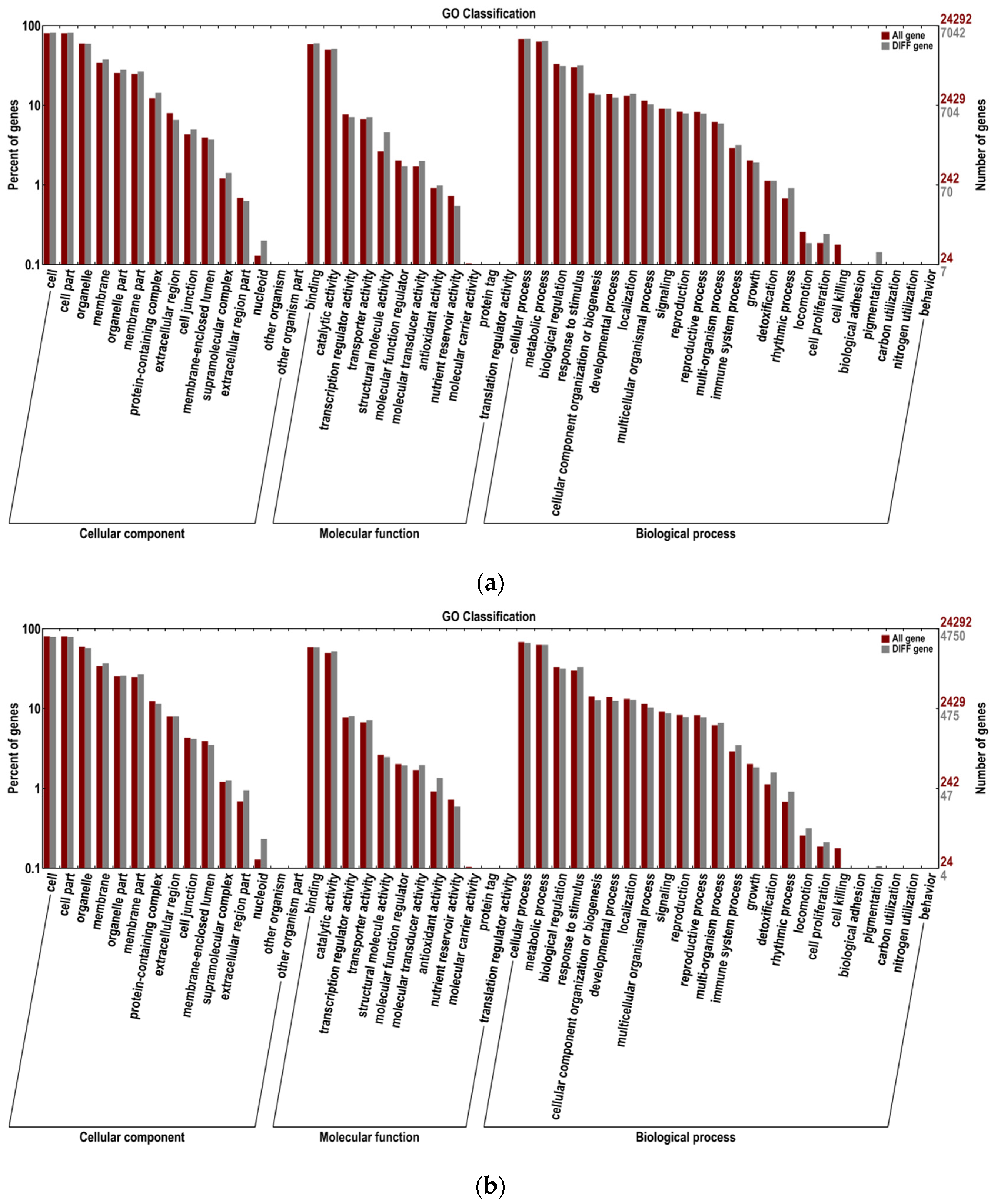
3.6. GO Functional Classification Analysis of Differentially Expressed Genes (DEGs)
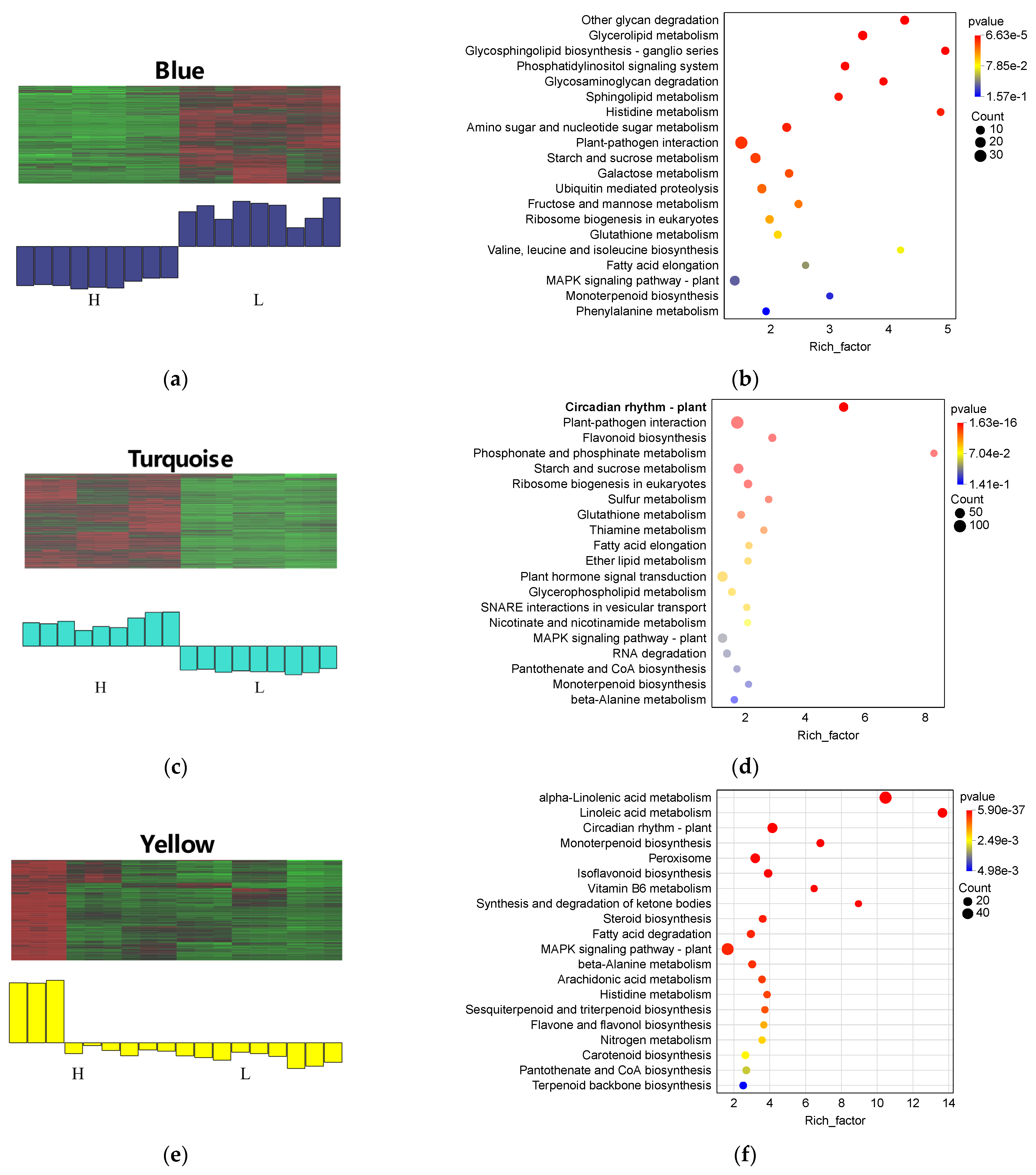
3.7. KEGG Pathway Enrichment Analysis of Differentially Expressed Genes (DEGs)
3.8. Construction and Analysis of Weighted Gene Co-Expression Network (WGCNA)
3.8.1. Construction of Gene Co-Expression Network and Its Correlation with Isoflavone Content
3.8.2. Expression Patterns of Key Modules in WGCNA
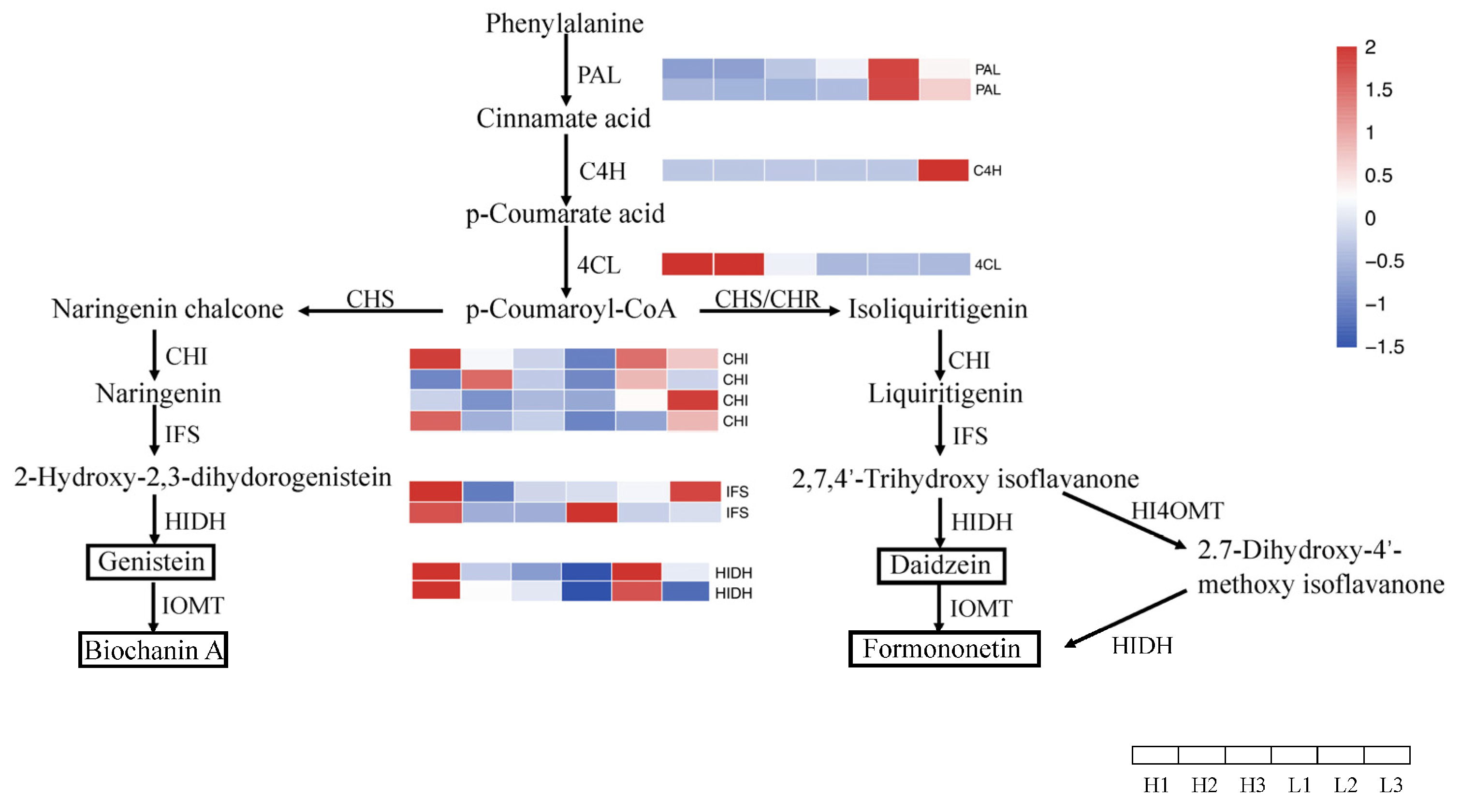
3.9. Expression Analysis of Key Genes in the Isoflavone Biosynthesis Pathway
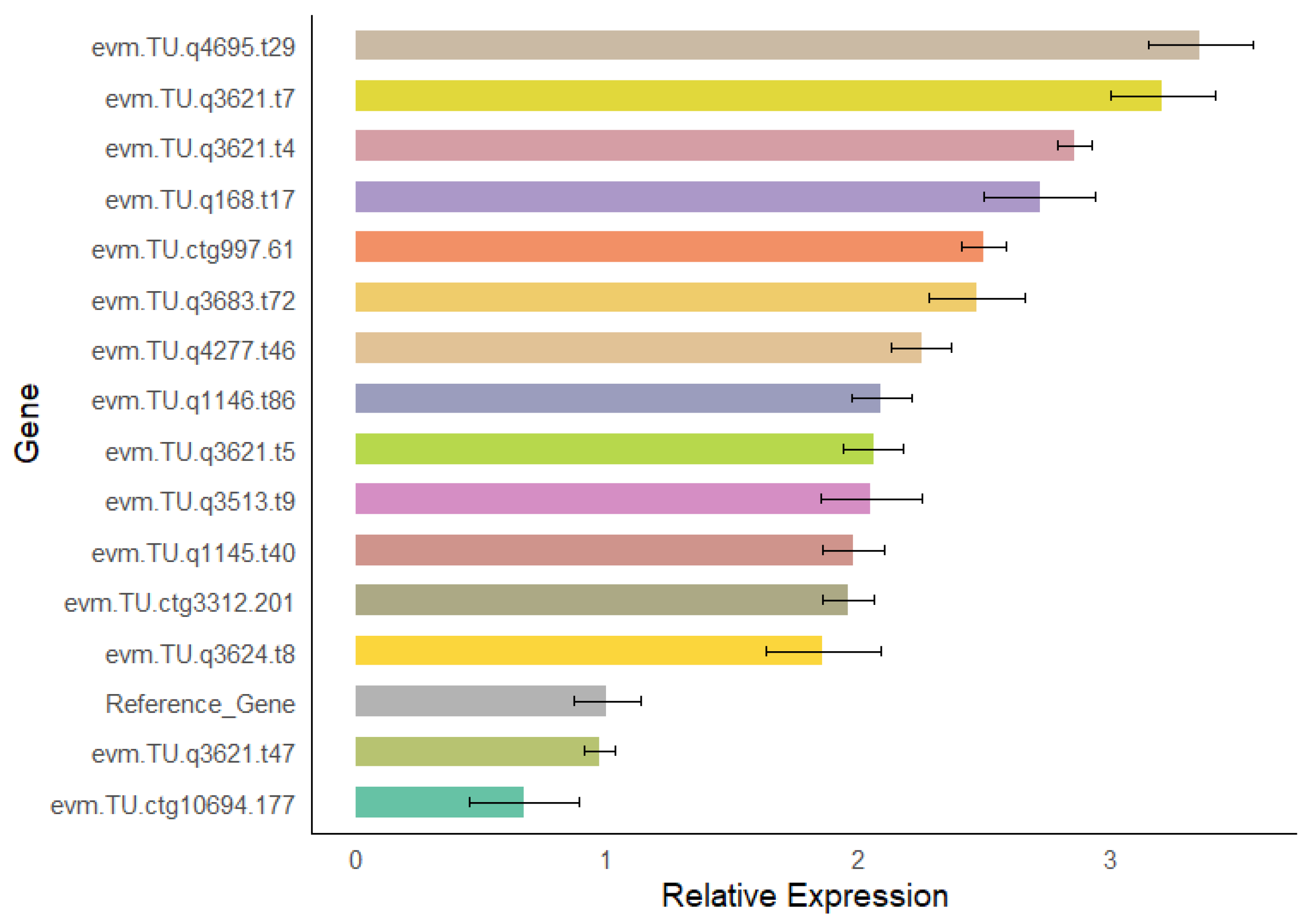
3.10. Validation of Differentially Expressed Genes in the Isoflavone Biosynthesis Pathway
4. Discussion
4.1. Genotypic Variation and Cluster Analysis of Isoflavone Accumulation in T. pratense
4.2. Quality Assessment of Transcriptome Sequencing Data and Sample Clustering Analysis
4.3. GO Functional Classification and KEGG Pathway Enrichment Analysis of Differentially Expressed Genes (DEGs)
4.4. WGCNA and Regulatory Mechanisms of Isoflavone Accumulation
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Huang, X.; Chu, G.; Wang, J.; Luo, H.; Yang, Z.; Sun, L.; Rong, W.; Wang, M. Integrated Metabolomic and Transcriptomic Analysis of Specialized Metabolites and Isoflavonoid Biosynthesis in Sophora alopecuroides L. under Different Degrees of Drought Stress. Ind. Crops Prod. 2023, 197, 116595. [Google Scholar] [CrossRef]
- Kim, J.M.; Lee, J.W.; Seo, J.S.; Ha, B.-K.; Kwon, S.-J. Differentially Expressed Genes Related to Isoflavone Biosynthesis in a Soybean Mutant Revealed by a Comparative Transcriptomic Analysis. Plants 2024, 13, 584. [Google Scholar] [CrossRef]
- Li, Z.; Geng, G.; Xie, H.; Zhou, L.; Wang, L.; Qiao, F. Metabolomic and Transcriptomic Reveal Flavonoid Biosynthesis and Regulation Mechanism in Phlomoides Rotata from Different Habitats. Genomics 2024, 116, 110850. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, S.; Li, J.; Muhammad, A.; Feng, Y.; Qi, J.; Sha, D.; Hao, Y.; Li, B.; Sun, J. An R2R3-type MYB Transcription Factor, GMMYB77, Negatively Regulates Isoflavone Accumulation in Soybean [Glycine max (L.) Merr.]. Plant Biotechnol. J. 2025, 23, 824–838. [Google Scholar] [CrossRef]
- Polturak, G.; Misra, R.C.; El-Demerdash, A.; Owen, C.; Steed, A.; McDonald, H.P.; Wang, J.; Saalbach, G.; Martins, C.; Chartrain, L.; et al. Discovery of Isoflavone Phytoalexins in Wheat Reveals an Alternative Route to Isoflavonoid Biosynthesis. Nat. Commun. 2023, 14, 6977. [Google Scholar] [CrossRef]
- Mandal, P.; Mortensen, D.A.; Brito, A.F.; Wallingford, A.K.; Lima, M.R.M.; Warren, N.D.; Smith, R.G. Water Stress Influences Phytoestrogen Levels in Red Clover (Trifolium pratense) but Not Kura Clover (T. ambiguum). J. Agric. Food Chem. 2024, 72, 10247–10256. [Google Scholar] [CrossRef]
- Meng, L.; Yang, Y.; Ma, Z.; Jiang, J.; Zhang, X.; Chen, Z.; Cui, G.; Yin, X. Integrated Physiological, Transcriptomic and Metabolomic Analysis of the Response of Trifolium pratense L. to Pb Toxicity. J. Hazard. Mater. 2022, 436, 129128. [Google Scholar] [CrossRef]
- Somasiri, S.C.; Kenyon, P.R.; Morel, P.C.H.; Morris, S.T.; Kemp, P.D. Selection by Lambs Grazing Plantain (Plantago lanceolata L.), Chicory (Cichorium intybus L.), White Clover (Trifolium repens L.), Red Clover (Trifolium pratense L.) and Perennial Ryegrass (Lolium perenne L.) across Seasons. Animals 2020, 10, 2292. [Google Scholar] [CrossRef]
- Wang, S.; Shao, T.; Li, J.; Zhao, J.; Dong, Z. Dynamics of Fermentation Profile, Bacterial Communities and Their Functional Characteristics in Red Clover. J. Agric. Sci. 2022, 160, 535–544. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Z.; Wu, Y.; Mu, M.; Jiang, J.; Nie, W.; Zhao, S.; Cui, G.; Yin, X. Genome-Wide Identification and Characterization of NAC Transcription Factor Family Members in Trifolium Pratense and Expression Analysis under Lead Stress. BMC Genomics 2024, 25, 128. [Google Scholar] [CrossRef]
- Vleugels, T.; Ruttink, T.; Ariza-Suarez, D.; Dubey, R.; Saleem, A.; Roldán-Ruiz, I.; Muylle, H. GWAS for Drought Resilience Traits in Red Clover (Trifolium pratense L.). Genes 2024, 15, 1347. [Google Scholar] [CrossRef]
- Vleugels, T.; Saleem, A.; Dubey, R.; Muylle, H.; Borra-Serrano, I.; Lootens, P.; De Swaef, T.; Roldán-Ruiz, I. Phenotypic Characterization of Drought Responses in Red Clover (Trifolium pratense L.). Front. Plant Sci. 2024, 14, 1304411. [Google Scholar] [CrossRef]
- Song, Z.; Zhao, F.; Chu, L.; Lin, H.; Xiao, Y.; Fang, Z.; Wang, X.; Dong, J.; Lyu, X.; Yu, D.; et al. The GmSTF1/2–GmBBX4 Negative Feedback Loop Acts Downstream of Blue-Light Photoreceptors to Regulate Isoflavonoid Biosynthesis in Soybean. Plant Commun. 2024, 5, 100730. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, J.; Zhang, Y.; Wang, Y.; Zhu, Y.; Liu, T.; Song, X.; He, Y.; Liu, Y.; Wang, Q.; et al. Overexpression of the R2R3-MYB Transcription Factor GmMYB3a Improves Isoflavone Accumulation in Soybean. Physiol. Plant. 2025, 177, e70120. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wu, X.; Zhu, Y.; Qu, Y.; Zhou, C.; Yuan, M.; Zhan, Y.; Li, Y.; Teng, W.; Zhao, X.; et al. Joint GWAS and WGCNA Identify Genes Regulating the Isoflavone Content in Soybean Seeds. J. Agric. Food Chem. 2024, 72, 18573–18584. [Google Scholar] [CrossRef]
- Zhan, Q.; Thakur, K.; Zhang, W.-W.; Feng, J.-Y.; Zhang, J.-G.; Khan, M.R.; Wei, Z.-J. Metabolic Changes and Isoflavone Biotransformation in Natto for Improved Nutritional Distribution and Bioavailability. Food Biosci. 2024, 59, 103937. [Google Scholar] [CrossRef]
- Dong, X.; Han, B.; Chen, J.; Luo, D.; Zhou, Q.; Liu, Z. Multiomics Analyses Reveal MsC3H29 Positively Regulates Flavonoid Biosynthesis to Improve Drought Resistance of Autotetraploid Cultivated Alfalfa (Medicago sativa L.). J. Agric. Food Chem. 2024, 72, 14448–14465. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.; Liu, X.; Wang, N.; An, Q.; Ye, X.M.; Zhao, Z.T.; Zhao, M.; Han, Y.; Ouyang, K.H.; et al. Investigation of Chemical Composition, Antioxidant Activity, and the Effects of Alfalfa Flavonoids on Growth Performance. Oxidative Med. Cell. Longev. 2020, 2020, 8569237. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Q.; Chen, Y.; Zhu, Y.; Zhou, X.; Li, B. Full-Length Transcriptome Sequencing Provides Insights into Flavonoid Biosynthesis in Camellia Nitidissima Petals. Gene 2023, 850, 146924. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, G. Isoflavonoid Metabolism in Leguminous Plants: An Update and Perspectives. Front. Plant Sci. 2024, 15, 1368870. [Google Scholar] [CrossRef]
- Naik, J.; Misra, P.; Trivedi, P.K.; Pandey, A. Molecular Components Associated with the Regulation of Flavonoid Biosynthesis. Plant Sci. 2022, 317, 111196. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Luo, Z.; Zhao, X.; Yang, M.; Liu, Z.; Liu, M. Transcriptome-Wide Expression Analysis of MYB Gene Family Leads to Functional Characterization of Flavonoid Biosynthesis in Fruit Coloration of Ziziphus Mill. Front. Plant Sci. 2023, 14, 1171288. [Google Scholar] [CrossRef]
- Xia, X.; Gong, R.; Zhang, C. Integrative Analysis of Transcriptome and Metabolome Reveals Flavonoid Biosynthesis Regulation in Rhododendron Pulchrum Petals. BMC Plant Biol. 2022, 22, 401. [Google Scholar] [CrossRef]
- Wang, J.; Su, H.; Wu, Z.; Wang, W.; Zhou, Y.; Li, M. Integrated Metabolites and Transcriptomics at Different Growth Stages Reveal Polysaccharide and Flavonoid Biosynthesis in Cynomorium Songaricum. IJMS 2022, 23, 10675. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, Q.; Rao, S.; Wang, A.; Zhang, H.; Wang, L.; Li, Y.; Chen, J. Metabolic Profiling and Gene Expression Analysis Provides Insights into Flavonoid and Anthocyanin Metabolism in Poplar. Tree Physiol. 2021, 41, 1046–1064. [Google Scholar] [CrossRef]
- Yao, D.; Xing, J.; Tang, Q.; Hou, Y.; Chen, B.; Yao, W.; Li, Z.; Wang, J.; Niu, Y.; Wang, D. Flavonoid Biosynthesis in Scutellaria Baicalensis Georgi: Metabolomics and Transcriptomics Analysis. Agronomy 2024, 14, 1494. [Google Scholar] [CrossRef]
- Yang, Z.; Wu, Y.; Dai, Z.; Chen, X.; Wangb, H.; Yang, S.; Xie, D.; Tang, Q.; Cheng, C.; Xu, Y.; et al. Comprehensive Transcriptome Analysis and Tissue-Specific Profiling of Gene Expression in Jute (Corchorus olitorius L.). Ind. Crops Prod. 2020, 146, 112101. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Zhang, J.; Xu, K.; Zheng, X.; Luo, J.; Lu, J. Integrative Physiology and Transcriptome Reveal Salt-Tolerance Differences between Two Licorice Species: Ion Transport, Casparian Strip Formation and Flavonoids Biosynthesis. BMC Plant Biol. 2024, 24, 272. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, Q.; Zhang, L.; Ren, F.; Zhang, Z.; Jia, Q. Integrated Metabolomics and Transcriptomics Analysis of Flavonoid Biosynthesis Pathway in Polygonatum Cyrtonema Hua. Molecules 2024, 29, 2248. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, X.; Wei, H.; Zhang, H.; Wu, Q.; Wang, L. Integrative Analysis of Transcriptome and Target Metabolites Uncovering Flavonoid Biosynthesis Regulation of Changing Petal Colors in Nymphaea ‘Feitian 2’. BMC Plant Biol. 2024, 24, 370. [Google Scholar] [CrossRef]
- Feng, Y.; Zhao, K.; Li, J.; Wang, M.; Yin, H.; Fan, Z.; Li, X.; Liu, W. Transcriptome and Pigment Analyses Provide Insights into Carotenoids and Flavonoids Biosynthesis in Camellia Nitidissima Stamens. Horticulturae 2024, 10, 420. [Google Scholar] [CrossRef]
- Li, Y.; Hei, J.; Wang, B.; Wang, S.; He, X. Unraveling the Molecular Mechanisms of Flavonoid Biosynthesis in Panax Notoginseng Flowers across Planting Patterns and Developmental Stages Using Integrated Metabolomics and Transcriptomics Analyses. Sci. Hortic. 2024, 335, 113362. [Google Scholar] [CrossRef]
- Song, S.; Tao, Y.; Gao, L.; Liang, H.; Tang, D.; Lin, J.; Wang, Y.; Gmitter, F.G.; Li, C. An Integrated Metabolome and Transcriptome Analysis Reveal the Regulation Mechanisms of Flavonoid Biosynthesis in a Purple Tea Plant Cultivar. Front. Plant Sci. 2022, 13, 880227. [Google Scholar] [CrossRef]
- Yuan, L.; Pan, K.; Li, Y.; Yi, B.; Gao, B. Comparative Transcriptome Analysis of Alpinia Oxyphylla Miq. Reveals Tissue-Specific Expression of Flavonoid Biosynthesis Genes. BMC Genom. Data 2021, 22, 19. [Google Scholar] [CrossRef]
- Tan, Z.; Lu, D.; Li, L.; Su, X.; Sun, Y.; Wang, L.; Yu, Y.; Wan, X.; Xu, L.; Li, C.; et al. Comprehensive Analysis of Safflower R2R3-MYBs Reveals the Regulation Mechanism of CtMYB76 on Flavonol Biosynthesis. Ind. Crops Prod. 2025, 227, 120795. [Google Scholar] [CrossRef]
- Du, Y.; Ma, H.; Liu, Y.; Gong, R.; Lan, Y.; Zhao, J.; Liu, G.; Lu, Y.; Wang, S.; Jia, H.; et al. Major Quality Regulation Network of Flavonoid Synthesis Governing the Bioactivity of Black Wolfberry. New Phytol. 2024, 242, 558–575. [Google Scholar] [CrossRef] [PubMed]
- Kicel, A.; Olszewska, M.A. Evaluation of Antioxidant Activity, and Quantitative Estimation of Flavonoids, Saponins and Phenols in Crude Extract and Dry Fractions of Medicago Lupulina Aerial Parts. Nat. Prod. Commun. 2015, 10, 1934578X1501000326. [Google Scholar] [CrossRef]
- Shan, R.; Zhang, Y.; You, X.; Kong, X.; Zhang, Y.; Li, X.; Wang, L.; Wang, X.; Chen, C. Revealing the Molecular Regulatory Mechanism of Flavonoid Accumulation in Tender Leaves of Tea Plants by Transcriptomic and Metabolomic Analyses. Plants 2025, 14, 625. [Google Scholar] [CrossRef]
- Huang, X.; He, Y.; Zhang, K.; Shi, Y.; Zhao, H.; Lai, D.; Lin, H.; Wang, X.; Yang, Z.; Xiao, Y.; et al. Evolution and Domestication of a Novel Biosynthetic Gene Cluster Contributing to the Flavonoid Metabolism and High-Altitude Adaptability of Plants in the Fagopyrum Genus. Adv. Sci. 2024, 11, 2403603. [Google Scholar] [CrossRef]
- Xie, N.; Guo, Q.; Li, H.; Yuan, G.; Gui, Q.; Xiao, Y.; Liao, M.; Yang, L. Integrated Transcriptomic and WGCNA Analyses Reveal Candidate Genes Regulating Mainly Flavonoid Biosynthesis in Litsea Coreana Var. Sinensis. BMC Plant Biol. 2024, 24, 231. [Google Scholar]
- Liu, Y.; Li, Y.; Liu, Z.; Wang, L.; Bi, Z.; Sun, C.; Yao, P.; Zhang, J.; Bai, J.; Zeng, Y. Integrated Transcriptomic and Metabolomic Analysis Revealed Altitude-Related Regulatory Mechanisms on Flavonoid Accumulation in Potato Tubers. Food Res. Int. 2023, 170, 112997. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Li, C.; Zhang, Y.; Wu, Z.; Li, W.; Zhang, Z.; Ye, W.; Guo, H.; Wang, C.; Long, T.; et al. Integrated Transcriptomic and Metabolomic Analyses Reveal the Molecular and Metabolic Basis of Flavonoids in Areca catechu L. J. Agric. Food Chem. 2023, 71, 4851–4862. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | Gene ID | Forward Primer | Reverse Primer |
|---|---|---|---|
| 1 | evm.TU.q168.t17 | TTTCAATTCACAAATTGGGT | TGGAGCATTCTCAGCTCTTT |
| 2 | evm.TU.q1145.t40 | ATGAGTTACTATAACCAACA | GCTGCTTGGCTGCTCTTT |
| 3 | evm.TU.q1146.t86 | TCCATCCACTTGTTTCCAC | GAGGTGAACTCTTCGCCAA |
| 4 | evm.TU.q3513.t9 | ATGAGTGTAGATCTGAAGAA | TGGTTTATTGTTCTGACGCA |
| 5 | evm.TU.q3621.t4 | ATGTCTTTGACAAATACAATT | GAGTCATTGATGACCATATGG |
| 6 | evm.TU.q3621.t5 | ATGTTTTGGCATATGATGCAG | CAGGAAGCGCTGCCGCCACC |
| 7 | evm.TU.q3621.t7 | ATGAGTTACTATAACCAACA | GCTGCTTGGCTGCTCTTT |
| 8 | evm.TU.q3621.t47 | ATGTCTTTGACAAATACAATT | GACGACTGAATTTTACTTTT |
| 9 | evm.TU.q3683.t72 | GTGCAAGTCAGCTTCAAAC | TTTGAGGCCAACAGACCAAA |
| 10 | evm.TU.q4277.t46 | ATGAATATGCATACCGGTCCA | AAGTGGTTGCCTAAAGGGAGTG |
| 11 | evm.TU.q3624.t8 | ATGTTTTGGCATATGATGCAG | GCAAGGATTCTATATGCAAC |
| 12 | evm.TU.q4695.t29 | ATGAAAATTCAGTGTGATGTG | TTAACTCGGTTAGTTCTTGAG |
| 13 | evm.TU.ctg997.61 | GCGATATTAATTAAACCAA | CATTGATGGTGGATCTGGA |
| 14 | evm.TU.ctg10694.177 | ATGGGCGATGGAGGTGTCGC | TGCTTCTGGATGGGGAAG |
| 15 | evm.TU.ctg3312.201 | GTTTTCGTGTCAAAATCTCAC | TGTTGGGAGCTATAGTTGACT |
| Population Number | Isoflavone Content | Average Isoflavone Content | Total Average Content |
|---|---|---|---|
| G1 | 29.99~36.33 mg·g−1 | 33.16 mg·g−1 | 25.21 mg·g−1 |
| G2 | 26.53~29.82 mg·g−1 | 28.18 mg·g−1 | |
| G3 | 25.14~26.38 mg·g−1 | 25.76 mg·g−1 | |
| G4 | 23.54~24.99 mg·g−1 | 24.27 mg·g−1 | |
| G5 | 15.65~18.77 mg·g−1 | 17.21 mg·g−1 | |
| G6 | 21.94~23.46 mg·g−1 | 22.70 mg·g−1 |
| Sample | ReadSum | BaseSum | GC (%) | Q20 (%) | CycleQ20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|
| H1.1 | 18,987,242 | 5,734,147,084 | 41.76 | 97.94 | 100 | 93.99 |
| H1.2 | 18,652,809 | 5,633,148,318 | 41.62 | 97.84 | 100 | 93.76 |
| H1.3 | 18,135,214 | 5,476,834,628 | 41.69 | 97.7 | 100 | 93.37 |
| H2.1 | 18,690,548 | 5,644,545,496 | 42.14 | 97.94 | 100 | 93.94 |
| H2.2 | 22,605,555 | 6,826,877,610 | 42.3 | 97.76 | 100 | 93.42 |
| H2.3 | 19,742,107 | 5,962,116,314 | 41.96 | 97.89 | 100 | 93.81 |
| H3.1 | 21,591,480 | 6,520,626,960 | 41.91 | 97.91 | 100 | 93.84 |
| H3.2 | 20,697,096 | 6,250,522,992 | 41.83 | 97.76 | 100 | 93.46 |
| H3.3 | 20,878,093 | 6,305,184,086 | 41.87 | 97.8 | 100 | 93.56 |
| L1.1 | 20,462,870 | 6,179,786,740 | 41.86 | 97.81 | 100 | 93.53 |
| L1.2 | 22,673,446 | 6,847,380,692 | 41.83 | 97.87 | 100 | 93.66 |
| L1.3 | 18,840,534 | 5,689,841,268 | 41.86 | 97.89 | 100 | 93.82 |
| L2.1 | 19,637,005 | 5,930,375,510 | 41.57 | 97.95 | 100 | 93.88 |
| L2.2 | 26,238,605 | 7,924,058,710 | 41.51 | 97.9 | 100 | 93.72 |
| L2.3 | 20,577,355 | 6,214,361,210 | 41.58 | 98.01 | 100 | 94.03 |
| L3.1 | 20,599,139 | 6,220,939,978 | 41.64 | 97.73 | 100 | 93.48 |
| L3.2 | 21,322,109 | 6,439,276,918 | 41.78 | 97.69 | 100 | 93.45 |
| L3.3 | 19,404,485 | 5,860,154,470 | 41.6 | 97.93 | 100 | 93.84 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, K.; Wang, S.; Zhang, H.; Ma, Y.; Wu, Q.; Huang, F.; Wang, M. Transcriptomic Regulatory Mechanisms of Isoflavone Biosynthesis in Trifolium pratense. Agronomy 2025, 15, 1061. https://doi.org/10.3390/agronomy15051061
Cao K, Wang S, Zhang H, Ma Y, Wu Q, Huang F, Wang M. Transcriptomic Regulatory Mechanisms of Isoflavone Biosynthesis in Trifolium pratense. Agronomy. 2025; 15(5):1061. https://doi.org/10.3390/agronomy15051061
Chicago/Turabian StyleCao, Kefan, Sijing Wang, Huimin Zhang, Yiming Ma, Qian Wu, Fan Huang, and Mingjiu Wang. 2025. "Transcriptomic Regulatory Mechanisms of Isoflavone Biosynthesis in Trifolium pratense" Agronomy 15, no. 5: 1061. https://doi.org/10.3390/agronomy15051061
APA StyleCao, K., Wang, S., Zhang, H., Ma, Y., Wu, Q., Huang, F., & Wang, M. (2025). Transcriptomic Regulatory Mechanisms of Isoflavone Biosynthesis in Trifolium pratense. Agronomy, 15(5), 1061. https://doi.org/10.3390/agronomy15051061