

Exploring Microbial Rhizosphere Communities in Asymptomatic and Symptomatic Apple Trees Using Amplicon Sequencing and Shotgun Metagenomics

 ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Description and Sample Collection
2.2. Total DNA Extraction and Sequencing
2.3. Amplicon Data Analysis
2.4. Metagenomic Data Analysis
3. Results
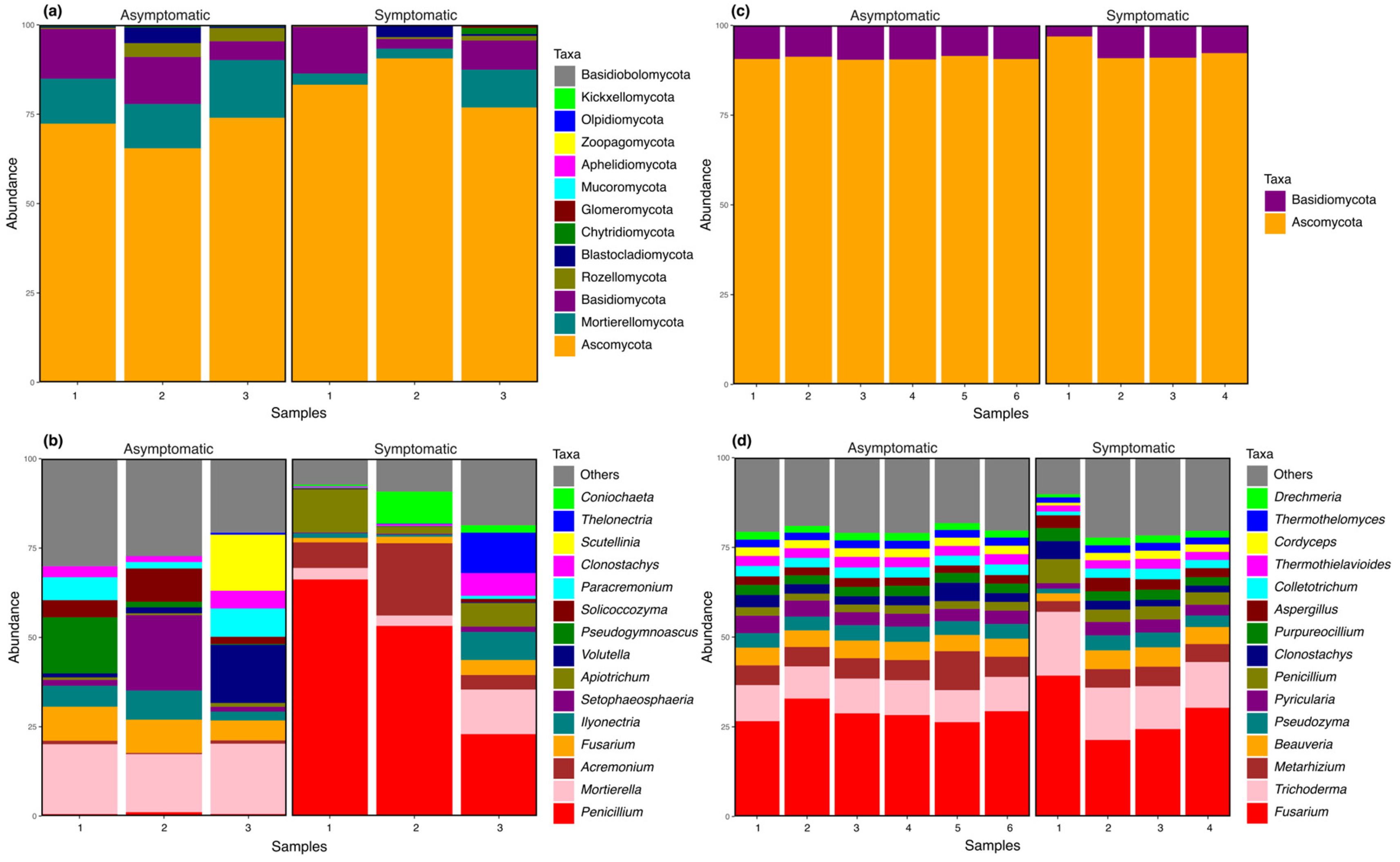
3.1. Composition and Abundance of Bacterial and Fungal Communities
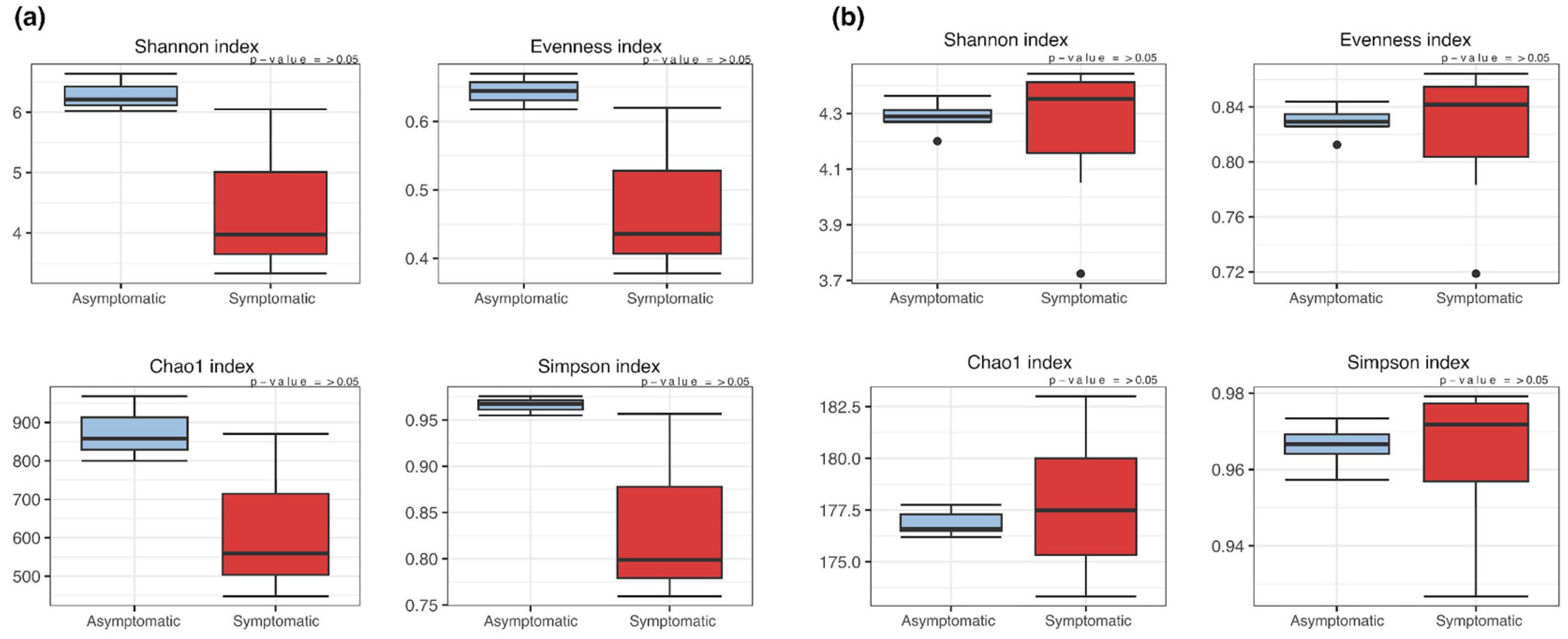
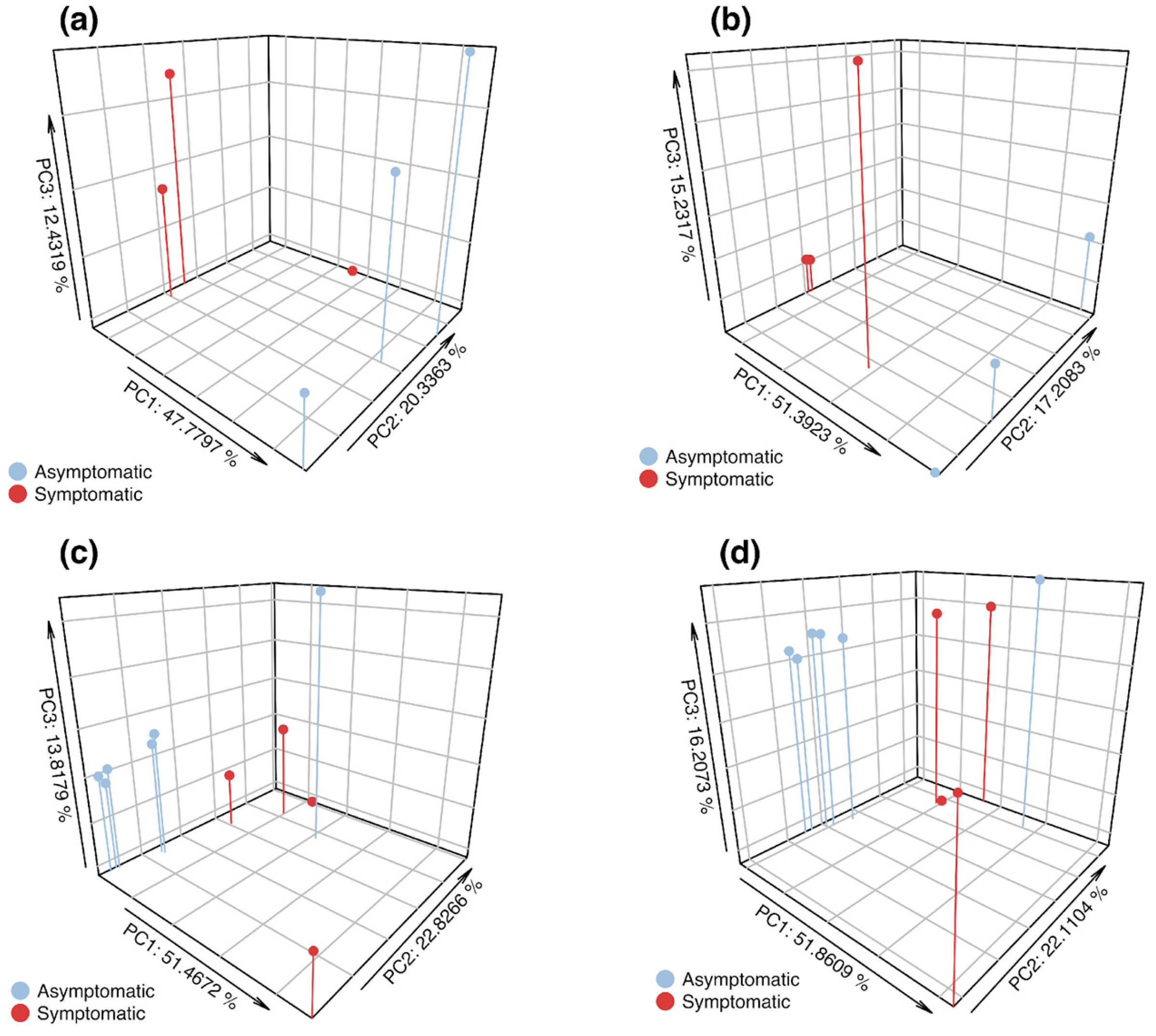
3.2. α- and β-Diversity of Asymptomatic and Symptomatic Apple Trees
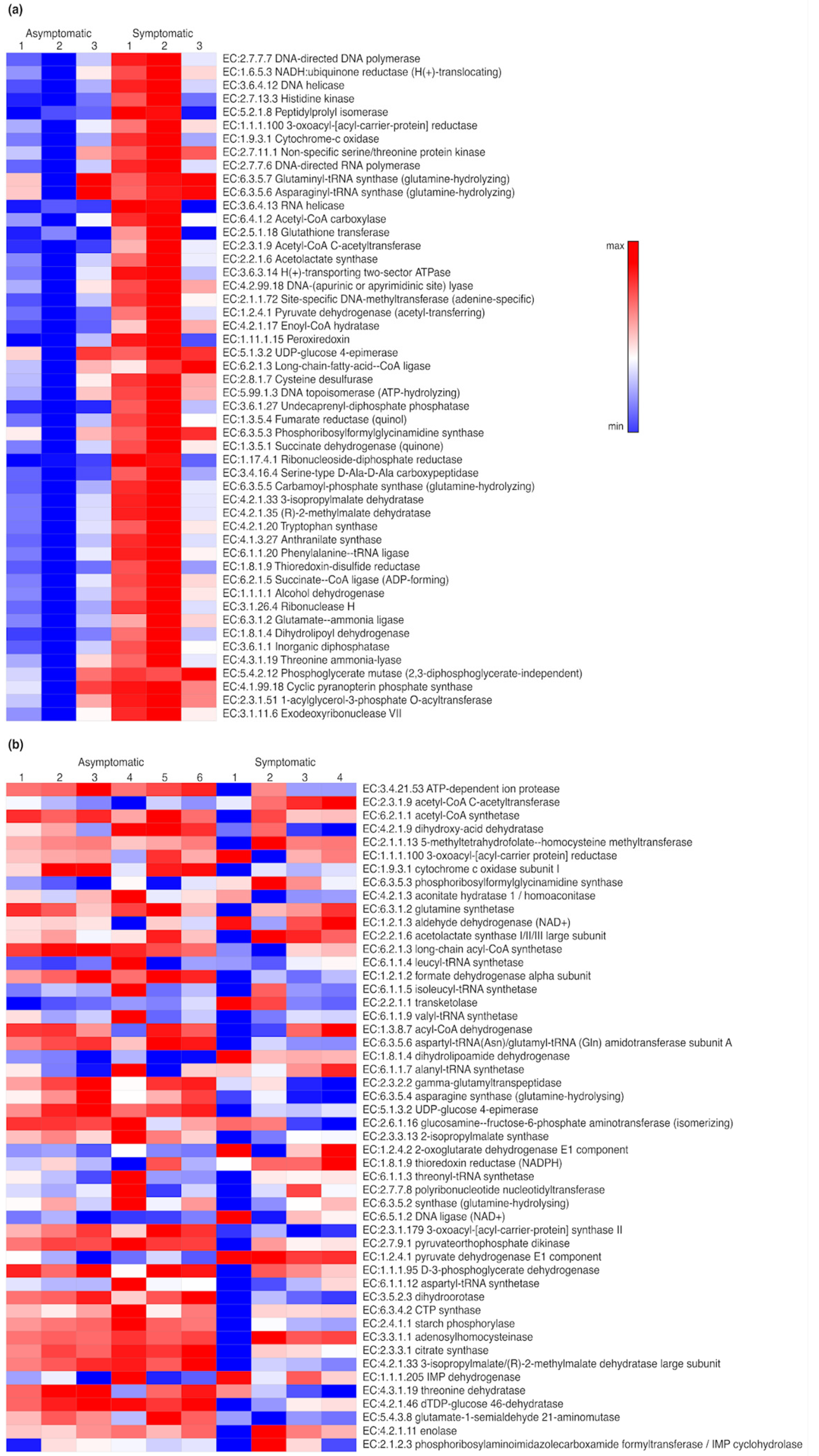
3.3. Functional Profiles of Microbial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neori, A.; Agami, M. The functioning of rhizosphere biota in wetlands—A review. Wetlands 2017, 37, 615–633. [Google Scholar] [CrossRef]
- Kumar, S.; Diksha; Sindhu, S.S.; Kumar, R. Biofertilizers: An ecofriendly technology for nutrient recycling and environmental sustainability. Curr. Res. Microb. Sci. 2021, 3, 100094. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Freilich, S.; Bartuv, R.; Zhimo, V.Y.; Kumar, A.; Biasi, A.; Salim, S.; Feygenberg, O.; Burchard, E.; Dardick, C.; et al. Global analysis of the apple fruit microbiome: Are all apples the same? Environ. Microbiol. 2021, 23, 6038–6055. [Google Scholar] [CrossRef]
- De Souza, R.S.; Okura, V.K.; Armanhi, J.S.; Jorrín, B.; Lozano, N.; da Silva, M.J.; González-Guerrero, M.; de Araújo, L.M.; Verza, N.C.; Bagheri, H.C.; et al. Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci. Rep. 2016, 30, 28774. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Luo, Y.; Gao, Y.; Gao, X.; Gao, J.; Wang, P.; Feng, B. Soil properties, bacterial and fungal community compositions and the key factors after 5-year continuous monocropping of three minor crops. PLoS ONE 2020, 15, e0237164. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Jiao, X.Y.; Chen, Q.; Wu, A.L.; Zheng, Y.; Lin, Y.X.; He, J.Z.; Hu, H.W. Microbial communities in crop phyllosphere and root endosphere are more resistant than soil microbiota to fertilization. Soil Biol. Biochem. 2021, 153, 108113. [Google Scholar] [CrossRef]
- Sun, R.; Zhang, W.; Liu, Y.; Yun, W.; Luo, B.; Chai, R.; Zhang, C.; Xian, X.; Su, X. Changes in phosphorus mobilization and community assembly of bacterial and fungal communities in rice rhizosphere under phosphate deficiency. Front. Microbiol. 2022, 13, 953340. [Google Scholar] [CrossRef]
- Bokszczanin, K.Ł.; Przybyłko, S.; Molska-Kawulok, K.; Wrona, D. Tree root-associated microbial communities depend on various floor management systems in an intensive apple (Malus × domestica Borkh.) orchard. Int. J. Mol. Sci. 2023, 24, 9898. [Google Scholar] [CrossRef]
- Jog, R.; Nareshkumar, G.; Rajkumar, S. Enhancing soil health and plant growth promotion by Actinomycetes. In Plant Growth Promoting Actinobacteria; Subramaniam, G., Arumugam, S., Rajendran, V., Eds.; Springer: Singapore, 2016. [Google Scholar] [CrossRef]
- Prasad, S.; Malav, L.C.; Choudhary, J.; Kannojiya, S.; Kundu, M.; Kumar, S.; Yadav, A.N. Soil microbiomes for healthy nutrient recycling. In Current Trends in Microbial Biotechnology for Sustainable Agriculture; Springer: Singapore, 2021; pp. 1–21. [Google Scholar] [CrossRef]
- Yadav, A.N.; Kour, D.; Kaur, T.; Devi, R.; Yadav, A.; Dikilitas, M.; Abdel-Azeem, A.M.; Ahluwalia, A.S.; Saxena, A.K. Biodiversity, and biotechnological contribution of beneficial soil microbiomes for nutrient cycling, plant growth improvement and nutrient uptake. Biocatal. Agric. Biotechnol. 2021, 33, 102009. [Google Scholar] [CrossRef]
- Peralta, A.L.; Sun, Y.; McDaniel, M.D.; Lennon, J.T. Crop rotational diversity increases disease suppressive capacity of soil microbiomes. Ecosphere 2018, 9, e02235. [Google Scholar] [CrossRef]
- Köhl, J.; Kolnaar, R.; Ravensberg, W.J. Mode of action of microbial biological control agents against plant diseases: Relevance beyond efficacy. Front. Plant Sci. 2019, 10, 845. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Yang, F.; Yao, E.; Raza, W.; Huang, Q.; Shen, Q. Application of bioorganic fertilizer significantly increased apple yields and shaped bacterial community structure in orchard soil. Microb. Ecol. 2017, 73, 404–416. [Google Scholar] [CrossRef]
- Vida, C.; de Vicente, A.; Cazorla, F.M. The role of organic amendments to soil for crop protection: Induction of suppression of soilborne pathogens. Ann. Appl. Biol. 2020, 176, 1–15. [Google Scholar] [CrossRef]
- Knief, C. Analysis of plant microbe interactions in the era of next generation sequencing technologies. Front. Plant Sci. 2014, 5, 216. [Google Scholar] [CrossRef] [PubMed]
- Kalam, S.; Anirban, B.; Podile, A.R. Difficult-to-culture bacteria in the rhizosphere: The underexplored signature microbial groups. Pedosphere 2022, 32, 75–89. [Google Scholar] [CrossRef]
- Ruiz-Cisneros, M.F.; Rios-Velasco, C.; Berlanga-Reyes, D.I.; Ornelas-Paz, J.D.J.; Acosta-Muñiz, C.H.; Romo-Chacón, A.; Zamudio-Flores, P.B.; Pérez-Corral, D.A.; Salas-Marina, M.Á.; Ibarra-Rendón, J.E.; et al. Incidence and causal agents of root diseases and its antagonists in apple orchards of Chihuahua, México. Rev. Mex. Fitopatol. 2017, 35, 437–462. [Google Scholar]
- Tewoldemedhin, Y.T.; Mazzola, M.; Botha, W.J.; Spies, C.F.; McLeod, A. Characterization of fungi (Fusarium and Rhizoctonia) and oomycetes (Phytophthora and Pythium) associated with apple orchards in South Africa. Eur. J. Plant Pathol. 2011, 130, 215–229. [Google Scholar] [CrossRef]
- Kelderer, M.; Manici, L.M.; Caputo, F.; Thalheimer, M. Planting in the ‘inter-row’ to overcome replant disease in apple orchards: A study on the effectiveness of the practice based on microbial indicators. Plant Soil 2012, 357, 381–393. [Google Scholar] [CrossRef]
- Duan, Y.N.; Jiang, W.T.; Zhang, R.; Chen, R.; Chen, X.S.; Yin, C.M.; Mao, Z.Q. Discovery of Fusarium proliferatum f. sp. malus domestica causing apple replant disease in China. Plant Dis. 2022, 106, 2958–2966. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, J.M.; Paulitz, T.C.; Steinberg, C.; Alabouvette, C.; Moënne-Loccoz, Y. The rhizosphere: A playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 2009, 321, 341–361. [Google Scholar] [CrossRef]
- Manici, L.M.; Kelderer, M.; Franke-Whittle, I.H.; Rühmer, T.; Baab, G.; Nicoletti, F.; Caputo, F.; Topp, A.; Insam, H.; Naef, A. Relationship between root-endophytic microbial communities and replant disease in specialized apple growing areas in Europe. Appl. Soil Ecol. 2013, 72, 207–214. [Google Scholar] [CrossRef]
- Yu, Y.; Lee, C.; Kim, J.; Hwang, S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 2005, 89, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an environmental DNA barcode for fungi: An in silico approach reveals potential PCR biases. BMC Microbiol. 2010, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio—PBC: Boston, MA, USA, 2021; Available online: http://www.rstudio.com/ (accessed on 10 November 2023).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-7. 2021. Available online: https://CRAN.R-project.org/package=vegan (accessed on 10 November 2023).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.A.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Bisanz, J.E. qiime2R: Importing QIIME2 Artifacts and Associated Data into R Sessions (v0.99); GitHub: San Francisco, CA, USA, 2018. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. J. Bioinform. 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Deakin, G.; Tilston, E.L.; Bennett, J.; Passey, T.; Harrison, N.; Fernández-Fernández, F.; Xu, X. Spatial structuring of soil microbial communities in commercial apple orchards. Appl. Soil Ecol. 2018, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Deakin, G.; Fernández-Fernández, F.; Bennett, J.; Passey, T.; Harrison, N.; Tilston, E.L.; Xu, X. The effect of rotating apple rootstock genotypes on apple replant disease and rhizosphere microbiome. Phytobiomes J. 2019, 3, 273–285. [Google Scholar] [CrossRef]
- Radl, V.; Winkler, J.B.; Kublik, S.; Yang, L.; Winkelmann, T.; Vestergaard, G.; Schröder, P.; Schloter, M. Reduced microbial potential for the degradation of phenolic compounds in the rhizosphere of apple plantlets grown in soils affected by replant disease. Environ. Microbiome 2019, 14, 8. [Google Scholar] [CrossRef]
- Singh, J.; Silva, K.J.P.; Fuchs, M.; Khan, A. Potential role of weather, soil and plant microbial communities in rapid decline of apple trees. PLoS ONE 2019, 14, e0213293. [Google Scholar] [CrossRef]
- Bartuv, R.; Berihu, M.; Medina, S.; Salim, S.; Feygenberg, O.; Faigenboim-Doron, A.; Zhimo, V.Y.; Abdelfattah, A.; Piombo, E.; Wisniewski, M.; et al. Functional analysis of the apple fruit microbiome based on shotgun metagenomic sequencing of conventional and organic orchard samples. Environ. Microbiol. 2023, 25, 1728–1746. [Google Scholar] [CrossRef] [PubMed]
- Caputo, F.; Nicoletti, F.; Picione, F.D.L.; Manici, L.M. Rhizospheric changes of fungal and bacterial communities in relation to soil health of multi-generation apple orchards. Biol. Control 2015, 88, 8–17. [Google Scholar] [CrossRef]
- Deng, S.; Wipf, H.M.; Pierroz, G.; Raab, T.K.; Khanna, R.; Coleman-Derr, D. A plant growth-promoting microbial soil amendment dynamically alters the strawberry root bacterial microbiome. Sci. Rep. 2019, 9, 17677. [Google Scholar] [CrossRef]
- Bulgari, D.; Bozkurt, A.I.; Casati, P.; Cağlayan, K.; Quaglino, F.; Bianco, P.A. Endophytic bacterial community living in roots of healthy and ‘Candidatus Phytoplasma mali’-infected apple (Malus domestica, Borkh.) trees. Antonie Leeuwenhoek 2012, 102, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Q.; Zhou, J.; Wei, Q. Illumina amplicon sequencing of 16S rRNA tag reveals bacterial community development in the rhizosphere of apple nurseries at a replant disease site and a new planting site. PLoS ONE 2014, 9, e111744. [Google Scholar] [CrossRef] [PubMed]
- Mahnkopp-Dirks, F.; Radl, V.; Kublik, S.; Gschwendtner, S.; Schloter, M.; Winkelmann, T. Molecular barcoding reveals the genus Streptomyces as associated root endophytes of apple (Malus domestica) plants grown in soils affected by apple replant disease. Phytobiomes J. 2021, 5, 177–189. [Google Scholar] [CrossRef]
- Xiong, W.; Li, R.; Ren, Y.; Liu, C.; Zhao, Q.; Wu, H.; Jousset, A.; Shen, Q. Distinct roles for soil fungal and bacterial communities associated with the suppression of vanilla Fusarium wilt disease. Soil Biol. Biochem. 2017, 107, 198–207. [Google Scholar] [CrossRef]
- Ding, T.; Yan, Z.; Zhang, W.; Duan, T. Green manure crops affected soil chemical properties and fungal diversity and community of apple orchard in the Loess Plateau of China. J. Soil Sci. Plant Nutr. 2021, 21, 1089–1102. [Google Scholar] [CrossRef]
- Lycus, P.; Lovise Bøthun, K.; Bergaust, L.; Peele Shapleigh, J.; Reier Bakken, L.; Frostegård, Å. Phenotypic and genotypic richness of denitrifiers revealed by a novel isolation strategy. ISME J. 2017, 11, 2219–2232. [Google Scholar] [CrossRef]
- Qi, G.; Chen, S.; Ke, L.; Ma, G.; Zhao, X. Cover crops restore declining soil properties and suppress bacterial wilt by regulating rhizosphere bacterial communities and improving soil nutrient contents. Microbiol. Res. 2020, 238, 126505. [Google Scholar] [CrossRef]
- Lahlali, R.; Ezrari, S.; Radouane, N.; Kenfaoui, J.; Esmaeel, Q.; El Hamss, H.; Belabess, Z.; Barka, E.A. Biological control of plant pathogens: A global perspective. Microorganisms 2022, 10, 596. [Google Scholar] [CrossRef] [PubMed]
- Khatri, S.; Sazinas, P.; Strube, M.L.; Ding, L.; Dubey, S.; Shivay, Y.S.; Sharma, S.; Jelsbak, L. Pseudomonas is a key player in conferring disease suppressiveness in organic farming. Plant Soil 2023, 1–20. [Google Scholar] [CrossRef]
- Sehrawat, A.; Phour, M.; Kumar, R.; Sindhu, S.S. Bioremediation of pesticides: An eco-friendly approach for environment sustainability. In Microbial Rejuvenation of Polluted Environment; Springer: Singapore, 2021; Volume 25, pp. 23–84. [Google Scholar] [CrossRef]
- Tao, J.; Chen, Q.; Chen, S.; Lu, P.; Chen, Y.; Jin, J.; Li, J.; Xu, Y.; He, W.; Long, T.; et al. Metagenomic insight into the microbial degradation of organic compounds in fermented plant leaves. Environ. Res. 2022, 214, 113902. [Google Scholar] [CrossRef]
- Grgas, D.; Rukavina, M.; Bešlo, D.; Štefanac, T.; Crnek, V.; Šikić, T.; Habuda-Stanić, M.; Landeka Dragičević, T. The bacterial degradation of lignin—A review. Water J. 2023, 15, 1272. [Google Scholar] [CrossRef]
- Etesami, H.; Glick, B.R. Halotolerant plant growth–promoting bacteria: Prospects for alleviating salinity stress in plants. Environ. Exp. Bot. 2020, 178, 104124. [Google Scholar] [CrossRef]
- Omar, A.F.; Abdelmageed, A.H.A.; Al-Turki, A.; Abdelhameid, N.M.; Sayyed, R.Z.; Rehan, M. Exploring the plant growth-promotion of four Streptomyces strains from rhizosphere soil to enhance cucumber growth and yield. Plants 2022, 11, 3316. [Google Scholar] [CrossRef] [PubMed]
- Rehan, M.; Al-Turki, A.; Abdelmageed, A.H.; Abdelhameid, N.M.; Omar, A.F. Performance of plant-growth-promoting rhizobacteria (PGPR) isolated from sandy soil on growth of tomato (Solanum lycopersicum L.). Plants 2023, 12, 1588. [Google Scholar] [CrossRef]
- Mu, H.M.; Wan, Y.Y.; Wu, B.C.; Tian, Y.; Dong, H.L.; Xian, C.G.; Li, Y. A rapid change in microbial communities of the shale gas drilling fluid from 3548 m depth to the above-ground storage tank. Sci. Total Environ. 2021, 784, 147009. [Google Scholar] [CrossRef] [PubMed]
- Higdon, S.M.; Pozzo, T.; Tibbett, E.J.; Chiu, C.; Jeannotte, R.; Weimer, B.C.; Bennett, A.B. Diazotrophic bacteria from maize exhibit multifaceted plant growth promotion traits in multiple hosts. PLoS ONE 2020, 15, e0239081. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Hayter, J.T.; Marlino, M.L.; Isakeit, T.; Chappell, T.M. Pathogenic and saprophytic growth rates of Fusarium oxysporum f. sp. vasinfectum interact to affect variation in inoculum density and interannual infection risk. J. Phytopathol. 2023, 113, 1447–1456. [Google Scholar] [CrossRef]
- Navasca, A.M.; Singh, J.; Rivera-Varas, V.; Geddes, B.; Secor, G.; Gill, U.; Baldwin, T. First report and draft genome resource of a unique opportunistic Fusarium solani pathogen associated with unknown dark galls in sugarbeet. PhytoFrontiers 2023, 3, 704–707. [Google Scholar] [CrossRef]
- El Hazzat, N.; Adnani, M.; Msairi, S.; El Alaoui, M.A.; Mouden, N.; Chliyeh, M.; Boughribil, S.; Selmaoui, K.; Ouazzani Touhami, A.; Douira, A. Fusarium equiseti as one of the main Fusarium species causing wilt and root rot of chickpeas in Morocco. Acta Mycol. 2022, 57, 1–10. [Google Scholar] [CrossRef]
- Ezrari, S.; Radouane, N.; Tahiri, A.; El Housni, Z.; Mokrini, F.; Özer, G.; Lazraq, A.; Belabess, Z.; Amiri, S.; Lahlali, R. Dry root rot disease, an emerging threat to citrus industry worldwide under climate change: A review. Physiol. Mol. Plant Pathol. 2022, 117, 101753. [Google Scholar] [CrossRef]
- Liu, Y.; Tian, Y.; Zhao, X.; Yue, L.; Uwaremwe, C.; Zhou, Q.; Wang, Y.; Zhang, Y.; Dun, Z.; Cui, Z.; et al. Identification of pathogenic Fusarium spp. responsible for root rot of Angelica sinensis and characterization of their biological enemies in Dingxi, China. Plant Dis. 2022, 106, 1898–1910. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, L. Fusarium species associated with diseases of major tropical fruit crops. Horticulturae 2023, 9, 322. [Google Scholar] [CrossRef]
- Mazzola, M.; Manici, L.M. Apple replant disease: Role of microbial ecology in cause and control. Annu. Rev. Phytopathol. 2012, 50, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Kanashiro, A.M.; Akiyama, D.Y.; Kupper, K.C.; Fill, T.P. Penicillium italicum: An underexplored postharvest pathogen. Front. Microbiol. 2020, 11, 606852. [Google Scholar] [CrossRef]
- Patel, D.; Andhare, P.; Menon, S.; Vadakan, S.; Goswami, D. Eccentricity in the behavior of Penicillium spp. as phytopathogen and phytoaugmentor. In Beneficial Microbes for Sustainable Agriculture and Environmental Management; CRC Press: Boca Raton, FL, USA, 2020; pp. 115–138. [Google Scholar]
- Morgunov, I.G.; Kamzolova, S.V.; Dedyukhina, E.G.; Chistyakova, T.I.; Lunina, J.N.; Mironov, A.A.; Stepanova, N.N.; Shemshura, O.N.; Vainshtein, M.B. Application of organic acids for plant protection against phytopathogens. Appl. Microbiol. Biotechnol. 2017, 101, 921–932. [Google Scholar] [CrossRef]
- Girvan, M.S.; Bullimore, J.; Pretty, J.N.; Osborn, A.M.; Ball, A.S. Soil type is the primary determinant of the composition of the total and active bacterial communities in arable soils. Appl. Environ. Microbiol. 2003, 69, 1800–1809. [Google Scholar] [CrossRef]
- Yang, M.; Yang, D.; Yu, X. Soil microbial communities and enzyme activities in sea-buckthorn (Hippophae rhamnoides) plantation at different ages. PLoS ONE 2018, 13, e0190959. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, B.; Zhu, Y.; Wang, J.; Zhang, H.; Wang, Z. Bacterial community diversity associated with the severity of bacterial wilt disease in tomato fields in southeast China. Can. J. Microbiol. 2019, 65, 538–549. [Google Scholar] [CrossRef] [PubMed]
- González-Escobedo, R.; Muñoz-Castellanos, L.N.; Muñoz-Ramirez, Z.Y.; Guigón-López, C.; Avila-Quezada, G.D. Rhizosphere bacterial and fungal communities of healthy and wilted pepper (Capsicum annuum L.) in an organic farming system. Ciênc. Rural 2023, 53, 0220072. [Google Scholar] [CrossRef]
- Wang, R.; Lv, F.; Lv, R.; Lin, H.; Zhang, Z.; Wei, L. Sampling period and disease severity of bacterial wilt significantly affected the bacterial community structure and functional prediction in the sesame rhizosphere soil. Rhizosphere 2023, 26, 100704. [Google Scholar] [CrossRef]
- Solís-García, I.A.; Ceballos-Luna, O.; Cortazar-Murillo, E.M.; Desgarennes, D.; Garay-Serrano, E.; Patiño-Conde, V.; Guevara-Avendaño, E.; Méndez-Bravo, A.; Reverchon, F. Phytophthora root rot modifies the composition of the avocado rhizosphere microbiome and increases the abundance of opportunistic fungal pathogens. Front. Microbiol. 2021, 11, 574110. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, G.; Schulz, S.; Schöler, A.; Schloter, M. Making big data smart—How to use metagenomics to understand soil quality. Biol. Fertil. Soils 2017, 53, 479–484. [Google Scholar] [CrossRef]
- Martin, C.C.S. Potential of compost tea for suppressing plant diseases. CABI Rev. 2014, 9, 1–38. [Google Scholar] [CrossRef]
- Ladygina, N.; Hedlund, K. Plant species influence microbial diversity and carbon allocation in the rhizosphere. Soil Biol. Biochem. 2010, 42, 162–168. [Google Scholar] [CrossRef]
- Shigyo, N.; Umeki, K.; Hirao, T. Seasonal dynamics of soil fungal and bacterial communities in cool-temperate montane forests. Front. Microbiol. 2019, 10, 1944. [Google Scholar] [CrossRef]
- Bei, Q.; Moser, G.; Müller, C.; Liesack, W. Seasonality affects function and complexity but not diversity of the rhizosphere microbiome in European temperate grassland. Sci. Total. Environ. 2021, 784, 147036. [Google Scholar] [CrossRef]
- Toole, D.R.; Zhao, J.; Martens-Habbena, W.; Strauss, S.L. Bacterial functional prediction tools detect but underestimate metabolic diversity compared to shotgun metagenomics in southwest Florida soils. Appl. Soil Ecol. 2021, 168, 104129. [Google Scholar] [CrossRef]
- Ayiti, O.E.; Ayangbenro, A.S.; Babalola, O.O. Relationship between nitrifying microorganisms and other microorganisms residing in the maize rhizosphere. Arch. Microbiol. 2022, 204, 246. [Google Scholar] [CrossRef] [PubMed]
- Sivaram, A.K.; Panneerselvan, L.; Mukunthan, K.; Megharaj, M. Effect of pyroligneous acid on the microbial community composition and plant growth-promoting bacteria (PGPB) in soils. Soil Syst. 2022, 6, 10. [Google Scholar] [CrossRef]
- Baz, L.; Abulfaraj, A.A.; Tashkandi, M.A.; Baeissa, H.M.; Refai, M.Y.; Barqawi, A.A.; Shami, A.; Abuauf, H.W.; Ashy, R.A.; Jalal, R.S. Predicted functional shifts due to type of soil microbiome and watering of two wild plants in western region of Saudi Arabia. Phyton 2022, 91, 2249–2268. [Google Scholar] [CrossRef]
- Kumar, A.; Dubey, A. Rhizosphere microbiome: Engineering bacterial competitiveness for enhancing crop production. J. Adv. Res. 2020, 24, 337–352. [Google Scholar] [CrossRef]
- Zhao, X.; Dong, Q.; Han, Y.; Zhang, K.; Shi, X.; Yang, X.; Yuan, Y.; Zhou, D.; Wang, K.; Wang, X.; et al. Maize/peanut intercropping improves nutrient uptake of side-row maize and system microbial community diversity. BMC Microbiol. 2022, 22, 14. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.M.; Sudhakar, P. Metagenomic Approaches for Studying Plant–Microbe Interactions. In Understanding the Microbiome Interactions in Agriculture and the Environment; Veera Bramhachari, P., Ed.; Springer: Singapore, 2022. [Google Scholar] [CrossRef]
- Wijayawardene, N.N.; Hyde, K.D.; Dai, D.Q. Outline of Ascomycota. Encycl. Mycol. 2021, 88, 246–254. [Google Scholar] [CrossRef]
- Gorshkov, V.; Tsers, I. Plant susceptible responses: The underestimated side of plant–pathogen interactions. Biol. Rev. 2022, 97, 45–66. [Google Scholar] [CrossRef]
- Hansen, E.; Delatour, C. Phytophthora species in oak forests of north-east France. Ann. For. Sci. 1999, 56, 539–547. [Google Scholar] [CrossRef]
- Khaliq, I.; Hardy, G.E.S.J.; McDougall, K.L.; Burgess, T.I. Phytophthora species isolated from alpine and sub-alpine regions of Australia, including the description of two new species; Phytophthora cacuminis sp. nov and Phytophthora oreophila sp. nov. Fungal Biol. 2019, 123, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Mannai, S.; Benfradj, N.; Boughalleb-M’Hamdi, N. Regional and seasonal variation of Fusarium and Oomycetes species associated with apple seedlings decline in Tunisian nurseries. Nov. Res. Microbiol. J. 2023, 7, 2015–2033. [Google Scholar] [CrossRef]
- Wannicke, N.; Brust, H. Inactivation of the plant pathogen Pythium ultimum by plasma-processed air (PPA). Appl. Sci. 2023, 13, 4511. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Ramírez, Z.Y.; González-Escobedo, R.; Avila-Quezada, G.D.; Ramírez-Sánchez, O.; Higareda-Alvear, V.M.; Zapata-Chávez, E.; Borrego-Loya, A.; Muñoz-Castellanos, L.N. Exploring Microbial Rhizosphere Communities in Asymptomatic and Symptomatic Apple Trees Using Amplicon Sequencing and Shotgun Metagenomics. Agronomy 2024, 14, 357. https://doi.org/10.3390/agronomy14020357
Muñoz-Ramírez ZY, González-Escobedo R, Avila-Quezada GD, Ramírez-Sánchez O, Higareda-Alvear VM, Zapata-Chávez E, Borrego-Loya A, Muñoz-Castellanos LN. Exploring Microbial Rhizosphere Communities in Asymptomatic and Symptomatic Apple Trees Using Amplicon Sequencing and Shotgun Metagenomics. Agronomy. 2024; 14(2):357. https://doi.org/10.3390/agronomy14020357
Chicago/Turabian StyleMuñoz-Ramírez, Zilia Y., Román González-Escobedo, Graciela D. Avila-Quezada, Obed Ramírez-Sánchez, Victor M. Higareda-Alvear, Emiliano Zapata-Chávez, Alejandra Borrego-Loya, and Laila N. Muñoz-Castellanos. 2024. "Exploring Microbial Rhizosphere Communities in Asymptomatic and Symptomatic Apple Trees Using Amplicon Sequencing and Shotgun Metagenomics" Agronomy 14, no. 2: 357. https://doi.org/10.3390/agronomy14020357
APA StyleMuñoz-Ramírez, Z. Y., González-Escobedo, R., Avila-Quezada, G. D., Ramírez-Sánchez, O., Higareda-Alvear, V. M., Zapata-Chávez, E., Borrego-Loya, A., & Muñoz-Castellanos, L. N. (2024). Exploring Microbial Rhizosphere Communities in Asymptomatic and Symptomatic Apple Trees Using Amplicon Sequencing and Shotgun Metagenomics. Agronomy, 14(2), 357. https://doi.org/10.3390/agronomy14020357