
Gene Mapping of a Yellow-to-Lethal Mutation Based on Bulked-Segregant Analysis-Seq in Soybean
 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Genetic Analysis
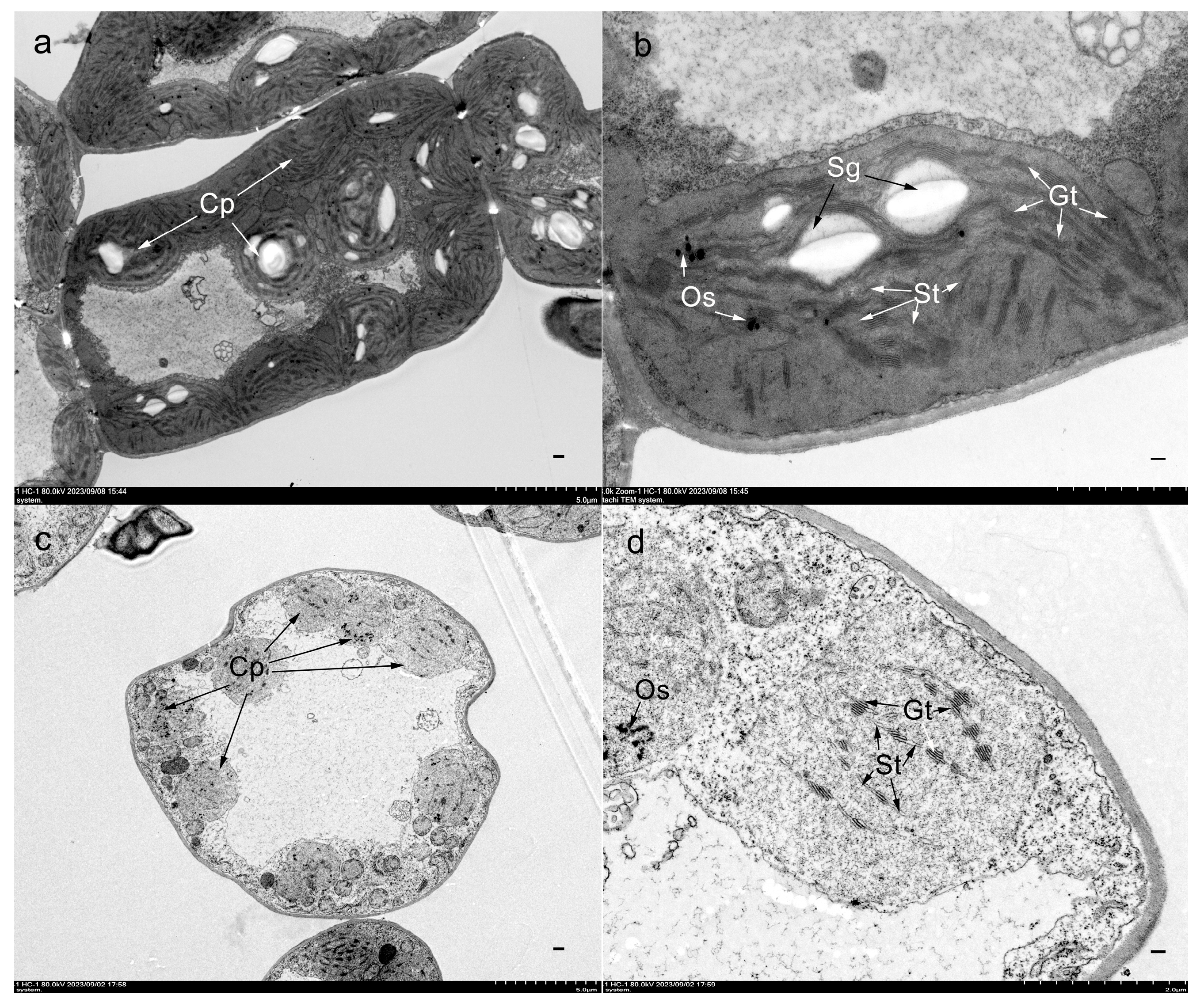
2.2. Chlorophyll Quantification and Transmission Electron Microscopy (TEM) Analysis
2.3. BSA-Seq
2.4. Quality Control and Analysis of Sequencing Data
2.5. Gene Mapping, Candidate Gene Prediction, and Clone
3. Results
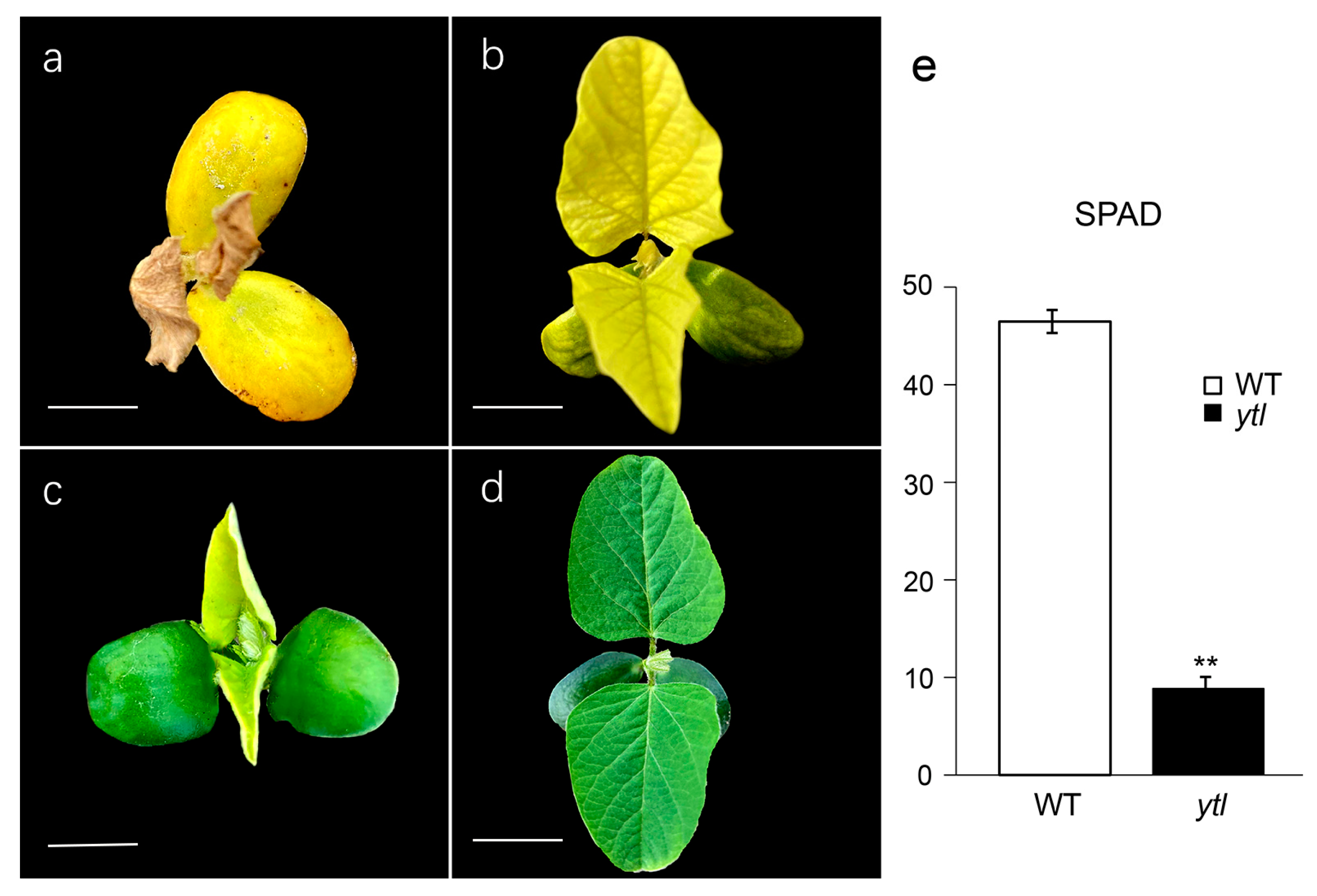
3.1. Characterization of the Mutant ytl
3.2. Genetic Analysis
3.3. Analysis of BSA-Seq Quality
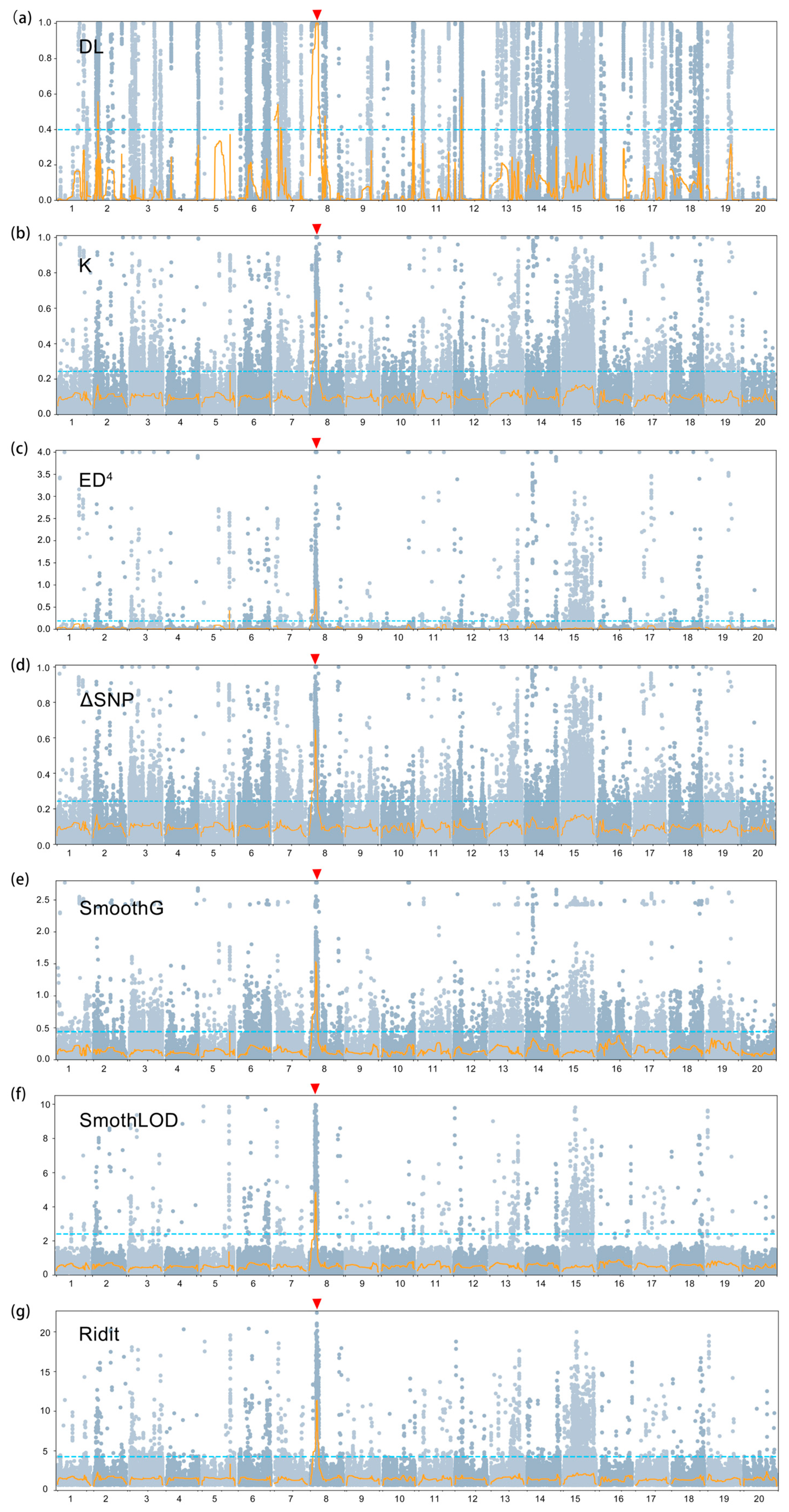
3.4. Preliminary Mapping Based on the BSA-Seq
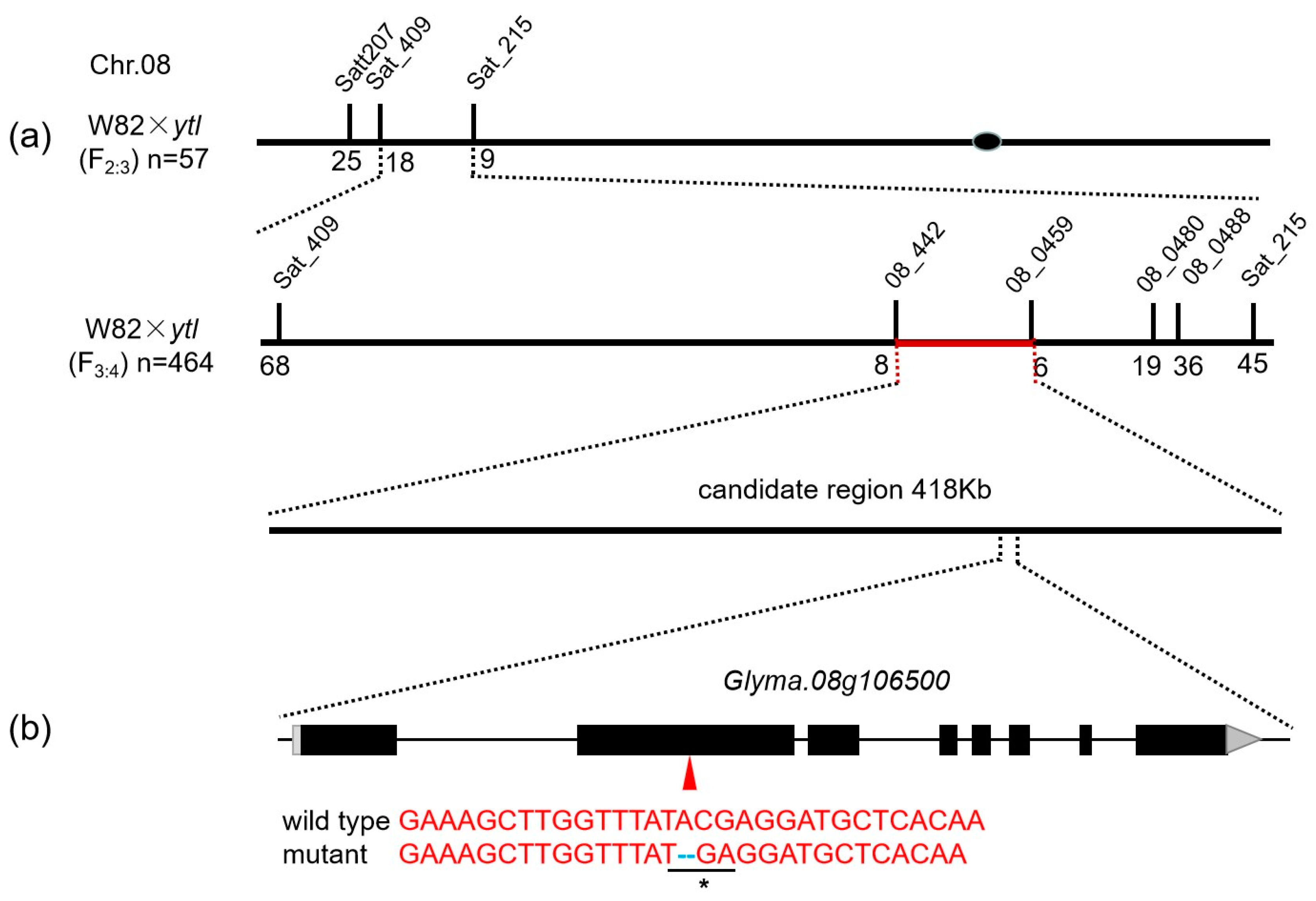
3.5. Fine Mapping by Using SSR Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sharrock, R.A.; Parks, B.M.; Koornneef, M.; Quail, P.H. Molecular analysis of the phytochrome deficiency in an aurea mutant of tomato. Mol. Gen. Genet. (MGG) 1988, 213, 9–14. [Google Scholar] [CrossRef]
- Kato, K.K.; Palmer, R.G. Duplicate chlorophyll-deficient loci in soybean. Genome 2004, 47, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.G.; Nelson, R.L.; Bernard, R.L.; Stelly, D.M. Genetics and linkage of three chlorophyll-deficient mutants in soybean: y19, y22, and y23. J. Hered. 1990, 81, 404–406. [Google Scholar] [CrossRef]
- Palmer, R.G.; Burzlaff, J.D.; Shoemaker, R.C. Genetic analyses of two independent chlorophyll-deficient mutants identified among the progeny of a single chimeric foliage soybean plant. J. Hered. 2000, 91, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.J.; Kurepa, J.; Vierstra, R.D. The Arabidopsis thaliana HY1 locus, required for phytochrome-chromophore biosynthesis, encodes a protein related to heme oxygenases. Proc. Natl. Acad. Sci. USA 1999, 96, 6541–6546. [Google Scholar] [CrossRef]
- Ki-Hong, J.; Junghe, H.; Choong-Hwan, R.; Youngju, C.; Yong-Yoon, C.; Akio, M.; Hirochika, H.; An, G. Characterization of a rice chlorophyll-deficient mutant using the T-DNA gene-trap system. Plant Cell Physiol. 2003, 44, 463–472. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Rahman, M.L.; Cho, S.H.; Kim, Y.S.; Koh, H.J.; Yoo, S.C.; Paek, N.C. The rice faded green leaf locus encodes protochlorophyllide oxidoreductaseb and is essential for chlorophyll synthesis under high light conditions. Plant J. 2013, 74, 122–133. [Google Scholar] [CrossRef]
- Gu, J.; Zhou, Z.; Li, Z.; Chen, Y.; Wang, Z.; Zhang, H. Rice (Oryza sativa L.) with reduced chlorophyll content exhibit higher photosynthetic rate and efficiency, improved canopy light distribution, and greater yields than normally pigmented plants. Field Crops Res. 2017, 200, 58–70. [Google Scholar] [CrossRef]
- Liu, M.; Wang, Y.; Nie, Z.; Gai, J.; Bhat, J.A.; Kong, J.J.; Zhao, T. Double mutation of two homologous genes yl1 and yl2 results in a leaf yellowing phenotype in soybean [Glycine max (L.) Merr]. Plant Mol. Biol. 2020, 103, 527–543. [Google Scholar] [CrossRef]
- Sandhu, D.; Coleman, Z.; Atkinson, T.; Krishan, M.R.; Mendu, V. Genetics and physiology of the nuclearly inherited yellow foliar mutants in soybean. Front. Plant Sci. 2018, 9, 471. [Google Scholar] [CrossRef]
- Weber, C.R.; Weiss, M.G. Chlorophyll mutant in soybeans provides teaching aid. J. Hered. 1959, 50, 53–54. [Google Scholar] [CrossRef]
- Campbell, B.W.; Mani, D.; Curtin, S.J.; Slattery, R.A.; Stupar, R.M. Identical substitutions in magnesium chelatase paralogs result in chlorophyll-deficient soybean mutants. G3-Genes Genom. Genet. 2015, 5, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.D.; Czarnota, C.D.; Cappy, J.J. Morphological and physiological characteristics of an achlorophyllous mutant soybean variety sustained to muturation via grafting. Am. J. Bot. 1977, 64, 1042–1045. [Google Scholar] [CrossRef]
- Reed, S.; Atkinson, T.; Gorecki, C.; Espinosa, K.; Przybylski, S.; Goggi, A.S.; Palmer, R.G.; Sandhu, D. Candidate gene identification for a lethal chlorophyll-deficient mutant in soybean. Agronomy 2014, 4, 462–469. [Google Scholar] [CrossRef]
- Sandhu, D.; Atkinson, T.; Noll, A.; Johnson, C.; Espinosa, K.; Boelter, J.; Abel, S.; Balpreet, K.; Dhatt, B.K.; Barta, T.; et al. Soybean proteins GmTic110 and GmPsbP are crucial for chloroplast development and function. Plant Sci. 2016, 252, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, K.; Ossowski, S.; Lanz, C.; Juul, T.; Petersen, A.H.; Nielsen, K.L.; Jrgensen, J.-E.; Weigel, D.; Andersen, S.U. SHOREmap: Simultaneous mapping and mutation identification by deep sequencing. Nat. Methods 2009, 6, 550–551. [Google Scholar] [CrossRef]
- Baek, G.; Kim, C.W.; Kim, S. Development of a molecular marker tightly linked to the C locus conferring a white bulb color in onion (Allium cepa L.) using bulked segregant analysis and RNA-Seq. Mol. Breed. 2017, 37, 94. [Google Scholar] [CrossRef]
- Yu, J.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. Genetic design and statistical power of nested association mapping in maize. Genetics 2008, 178, 539–551. [Google Scholar] [CrossRef]
- Kover, P.X.; Valdar, W.; Trakalo, J.; Scarcelli, N.; Ehrenreich, I.M.; Purugganan, M.D.; Durrant, C.; Mott, R. A multiparent advanced generation inter-cross to fine-map quantitative traits in Arabidopsis thaliana. PLoS Genet. 2009, 5, e1000551. [Google Scholar] [CrossRef]
- Zhang, S.; Abdelghany, A.M.; Azam, M.; Qi, J.; Li, J.; Feng, Y.; Liu, Y.; Feng, H.; Ma, C.; Gebregziabher, B.S.; et al. Mining candidate genes underlying seed oil content using BSA-seq in soybean. Ind. Crops Prod. 2023, 194, 116308. [Google Scholar] [CrossRef]
- Zou, C.; Wang, P.; Xu, Y. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol. J. 2016, 14, 1941–1955. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M.; et al. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef]
- Takagi, H.; Tamiru, M.; Abe, A.; Takagi, H.; Tamiru, M.; Abe, A.; Yoshida, K.; Uemura, A.; Yaegashi, H.; Obara, T.; et al. MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat. Biotechnol. 2015, 33, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Frouin, J.; Filloux, D.; Taillebois, J.; Grenier, C.; Montes, F.; Lamotte, F.D.; Verdeil, J.; Courtois, B.; Ahmadi, N. Positional cloning of the rice male sterility gene ms-IR36, widely used in the inter-crossing phase of recurrent selection schemes. Mol. Breed. 2014, 33, 555–567. [Google Scholar] [CrossRef]
- Rodriguez-Uribe, L.; Higbie, S.M.; Stewart, J.M.; Wilkins, T.; Lindemann, W.; Sengupta-Gopalan, C.; Zhang, J. Identification of salt responsive genes using comparative microarray analysis in Upland cotton (Gossypium hirsutum L.). Plant Sci. 2011, 180, 461–469. [Google Scholar] [CrossRef]
- Klein, H.; Xiao, Y.; Conklin, P.A.; Govindarajulu, R.; Kelly, J.A.; Scanlon, M.J.; Whipple, C.J.; Bartlett, M. 2018. Bulked-segregant analysis coupled to whole genome sequencing (BSA-Seq) for rapid gene cloning in maize. G3-Genes Genom. Genet. 2018, 8, 3583–3592. [Google Scholar] [CrossRef]
- Forrest, K.; Pujol, V.; Bulli, P.; Pumphrey, M.; Wellings, C.; Herrera-Foessel, S.; Huerta-Espino, J.; Singh, R.; Lagudah, E.; Hayden, M.; et al. Development of a SNP marker assay for the Lr67 gene of wheat using a genotyping by sequencing approach. Mol. Breed. 2014, 34, 2109–2118. [Google Scholar] [CrossRef]
- Livaja, M.; Wang, Y.; Wieckhorst, S.; Haseneyer, G.; Seidel, M.; Hahn, V.; Knapp, S.J.; Taudien, S.; Schön, C.-C.; Bauer, E. BSTA: A targeted approach combines bulked segregant analysis with next-generation sequencing and de novo transcriptome assembly for SNP discovery in sunflower. BMC Genom. 2013, 14, 628. [Google Scholar] [CrossRef]
- Al Amin, G.M.; Kong, K.; Sharmin, R.A.; Kong, J.; Bhat, J.A.; Zhao, T. Characterization and Rapid Gene-Mapping of Leaf Lesion Mimic Phenotype of spl-1 Mutant in Soybean (Glycine max (L.) Merr.). Int. J. Mol. Sci. 2019, 20, 2193. [Google Scholar] [CrossRef]
- Galya, K.; Yael, B.; Adi, F.D.; Abhinandan, P.; Ilan, H.; Ran, H. Fine-mapping the branching habit trait in cultivated peanut by combining bulked segregant analysis and high-throughput sequencing. Front. Plant Sci. 2017, 8, 467. [Google Scholar] [CrossRef][Green Version]
- Pearson, K.X. On the criterion that a given system of deviations from the probable in the case of a correlated system of variables is such that it can be reasonably supposed to have arisen from random sampling. Philos. Mag. 1900, 50, 157–175. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Ge, D.; Bhat, J.A.; Li, Y.; Kong, J.; Liu, K.; Zhao, T. Hydroperoxide lyase modulates defense response and confers lesion-mimic leaf phenotype in soybean (Glycine max (L.) merr.). Plant J. 2020, 104, 1315–1333. [Google Scholar] [CrossRef]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Ochar, K.; Bo-Hong, S.U.; Zhou, M.; Liu, Z.; Gao, H.; Flamlom, S.; Qiu, L. Identification of the genetic locus associated with the crinkled leaf phenotype in a soybean (Glycine max L.) mutant by BSA-Seq technology. J. Integr. Agric. 2022, 21, 3524–3539. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, Z.; Chen, X.; Shi, S.; Zhang, H.; Wang, X.; Chen, H.; Li, W.; Li, L. DeepBSA: A deep-learning algorithm improves bulked segregant analysis for dissecting complex traits. Mol. Plant 2022, 15, 1418–1427. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, W.; Zhang, Y.; Liu, M.; Kong, J.; Yu, Z.; Jaffer, A.M.; Gai, J.; Zhao, T. Identification of two duplicated loci controlling a disease-like rugose leaf phenotype in soybean. Crop Sci. 2016, 56, 1611–1618. [Google Scholar] [CrossRef]
- Garzon, L.N.; Blair, M.W. Development and mapping of SSR markers linked to resistance-gene homologue clusters in common bean. Crop J. 2014, 2, 183–194. [Google Scholar] [CrossRef]
- Song, Q.; Jia, G.; Zhu, Y.; Grant, D.; Nelson, R.T.; Hwang, E.Y.; Hyten, D.L.; Cregan, P.B. Abundance of SSR motifs and development of candidate polymorphic SSR markers (BARCSOYSSR_1.0) in soybean. Crop Sci. 2010, 50, 1950–1960. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, L.; Man, Y.; Li, X.; Xiao, J. Pdm4, a pentatricopeptide repeat protein, affects chloroplast gene expression and chloroplast development in Arabidopsis thaliana. Front. Plant Sci. 2020, 11, e1198. [Google Scholar] [CrossRef]
- Song, J.; Li, Z.; Liu, Z.; Guo, Y.; Qiu, L. Next-generation sequencing from bulked-segregant analysis accelerates the simultaneous identification of two qualitative genes in soybean. Front. Plant Sci. 2017, 8, e919. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Sun, S.; Li, Y.; Duan, C.; Zhu, Z. Next-generation sequencing to identify candidate genes and develop diagnostic markers for a novel phytophthora resistance gene, RpsHC18, in soybean. Theor. App. Genet. 2018, 131, 525–538. [Google Scholar] [CrossRef]
- Barneche, F.; Winter, V.; Crèvecoeur, M.; Rochaix, J.D. ATAB2 is a novel factor in the signalling pathway of light-controlled synthesis of photosystem proteins. Embo J. 2014, 25, 5907–5918. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Hiltbrunner, A.; Weibel, P.; Vidi, P.A.; Alvarez-Huerta, M.; Smith, M.D.; Schnell, D.J.; Kessler, F. Essential role of the G-domain in targeting of the protein import receptor atToc159 to the chloroplast outer membrane. J. Cell Biol. 2002, 159, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kim, S.J.; Lee, Y.J.; Jin, J.B.; Hwang, I. The M domain of atToc159 plays an essential role in the import of proteins into chloroplasts and chloroplast biogenesis. J. Biol. Chem. 2003, 278, 36794–36805. [Google Scholar] [CrossRef]
- Agne, B.; Infanger, S.; Wang, F.; Hofstetter, V.; Rahim, G.; Martin, M.; Lee, D.W.; Hwang, I.; Schnell, D.; Kessler, F. A Toc159 import receptor mutant, defective in hydrolysis of GTP, supports preprotein import into chloroplasts. J. Biol. Chem. 2009, 284, 8670–8679. [Google Scholar] [CrossRef]
- Moon, S.; Giglione, C.; Lee, D.Y.; An, S.; Jeong, D.H.; Meinnel, T.; An, G. Rice peptide deformylase PDF1B is crucial for development of chloroplasts. Plant Cell Physiol. 2008, 49, 1536–1546. [Google Scholar] [CrossRef]
- Barkan, A.; Small, I. Pentatricopeptide pepeat proteins in plants. Annu. Rev. Plant Biol. 2014, 65, 415–442. [Google Scholar] [CrossRef]
- Khrouchtchova, A.; Monde, R.A.; Barkan, A. A short PPR protein required for the splicing of specific group II introns in angiosperm chloroplasts. RNA 2012, 18, 1197–1209. [Google Scholar] [CrossRef]
- Cai, W.; Ji, D.; Peng, L.; Guo, J.; Ma, J.; Zou, M.; Lu, C.; Zhang, L. LPA66 is required for editing psbF chloroplast transcripts in Arabidopsis. Plant Physiol. 2009, 150, 1260–1271. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Total No. of Plants | No. of Green Leaf Plants | No. of Yellow-to-Lethal Plants | Expected Ratio | χ2 (3:1) | p |
|---|---|---|---|---|---|---|
| F2 (W82 × ytl) | 208 | 162 | 46 | 3:1 | 0.78 | 0.38 |
| Primer Name | Forward Primer Sequence (5′-3′) | Reverse Primer Sequence (5′-3′) |
|---|---|---|
| Satt207 | GCGTTTTTCTCATTTTGATTCCTAAAC | GCGATTGTGATTGTAGTCCCTAAA |
| Sat_409 | GCGGAGGTTTGTGCATTTCTAGGTCTTC | GCGACGCGTATGTACATAAAATATGCTGTT |
| Sat_215 | GCGTAGCAACAAAGCAATCTACAG | GCGTCCCATTTTATTCCACACTATGTAAT |
| BARCSOYSSR_08_0442 | GAAACGGTTGGAGAATAGCG | TCCATGCCCACTTAACAACA |
| BARCSOYSSR_08_0459 | CAACTTTCGCATCGGTTACA | CGGAAACTGCTTATGGTTGC |
| BARCSOYSSR_08_0480 | TCGTTGACTGCAATATGGTC | ACCGAATGAAGCGTAAAGGA |
| BARCSOYSSR_08_0488 | TCTCATGGGAACCTGGAAAA | CGAAACAGCAACGAATCAAA |
| Glyma.08g106500-1 | TTGGAGCTGGCAGAGTCTAATC | AATGAGCTGATGGCCACTGTA |
| Glyma.08g106500-2 | GCCAGATGGGGACGTCATAAG | TCAATTGATGGAGCAACACCG |
| Glyma.08g106500-3 | CAGGAAAACATCAGGAGGCAGA | GCTGCATTGCATCAAACGTGG |
| Gene ID | Gene Start (bp) | Gene End (bp) | Putative Function |
|---|---|---|---|
| Glyma.08G103100 | 7,902,820 | 7,907,892 | RING-type E3 ubiquitin transferase |
| Glyma.08G103200 | 7,918,306 | 7,932,242 | Mannosyl-oligosaccharide glucosidase |
| Glyma.08G103300 | 7,934,866 | 7,944,355 | Peptidase_M28 domain-containing protein |
| Glyma.08G103400 | 7,945,667 | 7,949,720 | F-box domain-containing protein |
| Glyma.08G103500 | 7,947,843 | 7,948,181 | Unknown |
| Glyma.08G103600 | 7,954,858 | 7,956,497 | Signal peptidase complex subunit 1 |
| Glyma.08G103700 | 7,959,250 | 7,962,513 | Large subunit ribosomal protein L17e |
| Glyma.08G103800 | 7,966,298 | 7,969,958 | Exostosin domain-containing protein |
| Glyma.08G103900 | 7,970,507 | 7,974,378 | Caffeoyl-CoA O-methyltransferase |
| Glyma.08G104000 | 7,976,043 | 7,980,358 | S-adenosyl-L-methionine-dependent methyltransferases |
| Glyma.08G104100 | 7,993,317 | 7,995,690 | Flavonol biosynthesis |
| Glyma.08G104200 | 8,004,960 | 8,006,787 | REF domain-containing protein |
| Glyma.08G104300 | 8,007,843 | 8,011,052 | Aerobic respiration I (cytochrome c) |
| Glyma.08G104400 | 8,016,766 | 8,018,606 | MYND-type domain-containing protein |
| Glyma.08G104500 | 8,041,175 | 8,044,478 | DUF493 |
| Glyma.08G104600 | 8,045,602 | 8,047,222 | Polysacc_synt_4 domain-containing protein |
| Glyma.08G104700 | 8,055,039 | 8,059,859 | Ribosome assembly protein RRB1 |
| Glyma.08G104800 | 8,063,060 | 8,066,966 | F-box domain-containing protein |
| Glyma.08G104900 | 8,067,568 | 8,069,734 | RAMP4 in endoplasmic reticulum |
| Glyma.08G105000 | 8,079,687 | 8,087,576 | Arabidopsis histidine kinase 2/3/4 (cytokinin receptor) |
| Glyma.08G105100 | 8,091,820 | 8,093,816 | NTF2 domain-containing protein |
| Glyma.08G105200 | 8,094,716 | 8,102,735 | TIG DNA binding |
| Glyma.08G105300 | 8,109,486 | 8,110,128 | DUF3339 |
| Glyma.08G105400 | 8,113,990 | 8,126,799 | K-box domain-containing protein |
| Glyma.08G105500 | 8,128,245 | 8,135,906 | K-box domain-containing protein |
| Glyma.08G105600 | 8,154,834 | 8,161,846 | DNA primase large subunit |
| Glyma.08G105700 | 8,166,200 | 8,168,551 | Rho GDP-dissociation inhibitor |
| Glyma.08G105800 | 8,171,582 | 8,177,075 | MACPF domain-containing protein |
| Glyma.08G105900 | 8,182,656 | 8,185,890 | U6 snRNA-associated Sm-like protein LSm7 |
| Glyma.08G106000 | 8,189,588 | 8,192,602 | Amidase domain-containing protein |
| Glyma.08G106100 | 8,194,006 | 8,194,604 | Unknown |
| Glyma.08G106200 | 8,194,864 | 8,196,276 | Amidase domain-containing protein |
| Glyma.08G106300 | 8,197,911 | 8,199,307 | PORR domain-containing protein |
| Glyma.08G106400 | 8,199,636 | 8,206,598 | kinesin family member 15 |
| Glyma.08G106500 | 8,208,987 | 8,215,162 | PPR_long domain-containing protein |
| Glyma.08G106600 | 8,215,384 | 8,215,533 | Unknown |
| Glyma.08G106700 | 8,216,338 | 8,219,979 | FK506-binding protein 4/5 |
| Glyma.08G106800 | 8,221,055 | 8,223,572 | Small subunit ribosomal protein S19 |
| Glyma.08G106900 | 8,228,380 | 8,228,919 | Unknown |
| Glyma.08G107000 | 8,229,694 | 8,231,371 | mTERF domain-containing protein |
| Glyma.08G107100 | 8,232,784 | 8,238,471 | C2H2-type domain-containing protein |
| Glyma.08G107200 | 8,242,346 | 8,245,459 | Tic22 domain-containing protein |
| Glyma.08G107300 | 8,250,677 | 8,253,307 | Xyloglucan:xyloglucosyl transferase |
| Glyma.08G107400 | 8,255,102 | 8,260,380 | Unknown |
| Glyma.08G107500 | 8,266,733 | 8,268,742 | Glycosyltransferase |
| Glyma.08G107600 | 8,276,759 | 8,278,195 | Glycosyltransferase |
| Glyma.08G107700 | 8,284,689 | 8,288,765 | Protein kinase domain-containing protein |
| Glyma.08G107800 | 8,296,196 | 8,307,328 | Bifunctional aspartokinase/homoserine dehydrogenase 1 |
| Glyma.08G107900 | 8,308,038 | 8,310,337 | Histone H3 |
| Glyma.08G108000 | 8,311,539 | 8,314,951 | Selenium-binding protein 1 |
| Glyma.08G108100 | 8,315,634 | 8,315,795 | RALF domain-containing protein |
| Glyma.08G108200 | 8,317,340 | 8,318,268 | RALF domain-containing protein |
| Glyma.08G108300 | 8,321,303 | 8,325,907 | Microfibrillar-associated protein 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Chang, F.; Al Amin, G.M.; Li, S.; Fu, M.; Yu, X.; Zhao, Z.; Xu, H.; Zhao, T. Gene Mapping of a Yellow-to-Lethal Mutation Based on Bulked-Segregant Analysis-Seq in Soybean. Agronomy 2024, 14, 185. https://doi.org/10.3390/agronomy14010185
Wang Y, Chang F, Al Amin GM, Li S, Fu M, Yu X, Zhao Z, Xu H, Zhao T. Gene Mapping of a Yellow-to-Lethal Mutation Based on Bulked-Segregant Analysis-Seq in Soybean. Agronomy. 2024; 14(1):185. https://doi.org/10.3390/agronomy14010185
Chicago/Turabian StyleWang, Yaqi, Fangguo Chang, G M Al Amin, Shuguang Li, Mengmeng Fu, Xiwen Yu, Zhixin Zhao, Haifeng Xu, and Tuanjie Zhao. 2024. "Gene Mapping of a Yellow-to-Lethal Mutation Based on Bulked-Segregant Analysis-Seq in Soybean" Agronomy 14, no. 1: 185. https://doi.org/10.3390/agronomy14010185
APA StyleWang, Y., Chang, F., Al Amin, G. M., Li, S., Fu, M., Yu, X., Zhao, Z., Xu, H., & Zhao, T. (2024). Gene Mapping of a Yellow-to-Lethal Mutation Based on Bulked-Segregant Analysis-Seq in Soybean. Agronomy, 14(1), 185. https://doi.org/10.3390/agronomy14010185