
Distinct Nitrogen Forms Shape Flavonoid Biosynthesis and Gene–Metabolite Networks in Erigeron breviscapus

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Agronomic Trait Measurements
2.3. Metabolite Extraction and Profiling
2.4. RNA Extraction, Sequencing, and Differential Expression Analysis
2.5. Statistical and Bioinformatics Analysis
2.6. Validation of Candidate Genes via qRT-PCR
3. Results
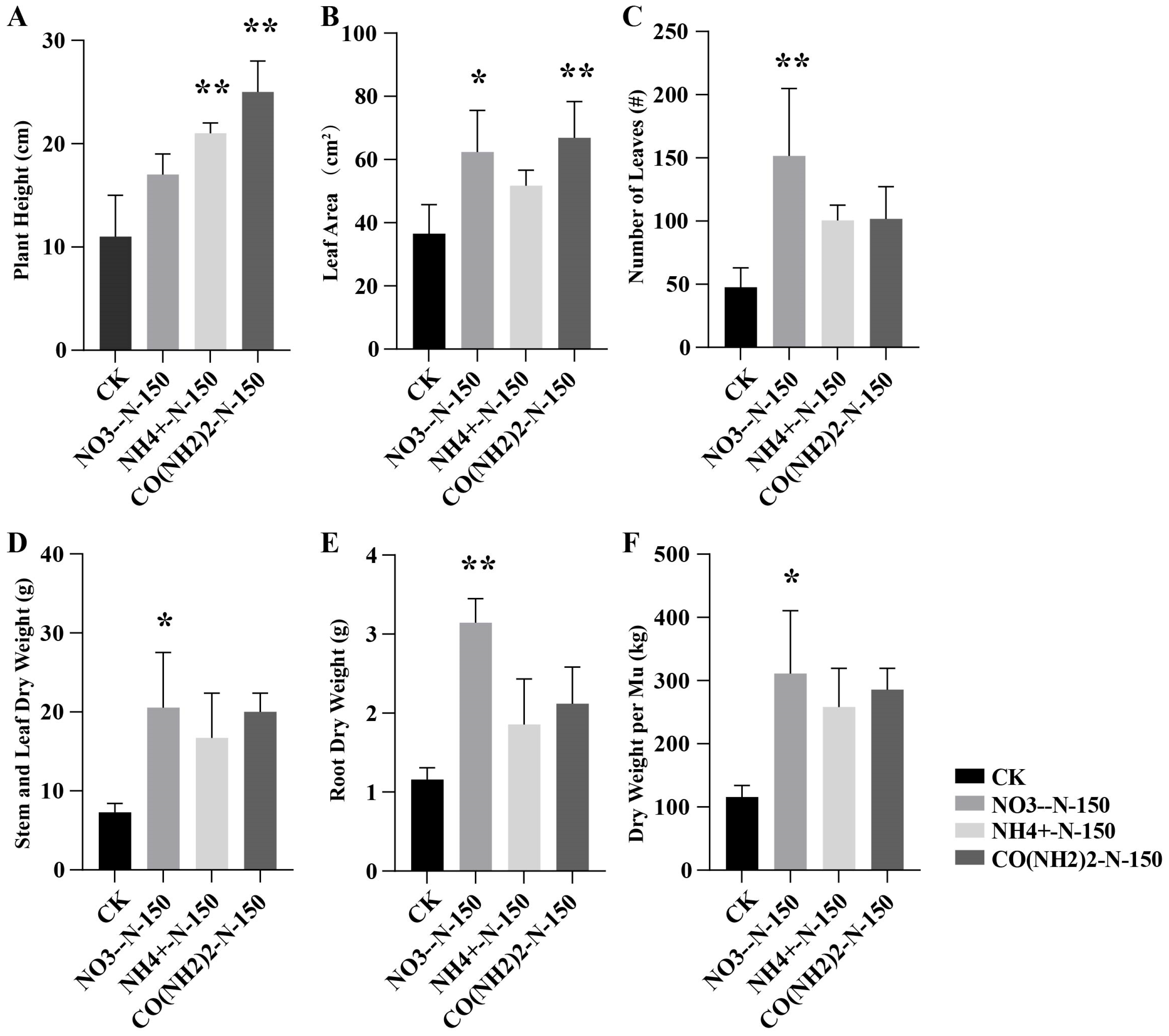
3.1. Agronomic Trait Analysis of E. breviscapus Stems and Leaves Under Four Different Nitrogen Treatments
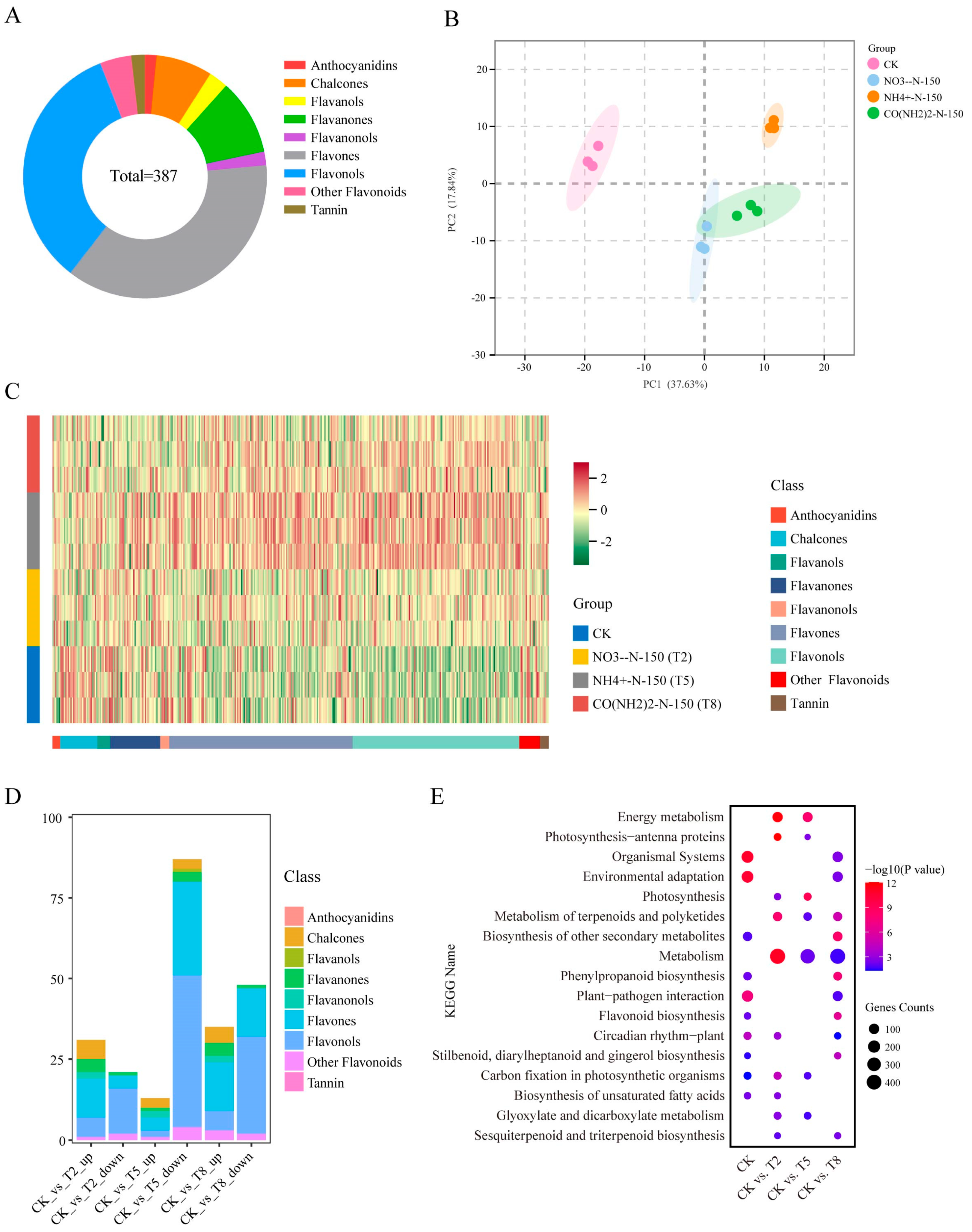
3.2. Metabolic Profiling of E. breviscapus Stems and Leaves Under Four Different Nitrogen Treatments
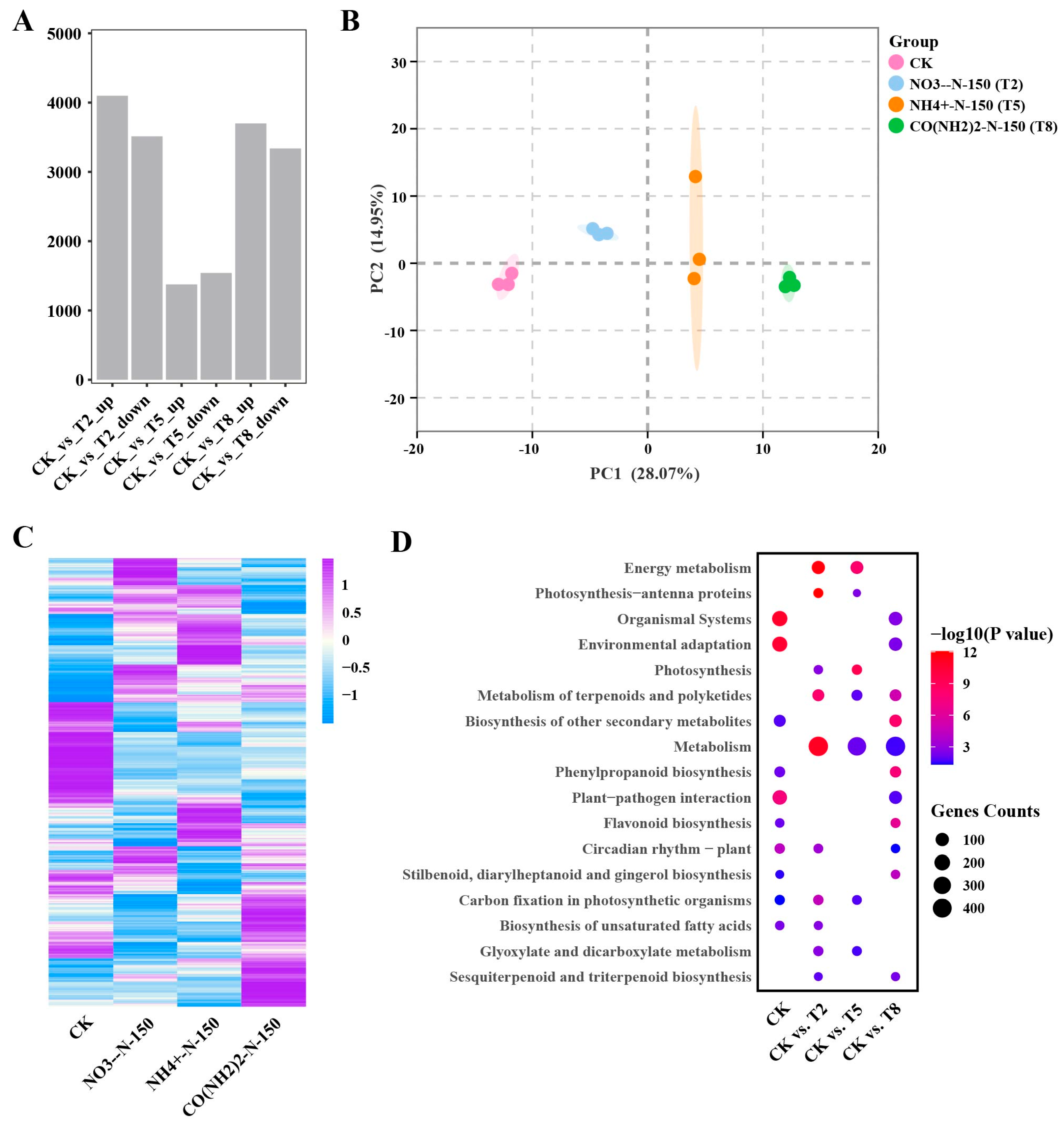
3.3. Transcriptomic Analysis of E. breviscapus Stems and Leaves Under Four Different Nitrogen Treatments
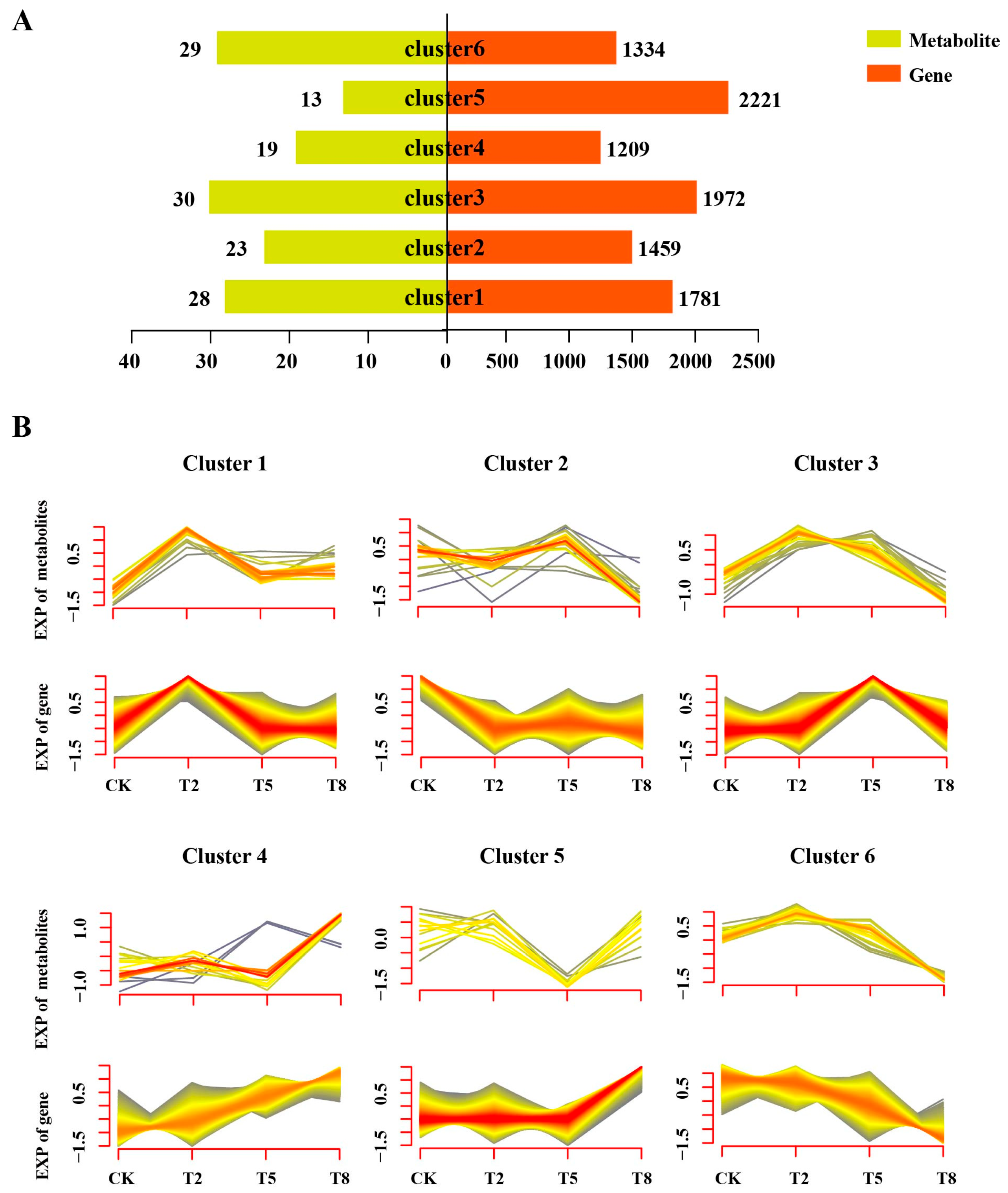
3.4. Integrated Transcriptomic and Metabolomic Analysis
3.5. Key Genes and Metabolites in Flavonoid Biosynthesis in E. breviscapus Under Nitrogen Treatment
4. Discussion
4.1. Impact of Nitrogen Treatments on Agronomic Traits and Flavonoid Biosynthesis
4.2. Metabolomic Insights into Flavonoid and Secondary Metabolism Under Nitrogen Treatments
4.3. Transcriptomic and Metabolomic Integration Reveals Key Regulatory Genes
4.4. Comparison with Previous Studies and Unique Aspects in E. breviscapus
4.5. Gene–Metabolite Networks and Future Directions
4.6. Mechanistic Insights into Nitrogen-Driven Growth Promotion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Peng, L.; Zhang, S.; Zhao, Q.; Yi, T. The relationships between chemical and genetic differentiation and environmental factors across the distribution of Erigeron breviscapus (Asteraceae). PLoS ONE 2013, 8, e74490. [Google Scholar]
- Song, K.; Wang, Y.; Yi, T.; Yang, Z. Karyological studies of Erigeron breviscapus and related species. Caryologia 2010, 63, 176–183. [Google Scholar]
- Shang, X.; He, X.; He, X.; Li, M.; Zhang, R.; Fan, P.; Zhang, Q.; Jia, Z. The genus Scutellaria an ethnopharmacological and phytochemical review. J. Ethnopharmacol. 2010, 128, 279–313. [Google Scholar] [PubMed]
- Fan, H.; Lin, P.; Kang, Q.; Zhao, Z.L.; Wang, J.; Cheng, J.Y. Metabolism and pharmacological mechanisms of active ingredients in Erigeron breviscapus. Curr. Drug Metab. 2021, 22, 24–39. [Google Scholar]
- Jo, H.G.; Baek, C.Y.; Lee, J.; Hwang, Y.; Baek, E.; Hwang, J.H.; Lee, D. Anti-inflammatory, analgesic, functional improvement, and chondroprotective effects of Erigeron breviscapus (Vant.) hand.-mazz. Extract in osteoarthritis: An in vivo and in vitro study. Nutrients 2024, 16, 1035. [Google Scholar] [CrossRef]
- Sharma, R.K.; Verma, N.; Jha, K.K.; Singh, N.K. Phytochemistry, pharmacological activity, traditional medicinal uses of erigeron species: A review. Int. J. Adv. Res. Inno. 2014, 2, 274–280. [Google Scholar]
- Pei, T.; Yan, M.; Huang, Y.; Wei, Y.; Martin, C.; Zhao, Q. Specific flavonoids and their biosynthetic pathway in Scutellaria baicalensis. Front. Plant Sci. 2022, 13, 866282. [Google Scholar]
- Pei, T.; Yan, M.; Li, T.; Li, X.; Yin, Y.; Cui, M.; Fang, Y.; Liu, J.; Kong, Y.; Xu, P.; et al. Characterization of UDP-glycosyltransferase family members reveals how major flavonoid glycoside accumulates in the roots of Scutellaria baicalensis. BMC Genom. 2022, 23, 169. [Google Scholar]
- Xu, H.; Park, N.I.; Li, X.; Kim, Y.K.; Lee, S.Y.; Park, S.U. Molecular cloning and characterization of phenylalanine ammonia-lyase, cinnamate 4-hydroxylase and genes involved in flavone biosynthesis in Scutellaria baicalensis. Bioresour Technol. 2010, 101, 9715–9722. [Google Scholar]
- Zhao, Q.; Cui, M.-Y.; Levsh, O.; Yang, D.; Liu, J.; Li, J.; Hill, L.; Yang, L.; Hu, Y.; Weng, J.K.; et al. Two CYP82D enzymes function as flavone hydroxylases in the biosynthesis of root-specific 4′-deoxyflavones in Scutellaria baicalensis. Mol. Plant 2018, 11, 135–148. [Google Scholar]
- Zhao, Q.; Yang, J.; Cui, M.Y.; Liu, J.; Fang, Y.; Yan, M.; Qiu, W.; Shang, H.; Xu, Z.; Yidiresi, R.; et al. The reference genome sequence of Scutellaria baicalensis provides insights into the evolution of wogonin biosynthesis. Mol. Plant 2019, 12, 935–950. [Google Scholar] [PubMed]
- Zhao, Q.; Zhang, Y.; Wang, G.; Hill, L.; Weng, J.K.; Chen, X.Y.; Xue, H.; Martin, C. A specialized flavone biosynthetic pathway has evolved in the medicinal plant, Scutellaria baicalensis. Sci. Adv. 2016, 2, e1501780. [Google Scholar] [PubMed]
- Shi, T.; Zhu, A.; Jia, J.; Hu, X.; Chen, J.; Liu, W.; Ren, X.; Sun, D.; Fernie, A.R.; Cui, F.; et al. Metabolomics analysis and metabolite-agronomic trait associations using kernels of wheat (Triticum aestivum) recombinant inbred lines. Plant J. 2020, 103, 279–292. [Google Scholar] [PubMed]
- He, S.; Dong, X.; Zhang, G.; Fan, W.; Duan, S.; Shi, H.; Li, D.; Li, R.; Chen, G.; Long, G.; et al. High quality genome of Erigeron breviscapus provides a reference for herbal plants in Asteraceae. Mol. Ecol. Resour. 2021, 1, 153–169. [Google Scholar]
- Yu, Y.; Ouyang, Z.; Guo, J.; Zeng, W.; Zhao, Y.; Huang, L. Complete chloroplast genome sequence of Erigeron breviscapus and characterization of chloroplast regulatory elements. Front. Plant Sci. 2021, 12, 758290. [Google Scholar]
- Gao, Q.; Li, X.; Xiang, C.; Li, R.; Xie, H.; Liu, J.; Li, X.; Zhang, G.; Yang, S.; Liang, Y.; et al. EbbHLH80 enhances salt responses by up-regulating flavonoid accumulation and modulating ROS levels. Int. J. Mol. Sci. 2023, 24, 11080. [Google Scholar] [CrossRef]
- Hao, D.; Luan, Y.; Wang, Y.; Xiao, P. Unveiling nitrogen fertilizer in medicinal plant cultivation. Agronomy 2024, 14, 1647. [Google Scholar] [CrossRef]
- Gao, Q.; Song, W.; Li, X.; Xiang, C.; Chen, G.; Xiang, G.; Liu, X.; Zhang, G.; Li, X.; Yang, S.; et al. Genome-wide identification of bHLH transcription factors: Discovery of a candidate regulator related to flavonoid biosynthesis in Erigeron breviscapus. Front. Plant Sci. 2022, 13, 977649. [Google Scholar]
- Song, W.; Zhang, S.; Li, Q.; Xiang, G.; Zhao, Y.; Wei, F.; Zhang, G.; Yang, S.; Hao, B. Genome-wide profiling of WRKY genes involved in flavonoid biosynthesis in Erigeron breviscapus. Front. Plant Sci. 2024, 15, 1412574. [Google Scholar]
- Yang, Y.; Liu, M.; Zhang, W.; Cao, Y.; Li, C.; Wang, W. Identification and characterization of two new 1-O-acyl-glucose-ester forming glucosyltransferases from Erigeron breviscapus. J. Agric. Food Chem. 2019, 67, 2848–2855. [Google Scholar]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [PubMed]
- Liu, J.; Osbourn, A.; Ma, P. MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol. Plant 2015, 8, 68–708. [Google Scholar]
- Jin, H.; Martin, C. Multifunctionality and diversity within the plant MYB-gene family. Plant Mol. Biol. 1999, 41, 577–585. [Google Scholar] [PubMed]
- Martin, C.; Paz-Ares, J. MYB transcription factors in plants. Trends Genet. 1997, 13, 67–73. [Google Scholar] [PubMed]
- Fang, Y.; Liu, J.; Zheng, M.; Zhu, S.; Pei, T.; Cui, M.; Chang, L.; Xiao, H.; Yang, J.; Martin, C.; et al. SbMYB3 transcription factor promotes root-specific flavone biosynthesis in Scutellaria baicalensis. Horti. Res. 2023, 10, 66. [Google Scholar]
- Chen, R.B.; Liu, J.H.; Xiao, Y.; Zhang, F.; Chen, J.; Ji, Q.; Tan, H.X.; Huang, X.; Feng, H.; Huang, B.K.; et al. Deep sequencing reveals the effect of MeJA on scutellarin biosynthesis in Erigeron breviscapus. PLoS ONE 2015, 10, e0143881. [Google Scholar]
- Shi, P.; Liu, M.; Yu, X.; Gielis, J.; Ratkowsky, D.A. Proportional relationship between leaf area and the product of leaf length and width of four types of special leaf shapes. Forests 2019, 10, 178. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Regev, A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hotelling, H. Analysis of a complex of statistical variables into principal components. J. Educ. Psychol. 1933, 24, 417–441. [Google Scholar] [CrossRef]
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. R Package 2015, 1, 790. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef]
- Student. The probable error of a mean. Biometrika 1908, 6, 1–25. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Bork, P. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Wickham, H. A layered grammar of graphics. J. Comput. Graph. Stat. 2010, 19, 3–28. [Google Scholar] [CrossRef]
- Wickham, H. dplyr: A Grammar of Data Manipulation (Version 1.0.2) [Computer Software]. 2020. Available online: https://cran.r-project.org/web/packages/dplyr/dplyr.pdf (accessed on 11 March 2025).
- Li, Q.F.; Sun, S.S.M.; Yuan, D.Y.; Yu, H.X.; Gu, M.H.; Liu, Q.Q. Validation of candidate reference genes for the accurate normalization of real-time quantitative RT-PCR data in rice during seed development. Plant Mol. Biol. Rep. 2010, 28, 49–57. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-△△CT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Li, S.; Yan, L.; Zhang, W.; Yi, C.; Haider, S.; Wang, C.; Liu, Y.; Shi, L.; Xu, F.; Ding, G. Nitrate alleviates ammonium toxicity in Brassica napus by coordinating rhizosphere and cell pH and ammonium assimilation. Plant J. 2024, 117, 786–804. [Google Scholar]
- Zhu, G.; Wang, S.; Huang, Z.; Zhang, S.; Liao, Q.; Zhang, C.; Lin, T.; Qin, M.; Peng, M.; Yang, C.; et al. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agronomic Trait | Plant Height (cm) | Leaf Area (cm2) | Number of Leaves | Stem and Leaf Dry Weight (g) | Root Dry Weight (g) | Dry Weight per Mu (kg) |
|---|---|---|---|---|---|---|
| CK | 11.67 ± 3.06 | 36.87 ± 2.62 | 48.33 ± 3.06 | 7.27 ± 0.37 | 1.16 ± 0.15 | 115.71 ± 3.77 |
| T2 | 18.00 ± 1.00 | 62.67 ± 2.35 | 152.67 ± 3.21 | 20.554 ± 2.46 | 3.15 ± 0.30 | 311.50 + 5.12 |
| T5 | 21 ± 1 | 51.53 ± 2.41 | 101.67 ± 4.04 | 16.71 ± 1.24 | 1.85 ± 0.19 | 258.19 ± 19.85 |
| T8 | 26 ± 1.73 | 66.77 ± 5.11 | 101.33 ± 5.69 | 20.03 ± 1.36 | 2.12 ± 0.23 | 285.07 ± 3.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Li, L.; Wang, X.; Yang, B.; Ma, W.; Jin, H.; Li, Y. Distinct Nitrogen Forms Shape Flavonoid Biosynthesis and Gene–Metabolite Networks in Erigeron breviscapus. Agronomy 2025, 15, 755. https://doi.org/10.3390/agronomy15030755
Yang Y, Li L, Wang X, Yang B, Ma W, Jin H, Li Y. Distinct Nitrogen Forms Shape Flavonoid Biosynthesis and Gene–Metabolite Networks in Erigeron breviscapus. Agronomy. 2025; 15(3):755. https://doi.org/10.3390/agronomy15030755
Chicago/Turabian StyleYang, Yan, Linyu Li, Xing Wang, Bin Yang, Weisi Ma, Hang Jin, and Yongmei Li. 2025. "Distinct Nitrogen Forms Shape Flavonoid Biosynthesis and Gene–Metabolite Networks in Erigeron breviscapus" Agronomy 15, no. 3: 755. https://doi.org/10.3390/agronomy15030755
APA StyleYang, Y., Li, L., Wang, X., Yang, B., Ma, W., Jin, H., & Li, Y. (2025). Distinct Nitrogen Forms Shape Flavonoid Biosynthesis and Gene–Metabolite Networks in Erigeron breviscapus. Agronomy, 15(3), 755. https://doi.org/10.3390/agronomy15030755